Targeting m6A RNA Modification in Tumor Therapeutics



Abstract
1. Introduction
2. Influence of m6A on mRNA Fate
3. Influence of m6A mRNA on Cell Fate
3.1. Influence of m6A mRNA on Normal Cell Fate
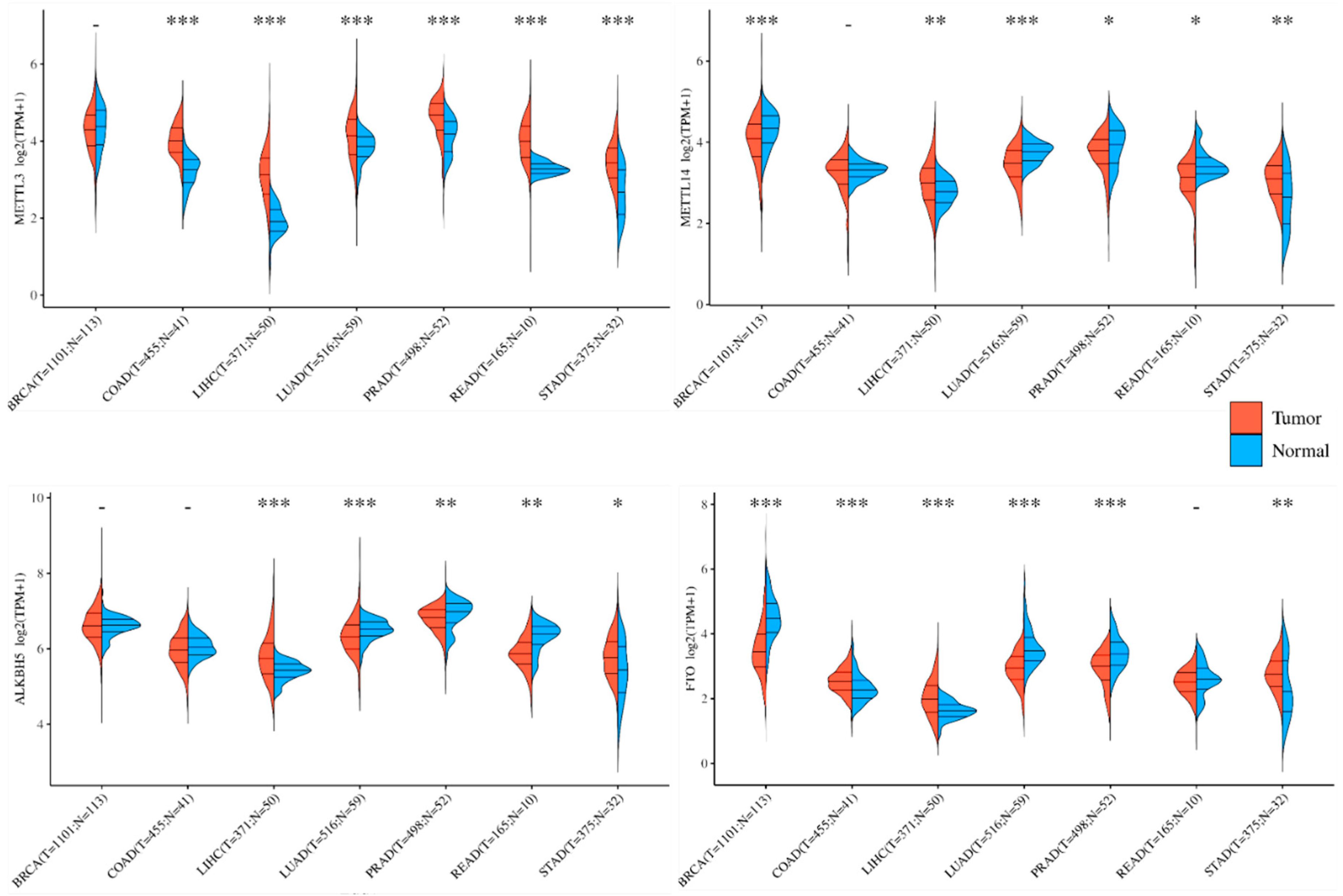
3.2. Influence of m6A mRNA on Tumor Cell Fate
4. Tumor Cell Fate via m6A mRNA Targeting
4.1. Effects of Decreasing m6A
4.2. Effects of Increasing m6A
4.3. Determiners of Targeting m6A mRNA in Tumors
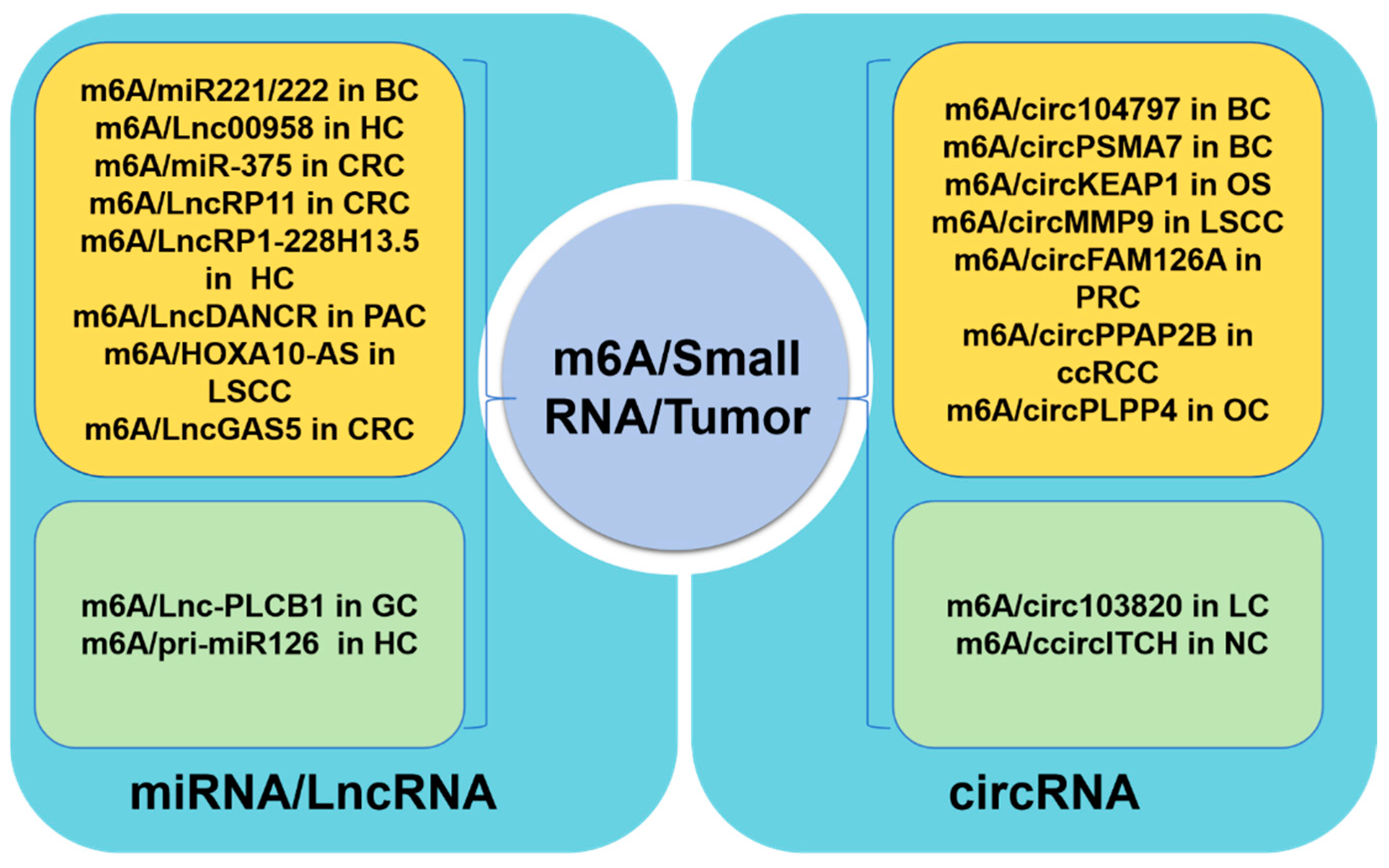
5. Contributions of m6A Non-Coding RNAs in Tumors
6. Perspectives on m6A RNA Targeting for Tumor Treatment
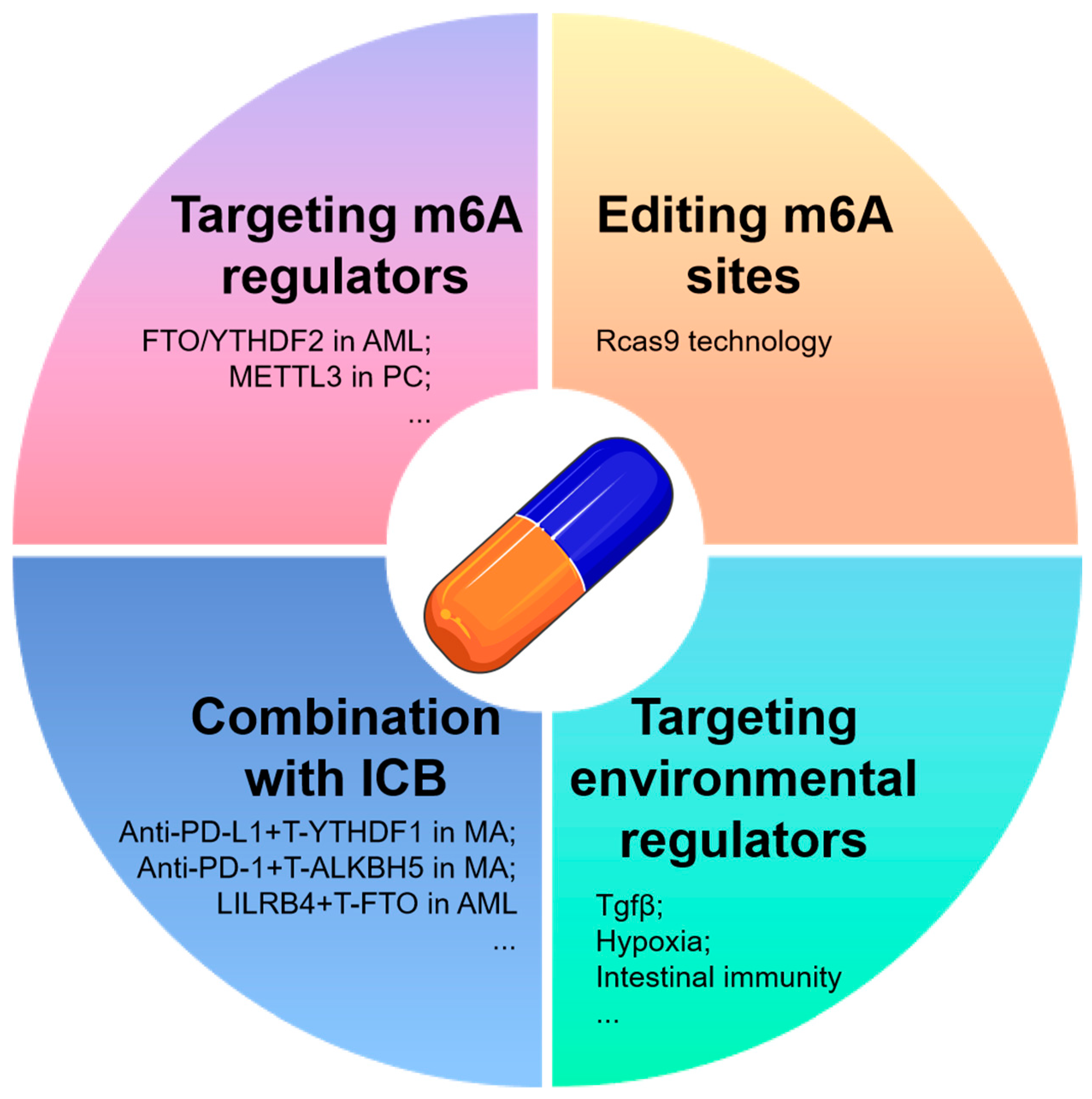
6.1. Strategies for Targeting m6A mRNA in Tumor Therapy
6.2. Clinical Trials and Drug Development for Tumor Therapy Targeting m6A
6.3. Challenges in the Development of Drugs Targeting m6A for Tumor Therapy
6.4. Causes of Heterogeneity of m6A Modification and Future Research Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Full Form/Description | Abbreviation | Full Form/Description |
| ALKBH5 | AlkB homolog 5 | NSCLC | Non-small-cell lung cancer |
| α-SMA | α-smooth muscle actin | OS | Osteosarcoma |
| AML | Acute myeloid leukemia | PC | Prostate cancer |
| BC | Bladder cancer | P2RX6 | Purinergic receptor P2X, ligand-gated ion channel 6 |
| BRCA | Breast cancer gene | PI3K-Akt | Phosphoinositide 3-kinase-Akt |
| CBX8 | Chromobox 8 | PDAC | Pancreatic ductal adenocarcinoma |
| CCNE1 | Cyclin E1 | PRAD | Prostate adenocarcinoma |
| CDK2 | Cyclin-dependent kinase 2 | PRC1 | Polycomb repressive complex 1 |
| COAD | Colon adenocarcinoma | RBM15 | RNA-binding motif protein 15 |
| CRC | Colorectal cancer | READ | Rectum adenocarcinoma |
| DCs | Dendritic cells | RCC | Renal cancer |
| EMT | Epithelial–mesenchymal transition | SRF | Serum response factor |
| ESCs | Embryonic stem cells | STAD | Stomach adenocarcinoma |
| FMRP | Fragile X mental retardation protein | TLR | Toll-like receptor |
| FTO | Fat mass and obesity-associated protein | Tregs | Regulatory T cells |
| GATA3 | GATA-binding protein 3 | USP7 | Ubiquitin-specific peptidase 7 |
| GC | Gastric cancer | WIF-1 | Wnt inhibitory factor 1 |
| GSCs | Glioblastoma stem cells | WTAP | Wilms tumor 1-associating protein |
| HB | Hepatoblastoma | Wnt | Wingless-related integration site |
| HCC | Hepatocellular carcinoma | YAP | Yes-associated protein |
| HDGF | Hepatoma-derived growth factor | YAP1 | Yes-associated protein 1 |
| HSPCs | Hematopoietic stem and progenitor cells | YTHDC1 | YTH domain containing 1 |
| HIF-1 | Hypoxia-inducible factor 1 | YTHDC2 | YTH domain containing 2 |
| HIF-1α | Hypoxia-inducible factor 1 alpha | YTHDF1 | YTH N6-methyladenosine RNA binding protein 1 |
| ICB | Immune checkpoint blockade | YTHDF2 | YTH N6-methyladenosine RNA binding protein 2 |
| IGF2BP1 | Insulin-like growth factor 2 mRNA binding protein 1 | YTHDF3 | YTH N6-methyladenosine RNA binding protein 3 |
| IGF2BP2 | Insulin-like growth factor 2 mRNA binding protein 2 | ZC3H13 | Zinc finger CCCH-type containing 13 |
| IGF2BP3 | Insulin-like growth factor 2 mRNA binding protein 3 | ccRCC | Clear-cell renal cell carcinoma |
| JAK/STAT | Janus kinase/signal transducer and activator of transcription | circRNA | Circular RNA |
| KIAA1429 | KIAA1429 gene | eIF3 | Eukaryotic translation initiation factor 3 |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 | m6A | N6-methyladenosine |
| LIHC | Liver hepatocellular carcinoma | mRNA | Messenger RNA |
| LUAD | Lung adenocarcinoma | mTOR | Mechanistic target of rapamycin |
| LUSC | Lung squamous cell carcinoma | miRNA | MicroRNA |
| LncRNA | Long non-coding RNA | ||
| LncXIST | Long non-coding RNA X-inactive specific transcript | ||
| MDSCs | Myeloid-derived suppressor cells | ||
| METTL14 | Methyltransferase-like protein 14 | ||
| METTL3 | Methyltransferase-like protein 3 | ||
