Papillary Tumor of the Pineal Region Identified by DNA Methylation Leads to the Incidental Finding of Germline Mutation PTEN G132D Associated with PTEN Hamartoma Tumor Syndrome: A Case Report and Systematic Review

 , and
, and 
Abstract
1. Introduction
2. Background on PTPRs and PHTS
3. Materials and Methods
3.1. Patient Sample Acquisition, Imaging, and Characterization
3.2. PTPR Literature Review
4. Results
4.1. Systematic Review of the Literatire Since 2020
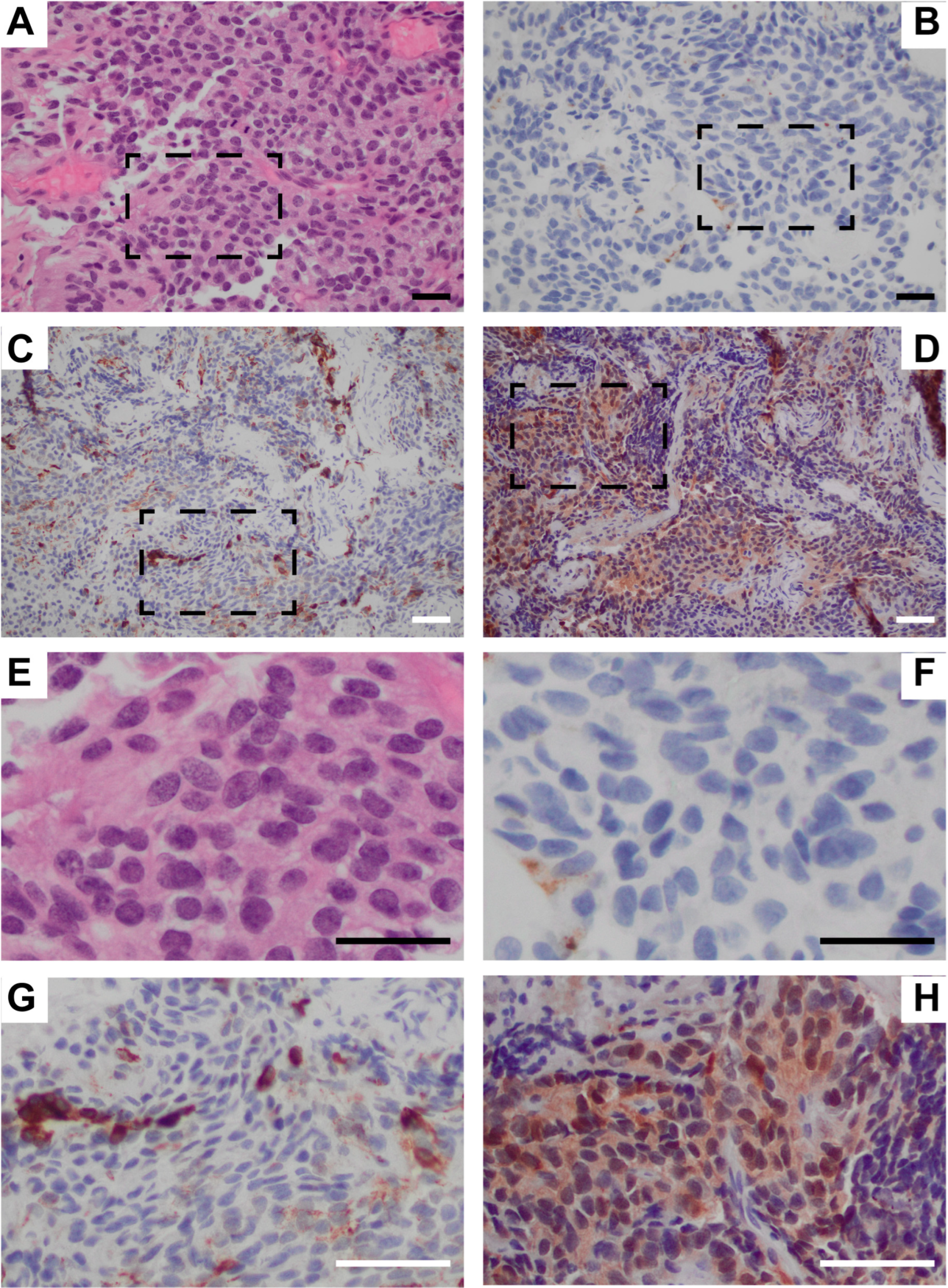
4.2. Detailed Case Report
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Karimi, S.; Zuccato, J.A.; Mamatjan, Y.; Mansouri, S.; Suppiah, S.; Nassiri, F.; Diamandis, P.; Munoz, D.G.; Aldape, K.D.; Zadeh, G. The central nervous system tumor methylation classifier changes neuro-oncology practice for challenging brain tumor diagnoses and directly impacts patient care. Clin. Epigenetics 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Heim, S.; Sill, M.; Jones, D.T.; Vasiljevic, A.; Jouvet, A.; Fèvre-Montange, M.; Wesseling, P.; Beschorner, R.; Mittelbronn, M.; Kohlhof, P.; et al. Papillary Tumor of the Pineal Region: A Distinct Molecular Entity. Brain Pathol. 2016, 26, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Jouvet, A.; Fauchon, F.; Liberski, P.; Saint-Pierre, G.; Didier-Bazes, M.; Heitzmann, A.; Delisle, M.B.; Biassette, H.A.; Vincent, S.; Mikol, J.; et al. Papillary tumor of the pineal region. Am. J. Surg. Pathol. 2003, 27, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Fèvre-Montange, M.; Hasselblatt, M.; Figarella-Branger, D.; Chauveinc, L.; Champier, J.; Saint-Pierre, G.; Taillandier, L.; Coulon, A.; Paulus, W.; Fauchon, F.; et al. Prognosis and histopathologic features in papillary tumors of the pineal region: A retrospective multicenter study of 31 cases. J. Neuropathol. Exp. Neurol. 2006, 65, 1004–1011. [Google Scholar] [CrossRef]
- Mobark, N.A.; Alharbi, M.; Alotabi, F.; Alshoumer, A.; Al Shakweer, W.; AlNaqib, Z.G.; AlSaad, A.N.; Balbaid, A.O.; Alsolme, E.; Abedalthagafi, M.S. Papillary Tumor of the Pineal Region Rare Pediatric CNS Tumor Case Series Treated in King Fahad Medical City (KFMC). Curr. Oncol. 2022, 29, 7558–7568. [Google Scholar] [CrossRef]
- Lancia, A.; Becherini, C.; Detti, B.; Bottero, M.; Baki, M.; Cancelli, A.; Ferlosio, A.; Scoccianti, S.; Sun, R.; Livi, L.; et al. Radiotherapy for papillary tumor of the pineal region: A systematic review of the literature. Clin. Neurol. Neurosurg. 2020, 190, 105646. [Google Scholar] [CrossRef]
- Assi, H.I.; Kakati, R.T.; Berro, J.; Saikali, I.; Youssef, B.; Hourany, R.; Alameh, I.; Tabbarah, A.; Khoury, J.; Darwish, H.; et al. PTEN R130Q Papillary Tumor of the Pineal Region (PTPR) with Chromosome 10 Loss Successfully Treated with Everolimus: A Case Report. Curr. Oncol. 2021, 28, 1274–1279. [Google Scholar] [CrossRef]
