Practical Management of Adult Ultra-Rare Primary Retroperitoneal Soft Tissue Sarcoma: A Focus on Perivascular Epithelioid Tumours and Extraosseous Ewing Sarcoma


Abstract
1. Introduction
1.1. Epidemiology of ‘Ultra-Rare’ Primary Retroperitoneal Sarcoma
1.2. Histology, Molecular Biology, and Genetics Drive Decision-Making for Retroperitoneal Sarcoma
1.3. The Challenge of Evidence-Based Medicine for Ultra-Rare Primary Retroperitoneal Sarcoma
2. Perivascular Epithelioid Family Tumours of the Retroperitoneum
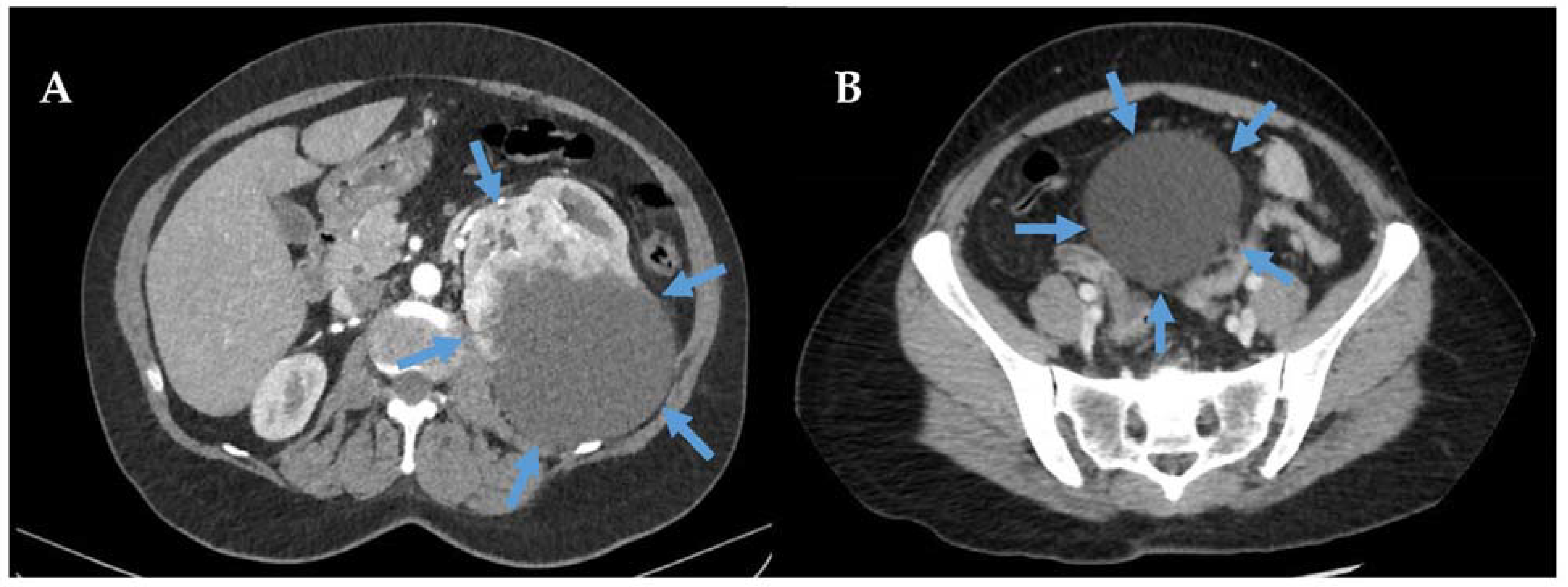
2.1. Overview
2.2. Prognosis of Perivascular Epithelioid Tumours
2.3. Differentiating Malignant PEComa-NOS from Benign AML
2.4. Genetics and Systemic Therapies Specific to PEComa-NOS
3. Extraosseous Ewing Sarcoma of the Retroperitoneum
3.1. Overview
3.2. Prognosis of Retroperitoneal Extraosseous Ewing Sarcoma
3.3. Multimodal Therapy for Retroperitoneal Extraosseous Ewing Sarcoma
4. A Note about Extraosseous Osteosarcoma and Adult Rhabdomyosarcoma
4.1. Extraosseous Osteosarcoma
4.2. Adult Rhabdomyosarcoma
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Soft Tissue and Bone Tumours, WHO Classification of Tumours, 5th ed.; WHO Classification of Tumours Editorial; World Health Organization: Geneva, Switzerland, 2020; Volume 3, ISBN 978-92-832-4502-5. [Google Scholar]
- Stiller, C.A.; Trama, A.; Serraino, D.; Rossi, S.; Navarro, C.; Chirlaque, M.D.; Casali, P.G. RARECARE Working Group Descriptive Epidemiology of Sarcomas in Europe: Report from the RARECARE Project. Eur. J. Cancer 2013, 49, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.V.; Maplethorpe, E.; Davis, F.G. Rare Cancers in Canada, 2006–2016: A Population-Based Surveillance Report and Comparison of Different Methods for Classifying Rare Cancers. Cancer Epidemiol. 2020, 67, 101721. [Google Scholar] [CrossRef]
- Gronchi, A.; Strauss, D.C.; Miceli, R.; Bonvalot, S.; Swallow, C.J.; Hohenberger, P.; Van Coevorden, F.; Rutkowski, P.; Callegaro, D.; Hayes, A.J.; et al. Variability in Patterns of Recurrence After Resection of Primary Retroperitoneal Sarcoma (RPS): A Report on 1007 Patients From the Multi-Institutional Collaborative RPS Working Group. Ann. Surg. 2016, 263, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Frezza, A.M.; Blay, J.-Y.; Baldini, E.H.; Bonvalot, S.; Bovée, J.V.M.G.; Callegaro, D.; Casali, P.G.; Chiang, R.C.; Demetri, G.D.; et al. Ultra-Rare Sarcomas: A Consensus Paper From the Connective Tissue Oncology Society Community of Experts on the Incidence Threshold and the List of Entities. Cancer 2021, 127, 2934–2942. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Baldi, G.G.; Morosi, C.; Gronchi, A.; Maestro, R. Extraskeletal Myxoid Chondrosarcoma: State of the Art and Current Research on Biology and Clinical Management. Cancers 2020, 12, 2703. [Google Scholar] [CrossRef] [PubMed]