| MYC | Myelocytomatosis oncogene | ||
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
References
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vagbo, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. MODOMICS: A database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Rottman, F.; Shatkin, A.J.; Perry, R.P. Sequences containing methylated nucleotides at the 5’ termini of messenger RNAs: Possible implications for processing. Cell 1974, 3, 197–199. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A.; Moss, B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 1975, 4, 379–386. [Google Scholar] [CrossRef]
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N6-Methyladenine DNA Modification in the Human Genome. Mol. Cell 2018, 71, 306–318.e307. [Google Scholar] [CrossRef]
- Ren, W.; Lu, J.; Huang, M.; Gao, L.; Li, D.; Wang, G.G.; Song, J. Structure and regulation of ZCCHC4 in mA-methylation of 28S rRNA. Nat. Commun. 2019, 10, 5042. [Google Scholar] [CrossRef]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e14. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hsu, P.J.; Chen, Y.S.; Yang, Y.G. Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018, 28, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 12626. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef]
- Zhou, K.I.; Shi, H.; Lyu, R.; Wylder, A.C.; Matuszek, Z.; Pan, J.N.; He, C.; Parisien, M.; Pan, T. Regulation of Co-transcriptional Pre-mRNA Splicing by m6A through the Low-Complexity Protein hnRNPG. Mol. Cell 2019, 76, 70–81.e79. [Google Scholar] [CrossRef]
- Ozkurede, U.; Kala, R.; Johnson, C.; Shen, Z.; Miller, R.A.; Garcia, G.G. Cap-independent mRNA translation is upregulated in long-lived endocrine mutant mice. J. Mol. Endocrinol. 2019, 63, 123–138. [Google Scholar] [CrossRef]
- Livneh, I.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G.; Dominissini, D. The m6A epitranscriptome: Transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 2020, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Shi, H.; Zhu, A.C.; Lu, Z.; Miller, N.; Edens, B.M.; Ma, Y.C.; He, C. The RNA-binding protein FMRP facilitates the nuclear export of N6-methyladenosine-containing mRNAs. J. Biol. Chem. 2019, 294, 19889–19895. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Dong, L.; Liu, X.M.; Guo, J.; Ma, H.; Shen, B.; Qian, S.B. m6A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 2019, 10, 5332. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5’ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.-B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Orouji, E.; Peitsch, W.K.; Orouji, A.; Houben, R.; Utikal, J. Oncogenic Role of an Epigenetic Reader of m6A RNA Modification: YTHDF1 in Merkel Cell Carcinoma. Cancers 2020, 12, 202. [Google Scholar] [CrossRef]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126. [Google Scholar] [CrossRef]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, Y.; Sun, B.; Wang, L.; Yang, Y.; Ma, D.; Lv, J.; Heng, J.; Ding, Y.; Xue, Y.; et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 2017, 549, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, Y.; Gao, S.; Zhang, C.; Chen, Y.; Li, W.; Yang, Y.G.; Zhou, Q.; Liu, F. Endothelial-specific m6A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018, 28, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Cirovic, B.; Schönheit, J.; Kowenz-Leutz, E.; Ivanovska, J.; Klement, C.; Pronina, N.; Bégay, V.; Leutz, A. C/EBP-Induced Transdifferentiation Reveals Granulocyte-Macrophage Precursor-like Plasticity of B Cells. Stem Cell Rep. 2017, 8, 346–359. [Google Scholar] [CrossRef]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef]
- Lin, Z.; Hsu, P.J.; Xing, X.; Fang, J.; Lu, Z.; Zou, Q.; Zhang, K.-J.; Zhang, X.; Zhou, Y.; Zhang, T.; et al. Mettl3-/Mettl14-mediated mRNA N-methyladenosine modulates murine spermatogenesis. Cell Res. 2017, 27, 1216–1230. [Google Scholar] [CrossRef]