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN mutations and activation of the PI3K/Akt/mTOR signaling pathway in papillary tumors of the pineal region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751. [Google Scholar] [CrossRef]
- Wu, Z.; Dazelle, K.; Abdullaev, Z.; Chung, H.J.; Dahiya, S.; Wood, M.; Lee, H.; Lucas, C.G.; Mao, Q.; Robinson, L.; et al. Papillary tumor of the pineal region: Analysis of DNA methylation profiles and clinical outcomes in 76 cases. Acta Neuropathol. Commun. 2024, 12, 117. [Google Scholar] [CrossRef]
- Cho, M.; Ahn, S.; Hong, M.; Bang, H.; Van Vrancken, M.; Kim, S.; Lee, J.; Hoon Park, S.; Oh Park, J.; Suk Park, Y.; et al. Tissue recommendations for precision cancer therapy using next generation sequencing: A comprehensive single cancer center’s experiences. Oncotarget 2017, 8, 42478. [Google Scholar] [CrossRef] [PubMed]
- Poulgrain, K.; Gurgo, R.; Winter, C.; Ong, B.; Lau, Q. Papillary tumour of the pineal region. J. Clin. Neurosci. 2011, 18, 1007–1017. [Google Scholar] [CrossRef]
- Matyja, E.; Grajkowska, W.; Nauman, P.; Bonicki, W. Histopathological patterns of papillary tumour of the pineal region. Folia Neuropathol. 2011, 49, 181–190. [Google Scholar] [PubMed]
- Fèvre Montange, M.; Vasiljevic, A.; Champier, J.; Jouvet, A. Papillary tumor of the pineal region: Histopathological characterization and review of the literature. Neurochirurgie 2015, 61, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Poliani, P.L.; Manara, R.; Berhouma, M.; Minniti, G.; Tabouret, E.; Razis, E.; Cerretti, G.; Zagonel, V.; Weller, M.; et al. Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview. Cancers 2022, 14, 3646. [Google Scholar] [CrossRef]
- Hasselblatt, M.; Blümcke, I.; Jeibmann, A.; Rickert, C.H.; Jouvet, A.; van de Nes, J.A.; Kuchelmeister, K.; Brunn, A.; Fevre-Montange, M.; Paulus, W. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Neuropathol. Appl. Neurobiol. 2006, 32, 278–283. [Google Scholar] [CrossRef]
- Coy, S.; Dubuc, A.M.; Dahiya, S.; Ligon, K.L.; Vasiljevic, A.; Santagata, S. Nuclear CRX and FOXJ1 Expression Differentiates Non-Germ Cell Pineal Region Tumors and Supports the Ependymal Differentiation of Papillary Tumor of the Pineal Region. Am. J. Surg. Pathol. 2017, 41, 1410–1421. [Google Scholar] [CrossRef]
- Bora, S.; Santhoor, H.A.; Kumar, A.; Das, S.; Sharma, M.C.; Mishra, S.; Singh, P.K.; Laythalling, R.K.; Kale, S.S. Papillary Tumors of Pineal Region: A Single-Center Experience in Management of 11 Cases. World Neurosurg. 2024, 184, e486–e493. [Google Scholar] [CrossRef]
- Li, J.; Recinos, P.F.; Orr, B.A.; Burger, P.C.; Jallo, G.I.; Recinos, V.R. Papillary tumor of the pineal region in a 15-month-old boy. J. Neurosurg. Pediatr. 2011, 7, 534–538. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, W.H. PTEN: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. International Statistical Classification of Diseases and Related Health Problems (ICD). Available online: https://www.who.int/standards/classifications/classification-of-diseases (accessed on 31 January 2025).