- Fiore, M.; Sambri, A.; Spinnato, P.; Zucchini, R.; Giannini, C.; Caldari, E.; Pirini, M.G.; De Paolis, M. The Biology of Synovial Sarcoma: State-of-the-Art and Future Perspectives. Curr. Treat. Options Oncol. 2021, 22, 109. [Google Scholar] [CrossRef]
- Sun, X.; Guo, W.; Shen, J.K.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Rhabdomyosarcoma: Advances in Molecular and Cellular Biology. Sarcoma 2015, 2015, 232010. [Google Scholar] [CrossRef]
- Domanski, H.A. The Small Round Cell Sarcomas Complexities and Desmoplastic Presentation. Acta Cytol. 2022, 66, 279–294. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Sobczuk, P.; Kostrzanowski, M.; Spalek, M.; Chojnacka, M.; Szumera-Cieckiewicz, A.; Rutkowski, P. Epithelioid Sarcoma—From Genetics to Clinical Practice. Cancers 2020, 12, 2112. [Google Scholar] [CrossRef]
- Smrke, A.; Frezza, A.M.; Giani, C.; Somaiah, N.; Brahmi, M.; Czarnecka, A.M.; Rutkowski, P.; der Graaf, W.V.; Baldi, G.G.; Connolly, E.; et al. Systemic Treatment of Advanced Clear Cell Sarcoma: Results from a Retrospective International Series from the World Sarcoma Network. ESMO Open 2022, 7, 100522. [Google Scholar] [CrossRef]
- Mack, T.; Purgina, B. Updates in Pathology for Retroperitoneal Soft Tissue Sarcoma. Curr. Oncol. 2022, 29, 6400–6418. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. Retroperitoneal Sarcomas: An Update on the Diagnostic Pathology Approach. Diagnostics 2020, 10, 642. [Google Scholar] [CrossRef]
- Young, V.; Wong, D.; Yan, M.; Maclean, F. NTRK-Fusion Associated Sarcoma: Identification of Two Cases with a Distinct Perineurioma-like Pattern. Hum. Pathol. Rep. 2022, 28, 300618. [Google Scholar] [CrossRef]
- Siozopoulou, V.; Smits, E.; De Winne, K.; Marcq, E.; Pauwels, P. NTRK Fusions in Sarcomas: Diagnostic Challenges and Clinical Aspects. Diagnostics 2021, 11, 478. [Google Scholar] [CrossRef]
- Arbajian, E.; Hofvander, J.; Magnusson, L.; Mertens, F. Deep Sequencing of Myxoinflammatory Fibroblastic Sarcoma. Genes Chromosom. Cancer 2020, 59, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.K.; Gupta, S.; Bansal, S.; Kumar, N. Low-Grade Myofibroblastic Sarcoma of Retroperitoneum: A Rare Case. Int. Surg. J. 2019, 6, 633–635. [Google Scholar] [CrossRef]
- Bahrami, A.; Folpe, A.L. Adult-Type Fibrosarcoma: A Reevaluation of 163 Putative Cases Diagnosed at a Single Institution over a 48-Year Period. Am. J. Surg. Pathol. 2010, 34, 1504–1513. [Google Scholar] [CrossRef]
- Mentzel, T.; Calonje, E.; Wadden, C.; Camplejohn, R.S.; Beham, A.; Smith, M.A.; Fletcher, C.D. Myxofibrosarcoma. Clinicopathologic Analysis of 75 Cases with Emphasis on the Low-Grade Variant. Am. J. Surg. Pathol. 1996, 20, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Yoshida, K.; Halik, A.; Kunitz, A.; Suzuki, H.; Kakiuchi, N.; Shiozawa, Y.; Yokoyama, A.; Inoue, Y.; Hirano, T.; et al. The Landscape of Genetic Aberrations in Myxofibrosarcoma. Int. J. Cancer 2022, 151, 565–577. [Google Scholar] [CrossRef]