- Xu, K.; Yang, Y.; Feng, G.H.; Sun, B.F.; Chen, J.Q.; Li, Y.F.; Chen, Y.S.; Zhang, X.X.; Wang, C.X.; Jiang, L.Y.; et al. Mettl3-mediated m6A regulates spermatogonial differentiation and meiosis initiation. Cell Res. 2017, 27, 1100–1114. [Google Scholar] [CrossRef]
- Ma, C.; Chang, M.; Lv, H.; Zhang, Z.-W.; Zhang, W.; He, X.; Wu, G.; Zhao, S.; Zhang, Y.; Wang, D.; et al. RNA m6A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. 2018, 19, 68. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Y.-C.; Huang, C.; Shen, H.; Sun, B.; Cheng, X.; Zhang, Y.-J.; Yang, Y.-G.; Shu, Q.; Yang, Y.; et al. m6A Regulates Neurogenesis and Neuronal Development by Modulating Histone Methyltransferase Ezh2. Genom. Proteom. Bioinform. 2019, 17, 154–168. [Google Scholar] [CrossRef]
- Xiang, Y.; Laurent, B.; Hsu, C.-H.; Nachtergaele, S.; Lu, Z.; Sheng, W.; Xu, C.; Chen, H.; Ouyang, J.; Wang, S.; et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 2017, 543, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Gao, Y.; Lv, D.; Wang, C.; Wang, D.; Li, Q. METTL3 mediated m6A modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating DeltaNp63. Biochem. Biophys. Res. Commun. 2019, 515, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Chen, J.; Jia, L.; Ma, J.; Song, D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem. Biophys. Res. Commun. 2019, 516, 719–725. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Si, Y.; Xu, J.; Lin, Y.; Wang, J.-Z.; Cao, M.; Sun, S.; Ding, Q.; Zhu, L.; Wei, J.-F. Methyltransferase like 3 promotes colorectal cancer proliferation by stabilizing CCNE1 mRNA in an m6A-dependent manner. J. Cell. Mol. Med. 2020, 24, 3521–3533. [Google Scholar] [CrossRef]
- Cheng, M.; Sheng, L.; Gao, Q.; Xiong, Q.; Zhang, H.; Wu, M.; Liang, Y.; Zhu, F.; Zhang, Y.; Zhang, X.; et al. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene 2019, 38, 3667–3680. [Google Scholar] [CrossRef]
- Liu, L.; Wang, J.; Sun, G.; Wu, Q.; Ma, J.; Zhang, X.; Huang, N.; Bian, Z.; Gu, S.; Xu, M.; et al. m6A mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol. Cancer 2019, 18, 188. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.-T.; Tsang, F.H.-C.; Shen, J.; Cheng, C.L.-H.; Tsang, L.-H.; Ho, D.W.-H.; Chiu, D.K.-C.; Lee, J.M.-F.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Lan, T.; Li, H.; Zhang, D.; Xu, L.; Liu, H.; Hao, X.; Yan, X.; Liao, H.; Chen, X.; Xie, K.; et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol. Cancer 2019, 18, 186. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Liu, T.; Yang, S.; Sui, J.; Xu, S.-Y.; Cheng, Y.-P.; Shen, B.; Zhang, Y.; Zhang, X.-M.; Yin, L.-H.; Pu, Y.-P.; et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J. Cell. Physiol. 2020, 235, 548–562. [Google Scholar] [CrossRef]
- Gong, D.; Zhang, J.; Chen, Y.; Xu, Y.; Ma, J.; Hu, G.; Huang, Y.; Zheng, J.; Zhai, W.; Xue, W. The m6A-suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP-induced Ca2+ influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Ge, S.; Huang, W.; Lin, X.; Gao, J.; Gong, J.; Shen, L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019, 8, 4766–4781. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677–691.E10. [Google Scholar] [CrossRef]
- Zou, D.; Dong, L.; Li, C.; Yin, Z.; Rao, S.; Zhou, Q. The m6A eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019, 19, 321. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Tang, B.; Yang, Y.; Kang, M.; Wang, Y.; Wang, Y.; Bi, Y.; He, S.; Shimamoto, F. m6A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 2020, 19, 3. [Google Scholar] [CrossRef]