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mester, J.; Eng, C. Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet. Med. 2012, 14, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Fernandez-Trujillo, L.; Castrillón, A.I.; Morales, E.I.; Diaz, Y.; Sua, L.F. Severe Central Airway Obstruction Secondary to a Giant Endobronchial Hamartoma: A Case Report. J. Investig. Med. High Impact. Case Rep. 2023, 11, 23247096231158951. [Google Scholar] [CrossRef]
- Gustafson, S.; Zbuk, K.M.; Scacheri, C.; Eng, C. Cowden syndrome. Semin. Oncol. 2007, 34, 428–434. [Google Scholar] [CrossRef]
- Yehia, L.; Plitt, G.; Tushar, A.M.; Joo, J.; Burke, C.A.; Campbell, S.C.; Heiden, K.; Jin, J.; Macaron, C.; Michener, C.M.; et al. Longitudinal Analysis of Cancer Risk in Children and Adults with Germline PTEN Variants. JAMA Netw. Open 2023, 6, e239705. [Google Scholar] [CrossRef]
- Kennedy, G.; Degueure, A.; Dai, M.; Cuevas-Ocampo, A.; Arevalo, O. An Unusual Finding: Papillary Tumor of the Pineal Region. Cureus 2023, 15, e34725. [Google Scholar] [CrossRef]
- Mehta, N.; Gupta, G.; Shaikh, S. Recurrent papillary tumor of pineal region misdiagnosed as pineocytoma 9-years ago. Asian J. Neurosurg. 2021, 16, 398–401. [Google Scholar] [CrossRef]
- Nemir, J.; Maric, L.S.; Trbojevic, T.; Zarkovic, K.; Jadrijević-Cvrlje, F. Papillary tumor of the pineal region in pediatric patient—A case report. Surg. Neurol. Int. 2022, 13, 488. [Google Scholar] [CrossRef]
- Gupta, K.; Khursheed, S.; Nayil, K.; Khursheed, S.; Makhdoomi, R. Papillary Tumor of Pineal Region in a 5-Year-Old Male Child: A Rare Entity. Asian J. Neurosurg. 2021, 16, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Damgacı, L.; Hayat, B.; Güreşçi, S. Papillary Tumor of the Pineal Region with Parinaud Syndrome: A Case Report. Curr. Med. Imaging 2020, 16, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Bechri, H.; Oudrhiri, M.Y.; Louraoui, S.M.; Melhaoui, A.; Sefiani, S.; Arkha, Y.; El Ouahabi, A. Papillary tumor of the pineal region: Is stereotactic radiosurgery efficient for this rare entity? Surg. Neurol. Int. 2021, 12, 386. [Google Scholar] [CrossRef]
- Schwartz, S.N.; Stamer, D.K.; Jiles, E.; Varma, H.; Vega, R.A.; White, B. DNA methylation profiling in a case of papillary tumor of the pineal region. J. Neuropathol. Exp. Neurol. 2025, 84, 158–160. [Google Scholar] [CrossRef]
- Marfia, G.; Ampollini, A.; Navone, S.E.; Di Vito, C.; Bornati, A.; Miozzo, M.; De Rezende, G.; Rampini, P.; Riboni, L.; Mancuso, M.E.; et al. Increased VEGF levels in a case of papillary tumor of the pineal region with intracranial hemorrhage: A potential surrogate indicator of tumor angiogenesis and aggressiveness? J. Neurosurg. Sci. 2020, 64, 107–112. [Google Scholar] [CrossRef]