- Ud Din, N.; Ahmad, Z.; Zreik, R.; Horvai, A.; Folpe, A.L.; Fritchie, K. Abdominopelvic and Retroperitoneal Low-Grade Fibromyxoid SarcomaA Clinicopathologic Study of 13 Cases. Am. J. Clin. Pathol. 2018, 149, 128–134. [Google Scholar] [CrossRef]
- Janczak, D.; Szydełko, T.; Janczak, D. Nine-Year Follow-Up of a Huge Retroperitoneal Alveolar Soft-Part Sarcoma: A Case Report. Am. J. Case Rep. 2021, 22, e932514. [Google Scholar] [CrossRef] [PubMed]
- Shim, E.; Yeom, S.K.; Um, J.W.; Kim, J.H.; Lee, S.H.; Cha, S.H.; Choi, J.-W. Malignant Extrarenal Rhabdoid Tumor in Adults: Three Case Reports Originating from the Ileum, Adrenal Gland and Uterus. J. Korean Soc. Radiol. 2014, 71, 20. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Miah, A.B.; Frezza, A.M.; Messiou, C.; Morosi, C.; Caraceni, A.; Antonescu, C.R.; Bajpai, J.; Baldini, E.; Bauer, S.; et al. Epithelioid Hemangioendothelioma, an Ultra-Rare Cancer: A Consensus Paper from the Community of Experts. ESMO Open 2021, 6, 100170. [Google Scholar] [CrossRef]
- Chen, G.; Folpe, A.L.; Colby, T.V.; Sittampalam, K.; Patey, M.; Chen, M.-G.; Chan, J.K.C. Angiomatoid Fibrous Histiocytoma: Unusual Sites and Unusual Morphology. Mod. Pathol. 2011, 24, 1560–1570. [Google Scholar] [CrossRef]
- Klunder, A.; Subik, K.; Ye, B.; Xu, H.; Li, F. Intimal Sarcoma of the Abdominal Aorta Presenting as a Retroperitoneal Mass. Pathology 2012, 44, 485–487. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D.M. Myoepithelial Tumors of Soft Tissue: A Clinicopathologic and Immunohistochemical Study of 101 Cases with Evaluation of Prognostic Parameters. Am. J. Surg. Pathol. 2003, 27, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Jo, V.Y.; Fletcher, C.D.M. Myoepithelial Neoplasms of Soft Tissue: An Updated Review of the Clinicopathologic, Immunophenotypic, and Genetic Features. Head. Neck Pathol. 2015, 9, 32–38. [Google Scholar] [CrossRef]
- Swallow, C.J.; Strauss, D.C.; Bonvalot, S.; Rutkowski, P.; Desai, A.; Gladdy, R.A.; Gonzalez, R.; Gyorki, D.E.; Fairweather, M.; van Houdt, W.J.; et al. Management of Primary Retroperitoneal Sarcoma (RPS) in the Adult: An Updated Consensus Approach from the Transatlantic Australasian RPS Working Group. Ann. Surg. Oncol. 2021, 28, 7873–7888. [Google Scholar] [CrossRef]
- Bonvalot, S.; Roland, C.; Raut, C.; Le Péchoux, C.; Tzanis, D.; Frezza, A.M.; Gronchi, A. Histology-Tailored Multidisciplinary Management of Primary Retroperitoneal Sarcomas. Eur. J. Surg. Oncol. 2022, 49, 1061–1067. [Google Scholar] [CrossRef]
- Strauss, D.C.; Qureshi, Y.A.; Hayes, A.J.; Thway, K.; Fisher, C.; Thomas, J.M. The Role of Core Needle Biopsy in the Diagnosis of Suspected Soft Tissue Tumours. J. Surg. Oncol. 2010, 102, 523–529. [Google Scholar] [CrossRef]