- Han, J.; Wang, J.-Z.; Yang, X.; Yu, H.; Zhou, R.; Lu, H.-C.; Yuan, W.-B.; Lu, J.-C.; Zhou, Z.-J.; Lu, Q.; et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 2019, 18, 110. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, S.; Zhao, T.; Dang, C. METTL3-mediated m6A modification of Bcl-2 mRNA promotes non-small cell lung cancer progression. Oncol. Rep. 2021, 46, 163. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, X.; Qian, F.; Zhu, Y.; He, G.; Yang, J.; Wu, X.; Zhang, H.; Yu, X.; Liu, X. Deubiquitinase USP19 modulates apoptotic calcium release and endoplasmic reticulum stress by deubiquitinating BAG6 in triple negative breast cancer. Clin. Transl. Med. 2023, 13, e1398. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, B.; Shi, J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene 2020, 722, 144076. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Xie, X.; Han, G.; Zhang, T.; Li, Y.; Li, Y.; Yin, R.; Wang, Q.; Zhang, T.; Wang, P.; et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m6A-dependent manner. Blood 2021, 138, 71–85. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, B.; Hu, X.; Ying, S.; Zhou, Q.; Xu, W.; Feng, L.; Hou, T.; Wang, X.; Zhu, L.; et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin. Transl. Med. 2022, 12, e703. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Jiang, X.; Li, D.; Jiang, X. HBXIP promotes gastric cancer via METTL3-mediated MYC mRNA m6A modification. Aging 2020, 12, 24967–24982. [Google Scholar] [CrossRef]
- Aziz, K.; Limzerwala, J.F.; Sturmlechner, I.; Hurley, E.; Zhang, C.; Jeganathan, K.B.; Nelson, G.; Bronk, S.; Fierro Velasco, R.O.; van Deursen, E.J.; et al. Ccne1 Overexpression Causes Chromosome Instability in Liver Cells and Liver Tumor Development in Mice. Gastroenterology 2019, 157, 210–226.e212. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Q.; Li, Y.; Wang, G.; Gao, S.; Zhang, X.; Yan, X.; Zhang, X.; Xie, J.; Wang, Y.; et al. Metformin targets a YAP1-TEAD4 complex via AMPKalpha to regulate CCNE1/2 in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 376. [Google Scholar] [CrossRef]
- Jin, D.; Guo, J.; Wu, Y.; Yang, L.; Wang, X.; Du, J.; Dai, J.; Chen, W.; Gong, K.; Miao, S.; et al. m6A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol. Cancer 2020, 19, 40. [Google Scholar] [CrossRef]
- Wei, Y.; Fu, J.; Zhang, H.; Ling, Y.; Tang, X.; Liu, S.; Yu, M.; Liu, F.; Zhuang, G.; Qian, H.; et al. N6-methyladenosine modification promotes hepatocarcinogenesis through circ-CDYL-enriched and EpCAM-positive liver tumor-initiating exosomes. iScience 2023, 26, 108022. [Google Scholar] [CrossRef]
- Müller, S.; Glaß, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019, 47, 375–390. [Google Scholar] [CrossRef]
- Tanabe, A.; Tanikawa, K.; Tsunetomi, M.; Takai, K.; Ikeda, H.; Konno, J.; Torigoe, T.; Maeda, H.; Kutomi, G.; Okita, K.; et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1alpha mRNA is translated. Cancer Lett. 2016, 376, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, F.; Zhan, H.; Situ, J.; Li, W.; Mao, Y.; Luo, Y. RNA m6A Methyltransferase METTL3 Promotes The Growth of Prostate Cancer By Regulating Hedgehog Pathway. OncoTargets Ther. 2019, 12, 9143–9152. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, C.; Sun, Y.; He, X.; Xue, D. m6A RNA modification modulates gene expression and cancer-related pathways in clear cell renal cell carcinoma. Epigenomics 2019, 12, 87–99. [Google Scholar] [CrossRef]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.-G.; et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- Marceau, A.H.; Brison, C.M.; Nerli, S.; Arsenault, H.E.; McShan, A.C.; Chen, E.; Lee, H.W.; Benanti, J.A.; Sgourakis, N.G.; Rubin, S.M. An order-to-disorder structural switch activates the FoxM1 transcription factor. elife 2019, 8, e46131. [Google Scholar] [CrossRef]
- Chao, Y.; Shang, J.; Ji, W. ALKBH5-m6A-FOXM1 signaling axis promotes proliferation and invasion of lung adenocarcinoma cells under intermittent hypoxia. Biochem. Biophys. Res. Commun. 2020, 521, 499–506. [Google Scholar] [CrossRef]