- Dhandapani, S.; Gendle, C. Supracerebellar Infratentorial Keyhole Approach in Sitting Position Using 3-Dimensional Exoscope and Angled Endoscope for a Giant Pineal Tumor. World Neurosurg. 2024, 190, 141. [Google Scholar] [CrossRef]
- Bromfield, M.; Profyris, C.; Mehtar, A.; Du Toit, M.; Wadee, R. Papillary tumour of the pineal region: A case report. Egypt. J. Neurol. Psychiatry Neurosurg. 2020, 56, 55. [Google Scholar] [CrossRef]
- Gupta, R.K.; Batra, V.V.; Srivastava, A.K.; Sharma, M.C. Papillary Tumor of the Pineal Region: A Case with Unique Immunohistochemical Keratin Expression Pattern. Indian J. Med. Paediatr. Oncol. 2020, 41, 412–414. [Google Scholar] [CrossRef]
- Anvari, K.; Rabiei, P.; Hashemian, H.; Farajirad, M.; Arastouei, S.; Pishevar Feizabad, Z. Papillary Tumor of the Pineal Region with Leptomeningeal Seeding: A Case Report and Literature Review. Middle East J. Cancer 2024, 15, 153–160. [Google Scholar] [CrossRef]
- Mathkour, M.; Hanna, J.; Ibrahim, N.; Scullen, T.; Kilgore, M.D.; Werner, C.; Cormier, I.; Spencer, P.; Keen, J.R.; Bui, C.J. Papillary tumor of the pineal region in pediatric populations: An additional case and systematic review of a rare tumor entity. Clin. Neurol. Neurosurg. 2021, 201, 106404. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, A.I.; Alanezi, T.; Aljofan, Z.F.; Alarabi, A.; Elwatidy, S. Lhermitte-Duclos disease: A systematic review. Surg. Neurol. Int. 2023, 14, 351. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Author, Year | Age | M/F | HCP | DX | TX | Outcome(s) |
|---|---|---|---|---|---|---|
| Kennedy et al., 2023 [31] | 61 | F | Yes | Imaging, histology, IHC, chromosomal microarray | Surgery×2 (STR, GTR), shunt | Residual/ recurrence; GTR, Stable 1 year post-op after 2nd surgery |
| * Mehta et al., 2021 [32] | 29 | M | Yes | Imaging, histology, IHC | Surgery×2 (STR), shunt | Tumor progression seen 6 months post-op after 2nd surgery |
| Nemir et al., 2022 [33] | 9 | F | Yes | Imaging, histology, IHC, tumor markers | Surgery (GTR), ETV, PRT | Stable, no recurrences 78 months post-op |
| Gutpa et al., 2021 [34] | 5 | M | Yes | Imaging, histology, IHC | Surgery (GTR) | Not reported |
| Damgacı et al., 2020 [35] | 17 | F | Yes | Imaging, histology | Surgery (GTR) | Not reported |
| Bechri et al., 2021 [36] | 26 | F | Yes | Imaging, histology, IHC, tumor markers, cytology | SRS, ETV | Stable, 60% reduction in tumor volume 1 year post-op |
| * Assi et al., 2021 [8] | 25 | M | Yes | Imaging, histology, IHC, next generation sequencing | Surgery×3; 1st GTR, RT, everolimus, dexamethasone 2nd ETV 3rd GTR | Multiple brain masses in cerebellum and pineal region excised, 43 months stable on everolimus |
| Schwartz et al., 2025 [37] | 52 | F | Yes | Imaging, histology, DNA methylation | Surgery, ETV | Not reported |