- Van Houdt, W.J.; Schrijver, A.M.; Cohen-Hallaleh, R.B.; Memos, N.; Fotiadis, N.; Smith, M.J.; Hayes, A.J.; Van Coevorden, F.; Strauss, D.C. Needle Tract Seeding Following Core Biopsies in Retroperitoneal Sarcoma. Eur. J. Surg. Oncol. 2017, 43, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Bonvalot, S.; Gronchi, A.; Le Péchoux, C.; Swallow, C.J.; Strauss, D.; Meeus, P.; van Coevorden, F.; Stoldt, S.; Stoeckle, E.; Rutkowski, P.; et al. Preoperative Radiotherapy plus Surgery versus Surgery Alone for Patients with Primary Retroperitoneal Sarcoma (EORTC-62092: STRASS): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2020, 21, 1366–1377. [Google Scholar] [CrossRef]
- Callegaro, D.; Raut, C.P.; Ajayi, T.; Strauss, D.; Bonvalot, S.; Ng, D.; Stoeckle, E.; Fairweather, M.; Rutkowski, P.; van Houdt, W.J.; et al. Preoperative Radiotherapy in Patients with Primary Retroperitoneal Sarcoma: EORTC-62092 Trial (STRASS) Versus Off-Trial (STREXIT) Results. Ann. Surg. 2022, 278, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Trufero, J.; Cruz Jurado, J.; Hernández-León, C.N.; Correa, R.; Asencio, J.M.; Bernabeu, D.; Alvarez, R.; Hindi, N.; Mata, C.; Marquina, G.; et al. Uncommon and Peculiar Soft Tissue Sarcomas: Multidisciplinary Review and Practical Recommendations. Spanish Group for Sarcoma Research (GEIS–GROUP). Part II. Cancer Treat. Rev. 2021, 99, 102260. [Google Scholar] [CrossRef] [PubMed]
- Vibert, J.; Watson, S. The Molecular Biology of Soft Tissue Sarcomas: Current Knowledge and Future Perspectives. Cancers 2022, 14, 2548. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Melot, T.; Garau, X.S.; Zucker, J.M.; Lenoir, G.M.; Ambros, P.F.; Sheer, D.; Turc-Carel, C.; Triche, T.J. The Ewing Family of Tumors--a Subgroup of Small-Round-Cell Tumors Defined by Specific Chimeric Transcripts. N. Engl. J. Med. 1994, 331, 294–299. [Google Scholar] [CrossRef]
- Dei Tos, A.P.; Doglioni, C.; Piccinin, S.; Sciot, R.; Furlanetto, A.; Boiocchi, M.; Dal Cin, P.; Maestro, R.; Fletcher, C.D.; Tallini, G. Coordinated Expression and Amplification of the MDM2, CDK4, and HMGI-C Genes in Atypical Lipomatous Tumours. J. Pathol. 2000, 190, 531–536. [Google Scholar] [CrossRef]
- Cote, G.M.; He, J.; Choy, E. Next-Generation Sequencing for Patients with Sarcoma: A Single Center Experience. Oncologist 2018, 23, 234–242. [Google Scholar] [CrossRef]
- van Houdt, W.J.; Raut, C.P.; Bonvalot, S.; Swallow, C.J.; Haas, R.; Gronchi, A. New Research Strategies in Retroperitoneal Sarcoma. The Case of TARPSWG, STRASS and RESAR: Making Progress through Collaboration. Curr. Opin. Oncol. 2019, 31, 310–316. [Google Scholar] [CrossRef]
- Gronchi, A.; Van Houdt, W.J. Surgery with or without Neoadjuvant Chemotherapy in High Risk RetroPeritoneal Sarcoma—Full Text View—ClinicalTrials.Gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04031677 (accessed on 2 January 2023).