- Li, J.; Han, Y.; Zhang, H.; Qian, Z.; Jia, W.; Gao, Y.; Zheng, H.; Li, B. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem. Biophys. Res. Commun. 2019, 512, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.-M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Kong, B.; Song, C.; Cong, J.; Hou, J.; Wang, S. Reduced m6A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget 2017, 8, 98918–98930. [Google Scholar] [CrossRef]
- Li, Q.; Ni, Y.; Zhang, L.; Jiang, R.; Xu, J.; Yang, H.; Hu, Y.; Qiu, J.; Pu, L.; Tang, J.; et al. HIF-1α-induced expression of m6A reader YTHDF1 drives hypoxia-induced autophagy and malignancy of hepatocellular carcinoma by promoting ATG2A and ATG14 translation. Signal Transduct. Target. Ther. 2021, 6, 76. [Google Scholar] [CrossRef]
- Zhang, C.; Samanta, D.; Lu, H.; Bullen, J.W.; Zhang, H.; Chen, I.; He, X.; Semenza, G.L. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m⁶A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E2047–E2056. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Zhang, Y.; Wang, Y.; Luo, Y.; Ding, H.; Li, P.; Ni, G. HIF-1α Regulated WTAP Overexpression Promoting the Warburg Effect of Ovarian Cancer by m6A-Dependent Manner. J. Immunol. Res. 2022, 2022, 6130806. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheng, C.; He, B.; Du, X.; Liu, J.; Xia, H.; Wang, P.; Wu, M.; Wu, H.; Liu, Q. Cigarette smoking, by accelerating the cell cycle, promotes the progression of non-small cell lung cancer through an HIF-1α-METTL3-m6A/CDK2AP2 axis. J. Hazard. Mater. 2023, 455, 131556. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, J.; Huang, Z.; Jin, P.; Peng, L.; Luo, M.; Zhang, Z.; Chen, Y.; Xie, N.; Gao, W.; et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m6A-mediated degradation of STEAP3 mRNA. Mol. Cancer 2022, 21, 168. [Google Scholar] [CrossRef]
- Xiong, J.; He, J.; Zhu, J.; Pan, J.; Liao, W.; Ye, H.; Wang, H.; Song, Y.; Du, Y.; Cui, B.; et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell 2022, 82, 1660–1677.E10. [Google Scholar] [CrossRef]
- Yu, J.; Chai, P.; Xie, M.; Ge, S.; Ruan, J.; Fan, X.; Jia, R. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021, 22, 85. [Google Scholar] [CrossRef]
- Wang, P.; Xie, D.; Xiao, T.; Cheng, C.; Wang, D.; Sun, J.; Wu, M.; Yang, Y.; Zhang, A.; Liu, Q. H3K18 lactylation promotes the progression of arsenite-related idiopathic pulmonary fibrosis via YTHDF1/m6A/NREP. J. Hazard. Mater. 2024, 461, 132582. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef]
- Du, F.; Yuan, P.; Wang, T.; Zhao, J.; Zhao, Z.; Luo, Y.; Xu, B. The Significance and Therapeutic Potential of GATA3 Expression and Mutation in Breast Cancer: A Systematic Review. Med. Res. Rev. 2015, 35, 1300–1315. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Chen, J.; Chen, D.; Yang, B.; He, B.; Hu, W.; Zhang, Y.; Liu, H.; Dai, L.; et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer 2019, 18, 127. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; He, C. Fate by RNA methylation: m6A steers stem cell pluripotency. Genome Biol. 2015, 16, 43. [Google Scholar] [CrossRef] [PubMed]
- Roignant, J.Y.; Soller, M. m6A in mRNA: An Ancient Mechanism for Fine-Tuning Gene Expression. Trends Genet. TIG 2017, 33, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Zuo, X.; Chen, Z.; Gao, W.; Zhang, Y.; Wang, J.; Wang, J.; Cao, M.; Cai, J.; Wu, J.; Wang, X. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J. Hematol. Oncol. 2020, 13, 5. [Google Scholar] [CrossRef]
- Xu, J.; Liu, C.; Qu, K.; Zhang, J.; Liu, S.; Meng, F.; Wan, Y. m6A methyltransferase METTL14-mediated RP1-228H13.5 promotes the occurrence of liver cancer by targeting hsa-miR-205/ZIK1. Oncol. Rep. 2024, 51, 59. [Google Scholar] [CrossRef]