| Marfia et al., 2020 [38] | 40 | M | Yes | Imaging, histology, IHC | Surgery, ETV, RT | Stable, reduced tumor size 20 months post-op |
| Dhandapani et al., 2024 [39] | 16 | F | Yes | Imaging, histology | Surgery (GTR), ETV, RT | Stable, no neurological deficits |
| Bromfield et al., 2020 [40] | 24 | M | Yes | Imaging, histology, IHC | Surgery (STR), ETV | Death at 3 months post-op, no post mortem exam |
| Gupta et al., 2020 [41] | 25 | M | Yes | Imaging, histology, IHC | Surgery | Death at 4 days post-op via cardiac arrest |
| Anvari et al., 2024 [42] | 46 | M | Yes | Imaging, histology, IHC | Shunt, surgery×2 (STR), vincristine, carboplatin, | 1st surgery; stable 14 months post-RT, 2nd surgery; disseminated disease, stable 6 months post-op after chemotherapy |
| Mathkour et al., 2021 [43] | 10 | F | Yes | Imaging, histology, IHC, cytology | Shunt after ETV failed, surgery (GTR) | Stable, no recurrences at 3 years post-op |
| Variable Analyzed | Values |
|---|---|
| Age | Mean: 27.5 years |
| Gender (M/F) | Ratio: 50/50 |
| Hydrocephalus | 100% |
| DX | Imaging: 100% Histology: 100% IHC: (3) 21.42% Cytology: (2) 14.29% Tumor markers: (2) 14.29% Molecular: (3) 21.42% |
| TX | Surgery: 93% >1 surgery: 28.57% Shunt: 28.57% ETV: 57.14% RT: 35.71% Chemotherapy/kinase inhibitor: 14.29% |
| Outcomes | Stable: 57.14% Ongoing/progressive disease: 35.71% Death: 14.29% Not reported: 21.42% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neal, N.; Goold, E.; Zarei Haji Abadi, F.; Okojie, J.; Barrott, J. Papillary Tumor of the Pineal Region Identified by DNA Methylation Leads to the Incidental Finding of Germline Mutation PTEN G132D Associated with PTEN Hamartoma Tumor Syndrome: A Case Report and Systematic Review. Curr. Oncol. 2025, 32, 172. https://doi.org/10.3390/curroncol32030172
O’Neal N, Goold E, Zarei Haji Abadi F, Okojie J, Barrott J. Papillary Tumor of the Pineal Region Identified by DNA Methylation Leads to the Incidental Finding of Germline Mutation PTEN G132D Associated with PTEN Hamartoma Tumor Syndrome: A Case Report and Systematic Review. Current Oncology. 2025; 32(3):172. https://doi.org/10.3390/curroncol32030172
Chicago/Turabian StyleO’Neal, Nikole, Eric Goold, Fatemeh Zarei Haji Abadi, Jeffrey Okojie, and Jared Barrott. 2025. "Papillary Tumor of the Pineal Region Identified by DNA Methylation Leads to the Incidental Finding of Germline Mutation PTEN G132D Associated with PTEN Hamartoma Tumor Syndrome: A Case Report and Systematic Review" Current Oncology 32, no. 3: 172. https://doi.org/10.3390/curroncol32030172
APA StyleO’Neal, N., Goold, E., Zarei Haji Abadi, F., Okojie, J., & Barrott, J. (2025). Papillary Tumor of the Pineal Region Identified by DNA Methylation Leads to the Incidental Finding of Germline Mutation PTEN G132D Associated with PTEN Hamartoma Tumor Syndrome: A Case Report and Systematic Review. Current Oncology, 32(3), 172. https://doi.org/10.3390/curroncol32030172