- Vyse, S.; Thway, K.; Huang, P.H.; Jones, R.L. Next-Generation Sequencing for the Management of Sarcomas with No Known Driver Mutations. Curr. Opin. Oncol. 2021, 33, 315–322. [Google Scholar] [CrossRef]
- Bonetti, F.; Pea, M.; Martignoni, G.; Zamboni, G. PEC and Sugar. Am. J. Surg. Pathol. 1992, 16, 307–308. [Google Scholar] [CrossRef]
- Bonetti, F.; Pea, M.; Martignoni, G.; Zamboni, G.; Iuzzolino, P. Cellular Heterogeneity in Lymphangiomyomatosis of the Lung. Hum. Pathol. 1991, 22, 727–728. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D.M. PEComa: What Do We Know so Far? Histopathology 2006, 48, 75–82. [Google Scholar] [CrossRef]
- Martignoni, G.; Bonetti, F.; Chilosi, M.; Brunelli, M.; Segala, D.; Amin, M.B.; Argani, P.; Eble, J.N.; Gobbo, S.; Pea, M. Cathepsin K Expression in the Spectrum of Perivascular Epithelioid Cell (PEC) Lesions of the Kidney. Mod. Pathol. 2012, 25, 100–111. [Google Scholar] [CrossRef]
- Rutkowski, P.L.; Mullen, J.T. Management of the “Other” Retroperitoneal Sarcomas. J. Surg. Oncol. 2018, 117, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, G.; Pea, M.; Martignoni, G.; Zancanaro, C.; Faccioli, G.; Gilioli, E.; Pederzoli, P.; Bonetti, F. Clear Cell “Sugar” Tumor of the Pancreas. A Novel Member of the Family of Lesions Characterized by the Presence of Perivascular Epithelioid Cells. Am. J. Surg. Pathol. 1996, 20, 722–730. [Google Scholar] [CrossRef]
- Touloumis, Z.; Giannakou, N.; Sioros, C.; Trigka, A.; Cheilakea, M.; Dimitriou, N.; Griniatsos, J. Retroperitoneal Perivascular Epithelioid Cell Tumours: A Case Report and Review of Literature. World J. Clin. Cases 2019, 7, 3524–3534. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Moss, J. Lymphangioleiomyomatosis. Am. J. Med. Sci. 2001, 321, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Avila, N.A.; Barnes, P.M.; Litzenberger, R.A.; Bechtle, J.; Brooks, P.G.; Hedin, C.J.; Hunsberger, S.; Kristof, A.S. Prevalence and Clinical Characteristics of Lymphangioleiomyomatosis (LAM) in Patients with Tuberous Sclerosis Complex. Am. J. Respir. Crit. Care Med. 2001, 164, 669–671. [Google Scholar] [CrossRef]
- Aydin, H.; Magi-Galluzzi, C.; Lane, B.R.; Sercia, L.; Lopez, J.I.; Rini, B.I.; Zhou, M. Renal Angiomyolipoma: Clinicopathologic Study of 194 Cases with Emphasis on the Epithelioid Histology and Tuberous Sclerosis Association. Am. J. Surg. Pathol. 2009, 33, 289–297. [Google Scholar] [CrossRef]
- He, W.; Cheville, J.C.; Sadow, P.M.; Gopalan, A.; Fine, S.W.; Al-Ahmadie, H.A.; Chen, Y.-B.; Oliva, E.; Russo, P.; Reuter, V.E.; et al. Epithelioid Angiomyolipoma of the Kidney: Pathological Features and Clinical Outcome in a Series of Consecutively Resected Tumors. Mod. Pathol. 2013, 26, 1355–1364. [Google Scholar] [CrossRef]
- Brimo, F.; Robinson, B.; Guo, C.; Zhou, M.; Latour, M.; Epstein, J.I. Renal Epithelioid Angiomyolipoma with Atypia: A Series of 40 Cases with Emphasis on Clinicopathologic Prognostic Indicators of Malignancy. Am. J. Surg. Pathol. 2010, 34, 715–722. [Google Scholar] [CrossRef]
- Bourgmayer, A.; Nannini, S.; Bonjean, P.; Kurtz, J.-E.; Malouf, G.G.; Gantzer, J. Natural History and Treatment Strategies of Advanced PEComas: A Systematic Review. Cancers 2021, 13, 5227. [Google Scholar] [CrossRef]