- Ni, W.; Yao, S.; Zhou, Y.; Liu, Y.; Huang, P.; Zhou, A.; Liu, J.; Che, L.; Li, J. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m6A reader YTHDF3. Mol. Cancer 2019, 18, 143. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Chen, Z.; Tian, L.; Jiang, G.; Chen, F.; Li, J.; An, P.; Lu, L.; Luo, N.; et al. m6A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer 2019, 18, 87. [Google Scholar] [CrossRef]
- Hu, X.; Peng, W.-X.; Zhou, H.; Jiang, J.; Zhou, X.; Huang, D.; Mo, Y.-Y.; Yang, L. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2019, 27, 1782–1794. [Google Scholar] [CrossRef]
- Zhao, K.; Chen, L.; Xie, Y.; Ren, N.; Li, J.; Zhai, X.; Zheng, S.; Liu, K.; Wang, C.; Qiu, Q.; et al. m6A/HOXA10-AS/ITGA6 axis aggravates oxidative resistance and malignant progression of laryngeal squamous cell carcinoma through regulating Notch and Keap1/Nrf2 pathways. Cancer Lett. 2024, 587, 216735. [Google Scholar] [CrossRef]
- Zhang, L.; Hou, C.; Chen, C.; Guo, Y.; Yuan, W.; Yin, D.; Liu, J.; Sun, Z. The role of N6-methyladenosine (m6A) modification in the regulation of circRNAs. Mol. Cancer 2020, 19, 105. [Google Scholar] [CrossRef]
- Li, H.; Lin, R.; Zhang, Y.; Zhu, Y.; Huang, S.; Lan, J.; Lu, N.; Xie, C.; He, S.; Zhang, W. N6-methyladenosine-modified circPLPP4 sustains cisplatin resistance in ovarian cancer cells via PIK3R1 upregulation. Mol. Cancer 2024, 23, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhou, J.; Zhang, X.; Kang, X.; Liu, S.; Song, M.; Chang, C.; Lin, Y.; Wang, Y. N6-methyladenosine-modified circ_104797 sustains cisplatin resistance in bladder cancer through acting as RNA sponges. Cell Mol. Biol. Lett. 2024, 29, 28. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Ma, X.; Ying, Y.; Liu, Z.; Tang, Y.; Shu, X.; Sun, J.; Wu, Y.; Lu, D.; Wang, X.; et al. N6-methyladenosine-modified CircPSMA7 enhances bladder cancer malignancy through the miR-128-3p/MAPK1 axis. Cancer Lett. 2024, 585, 216613. [Google Scholar] [CrossRef]
- Zheng, Z.; Zeng, X.; Zhu, Y.; Leng, M.; Zhang, Z.; Wang, Q.; Liu, X.; Zeng, S.; Xiao, Y.; Hu, C.; et al. CircPPAP2B controls metastasis of clear cell renal cell carcinoma via HNRNPC-dependent alternative splicing and targeting the miR-182-5p/CYP1B1 axis. Mol. Cancer 2024, 23, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yao, L.; Su, Y.; Tian, L. IGF2BP3 loss inhibits cell progression by upregulating has_circRNA_103820, and hsa_circRNA_103820-encoded peptide inhibits cell progression by inactivating the AKT pathway in lung cancer. Chem. Biol. Drug Des. 2024, 103, e14473. [Google Scholar] [CrossRef]
- Li, J.; Cao, H.; Yang, J.; Wang, B. IGF2BP2-m6A-circMMP9 axis recruits ETS1 to promote TRIM59 transcription in laryngeal squamous cell carcinoma. Sci. Rep. 2024, 14, 3014. [Google Scholar] [CrossRef]
- Zhou, Q.; Song, W.; Li, X.; Lin, J.; Zhu, C.; Cao, L.; Li, W.; Lin, S. N6-Methyladenosine reader HNRNPC-mediated downregulation of circITCH prevents miR-224-3p sequestering and contributes to tumorigenesis in nasopharyngeal carcinoma. Environ. Toxicol. 2024, 39, 2893–2907. [Google Scholar] [CrossRef]
- Luo, L.; Li, P.; Xie, Q.; Wu, Y.; Qin, F.; Liao, D.; Zeng, K.; Wang, K. n6-methyladenosine-modified circular RNA family with sequence similarity 126, member A affects cholesterol synthesis and malignant progression of prostate cancer cells by targeting microRNA-505-3p to mediate calnexin. J. Cancer 2024, 15, 966–980. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, K.; Shou, Y.; Li, S.; Wang, J.; Zhang, Q.; Huang, Z.; Xu, J.; Li, M.; Liu, D.; et al. circRARS synergises with IGF2BP3 to regulate RNA methylation recognition to promote tumour progression in renal cell carcinoma. Clin. Transl. Med. 2023, 13, e1512. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.; Morgan, M.; Campos, J.; Spencer, G.J.; Shmakova, A.; Ivanova, I.; Mapperley, C.; Lawson, H.; Wotherspoon, D.A.; Sepulveda, C.; et al. Targeting the RNA m6A Reader YTHDF2 Selectively Compromises Cancer Stem Cells in Acute Myeloid Leukemia. Cell Stem Cell 2019, 25, 137–148.e136. [Google Scholar] [CrossRef]
- Taketo, K.; Konno, M.; Asai, A.; Koseki, J.; Toratani, M.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H.; Ogawa, K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018, 52, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/mA/MYC/CEBPA Signaling. Cell 2018, 172, 90–105.E23. [Google Scholar] [CrossRef] [PubMed]
- Rau, K.; Roesner, L.; Rentmeister, A. Sequence-specific m6A demethylation in RNA by FTO fused to RCas9. RNA 2019, 25, 1311–1323. [Google Scholar] [CrossRef]
- Liu, Q.; Li, M.; Jiang, L.; Jiang, R.; Fu, B. METTL3 promotes experimental osteoarthritis development by regulating inflammatory response and apoptosis in chondrocyte. Biochem. Biophys. Res. Commun. 2019, 516, 22–27. [Google Scholar] [CrossRef]
- Jia, R.; Chai, P.; Wang, S.; Sun, B.; Xu, Y.; Yang, Y.; Ge, S.; Jia, R.; Yang, Y.G.; Fan, X. m6A modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol. Cancer 2019, 18, 161. [Google Scholar] [CrossRef]
- Hou, J.; Zhang, H.; Liu, J.; Zhao, Z.; Wang, J.; Lu, Z.; Hu, B.; Zhou, J.; Zhao, Z.; Feng, M.; et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol. Cancer 2019, 18, 163. [Google Scholar] [CrossRef]
- Bertero, A.; Brown, S.; Madrigal, P.; Osnato, A.; Ortmann, D.; Yiangou, L.; Kadiwala, J.; Hubner, N.C.; de Los Mozos, I.R.; Sadee, C.; et al. The SMAD2/3 interactome reveals that TGFbeta controls m6A mRNA methylation in pluripotency. Nature 2018, 555, 256–259. [Google Scholar] [CrossRef]
- Liao, Z.; Zhang, H.; Liu, F.; Wang, W.; Liu, Y.; Su, C.; Zhu, H.; Chen, X.; Zhang, B.; Zhang, Z. m6A-Dependent ITIH1 Regulated by TGF-β Acts as a Target for Hepatocellular Carcinoma Progression. Adv. Sci. 2024, 11, e2401013. [Google Scholar] [CrossRef]
- Wu, Z.; Ke, Q.; Jiang, L.; Hong, H.; Pan, W.; Chen, W.; Abudukeremu, X.; She, F.; Chen, Y. TGF-β1 facilitates gallbladder carcinoma metastasis by regulating FOXA1 translation efficiency through m6A modification. Cell Death Dis. 2024, 15, 422. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Yu, B.; Tao, D.; Xu, X.; Xu, Y.; Wang, J.; Jiao, Y.; Wang, L. The role of m6A methylation in therapy resistance in cancer. Mol. Cancer 2023, 22, 91. [Google Scholar] [CrossRef]
- Lin, H.; Wang, Y.; Wang, P.; Long, F.; Wang, T. Mutual regulation between N6-methyladenosine (m6A) modification and circular RNAs in cancer: Impacts on therapeutic resistance. Mol. Cancer 2022, 21, 148. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96.E11. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, H.; Chen, J.; Lin, L.; Chen, Y. Immune signature of T follicular helper cells predicts clinical prognostic and therapeutic impact in lung squamous cell carcinoma. Int. Immunopharmacol. 2020, 81, 105932. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Ronnblom, L.; Eloranta, M.L.; Alm, G.V. Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity 2003, 36, 463–472. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Lluch, S.; Blanco, E.; Tilgner, H.; Curado, J.; Ruiz-Romero, M.; Corominas, M.; Guigó, R. Absence of canonical marks of active chromatin in developmentally regulated genes. Nat. Genet. 2015, 47, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Smith, A.R.; Qian, Z.; Zheng, G. Patent landscape of small molecule inhibitors of METTL3 (2020-present). Expert. Opin. Ther. Pat. 2024, 35, 305–320. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m6A Enzyme | Nucleic Acid Type | Ref. |
|---|---|---|
| N6AMT1 (Me) ALKBH1 (D-me) | DNA | [8] |
| ZCCHC4 (Me) | 28S rRNA | [9] |
| METTL5 | 18S rRNA | [10] |
| METTL16 | snRNA | [11] |
| METTL3/METTL14 (Me) | mRNA | [12] |
| ALKBH5/FTO (D-me) | [13] |
| Effect | Enzyme | Disease | Organ | Genes/Signaling | Ref. |
|---|---|---|---|---|---|
| Promote | METTL3 ↑ | Osteosarcoma (OS) | Bone | LEF1↑ and the activity of Wnt/β-catenin signaling pathway ↑ | [44] |
| Promote | METTL3 ↑ | Colorectal cancer (CRC) | Colorectum | Cyclin E1 ↑ | [45] |
| Promote | METTL3 ↑ | Gastric cancer (GC) | Stomach | P300-H3K27 acetylation ↑- METTL3 ↑- HDGF stability ↑ (IGF2BP3)- GLUT4 and ENO2 ↑ | [50] |
| Promote | METTL3 ↑ | GC | Stomach | GFI-1 (related to EMT) and α-SMA ↑ | [51] |
| Promote | METTL3 ↑ | Prostate cancer (PC) | Prostate | Hedgehog pathway | [72] |
| Promote | ALKBH5 ↓ | PC | Pancreas | WIF-1/ Wnt signaling ↑ | [58] |
| Promote | METTL3 ↑ | Bladder cancer (BC) | bladder | AFF4/NF-κB/MYC ↑ | [46] |
| Promote | METTL3 ↑ | Osteoarthritis | Joint | NF-kB signaling and ECM synthesis | [116] |
| Promote | METTL3 ↑ | Hepatoblastoma (HB) | Liver | Wnt/β-catenin pathway ↑ | [47] |
| Promote | METTL3 ↑ YTHDF2 | Hepatocellular carcinoma (HCC) | Liver | SOCS2/JAK/STAT pathway ↑ | [48] |
| Promote | KIAA1429↑ | GATA3 ↓ | [49] | ||
| Promote | YTHDF1 ↑ | Merkel cell carcinoma (MCC) | Skin | --- | [29] |
| Inhibit | METTL14 | Renal cancer (RCC) | Kidney | P2RX6/Ca2+/p-ERK1/2/MMP9 signaling ↓ | [52] |
| Inhibit | METTL3/14 FTO | Glioblastoma | Brain | Cell proliferation, differentiation, and DNA damage response | [74] |
| Inhibit | ALKBH5/FTO ↑ METTL3/14 ↓ YTHDF1-3 ↓ | GC | Stomach | Wnt and PI3K-Akt signaling ↑ | [53] |
| Inhibit | FTO ↑ | Acute myeloid leukemia (AML) | Bone | P53/apoptotic pathway ↓ | [54] |
| Inhibit | FTO ↑ | Cervical cancer | Cervix | E2F1 and MYC | [55] |
| Inhibit | YTHDF1 | Ocular melanoma | Uveal or conjunctival | HINT2 ↑ | [117] |
| Registration Number | Drug | Target | Indications | Stage | Country |
|---|---|---|---|---|---|
| NCT04989335 | CS1, fludarabine, clofarabine [126] | FTO | Relapsed or refractory AML | II | Israel |
| NCT05584111 | STC-15 [133] | METTL3 | AML, advanced solid tumor, hematological neoplasm | I | United States |
| NCT03384914 | pUMVC3-IGFBP2-HER2-IGF1R | IGFBP2 | Breast cancer | II | United States |
| NCT05794659 | AST-201 | IGFBP2 | Peritoneal neoplasm, ovarian neoplasm, fallopian tube cancer, metastatic ovarian cancer | II | United States |
| METTL3 inhibitor | METTL3 | AML | Preclinical | United States | |
| EP-102 | METTL3 | Non-small-cell lung cancer, acute myeloid leukemia, squamous cell carcinoma, ovarian tumor | Preclinical | Belgium | |
| Next-generation CS1 | FTO | Malignant neoplasm | Preclinical | Australia |
| Drug | Target | Molecular Formula | Indications | Country |
|---|---|---|---|---|
| MV-1035 | ALKBH5 | C14 H14 N2 O S | Glioblastoma | United States |
| RSM-3 | METTL3 | C124 H214 N54 O26 S3 | Malignant tumor | China |
| UZH-2 | METTL3 | C27 H37 F2 N7 O | Malignant tumor | Switzerland |
| UZH-1a | METTL3 | C32 H42 N6 O3 | AML | United States |
| STM-3480 | METTL3 | C24 H25 N5 O2 | Malignant tumor | Britain |
| D272-0843 | METTL3 | C21 H17 Cl F N3 O2 | Malignant tumor | China |
| EP-201 | IGFBP2 | -- | Metastatic ovarian cancer | United States |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Z.; Li, M.; Wang, S. Targeting m6A RNA Modification in Tumor Therapeutics. Curr. Oncol. 2025, 32, 159. https://doi.org/10.3390/curroncol32030159
Mao Z, Li M, Wang S. Targeting m6A RNA Modification in Tumor Therapeutics. Current Oncology. 2025; 32(3):159. https://doi.org/10.3390/curroncol32030159
Chicago/Turabian StyleMao, Zhenwei, Min Li, and Shengjun Wang. 2025. "Targeting m6A RNA Modification in Tumor Therapeutics" Current Oncology 32, no. 3: 159. https://doi.org/10.3390/curroncol32030159
APA StyleMao, Z., Li, M., & Wang, S. (2025). Targeting m6A RNA Modification in Tumor Therapeutics. Current Oncology, 32(3), 159. https://doi.org/10.3390/curroncol32030159