- Bleeker, J.S.; Quevedo, J.F.; Folpe, A.L. “Malignant” Perivascular Epithelioid Cell Neoplasm: Risk Stratification and Treatment Strategies. Sarcoma 2012, 2012, 541626. [Google Scholar] [CrossRef] [PubMed]
- Folpe, A.L.; Goodman, Z.D.; Ishak, K.G.; Paulino, A.F.; Taboada, E.M.; Meehan, S.A.; Weiss, S.W. Clear Cell Myomelanocytic Tumor of the Falciform Ligament/Ligamentum Teres: A Novel Member of the Perivascular Epithelioid Clear Cell Family of Tumors with a Predilection for Children and Young Adults. Am. J. Surg. Pathol. 2000, 24, 1239–1246. [Google Scholar] [CrossRef]
- Folpe, A.L.; Mentzel, T.; Lehr, H.-A.; Fisher, C.; Balzer, B.L.; Weiss, S.W. Perivascular Epithelioid Cell Neoplasms of Soft Tissue and Gynecologic Origin: A Clinicopathologic Study of 26 Cases and Review of the Literature. Am. J. Surg. Pathol. 2005, 29, 1558–1575. [Google Scholar] [CrossRef] [PubMed]
- Ditonno, P.; Smith, R.B.; Koyle, M.A.; Hannah, J.; Belldegrun, A. Extrarenal Angiomyolipomas of the Perinephric Space. J. Urol. 1992, 147, 447–450. [Google Scholar] [CrossRef]
- Minja, E.J.; Pellerin, M.; Saviano, N.; Chamberlain, R.S. Retroperitoneal Extrarenal Angiomyolipomas: An Evidence-Based Approach to a Rare Clinical Entity. Case Rep. Nephrol. 2012, 2012, 374107. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.J. Lymphangioleiomyomatosis: A Review. Chest 1998, 114, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Zekry, N.; Rettenmaier, M.A.; Abaid, L.N.; John, C.R.; Micha, J.P.; Brown, J.V.; Goldstein, B.H. Perivascular Epithelioid Cell Neoplasms: A Systematic Review of Prognostic Factors. J. Minim. Invasive Gynecol. 2009, 16, 527–532. [Google Scholar] [CrossRef]
- Tirumani, S.H.; Shinagare, A.B.; Hargreaves, J.; Jagannathan, J.P.; Hornick, J.L.; Wagner, A.J.; Ramaiya, N.H. Imaging Features of Primary and Metastatic Malignant Perivascular Epithelioid Cell Tumors. AJR Am. J. Roentgenol. 2014, 202, 252–258. [Google Scholar] [CrossRef]
- Nese, N.; Martignoni, G.; Fletcher, C.D.; Gupta, R.; Pan, C.-C.; Kim, H.; Ro, J.Y.; Hwang, I.S.; Sato, K.; Bonetti, F.; et al. Pure Epithelioid PEComas (so-Called Epithelioid Angiomyolipoma) of the Kidney: A Clinicopathologic Study of 41 Cases: Detailed Assessment of Morphology and Risk Stratification. Am. J. Surg. Pathol. 2011, 35, 161–176. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D.M. Sclerosing PEComa: Clinicopathologic Analysis of a Distinctive Variant with a Predilection for the Retroperitoneum. Am. J. Surg. Pathol. 2008, 32, 493–501. [Google Scholar] [CrossRef]
- Armah, H.B.; Parwani, A.V. Malignant Perivascular Epithelioid Cell Tumor (PEComa) of the Uterus with Late Renal and Pulmonary Metastases: A Case Report with Review of the Literature. Diagn. Pathol. 2007, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Parra-Medina, R.; Morales, S.D. Cutaneous Perivascular Epithelioid Cell Tumor of Gynecologic Origin Metastatic to Skin, Lung, Stomach, and Brain. Am. J. Derm. 2017, 39, 157–159. [Google Scholar] [CrossRef]
- Parfitt, J.R.; Bella, A.J.; Izawa, J.I.; Wehrli, B.M. Malignant Neoplasm of Perivascular Epithelioid Cells of the Liver. Arch. Pathol. Lab. Med. 2006, 130, 1219–1222. [Google Scholar] [CrossRef]
- Sobiborowicz, A.; Świtaj, T.; Teterycz, P.; Spałek, M.J.; Szumera-Ciećkiewicz, A.; Wągrodzki, M.; Zdzienicki, M.; Czarnecka, A.M.; Rutkowski, P. Feasibility and Long-Term Efficacy of PEComa Treatment-20 Years of Experience. J. Clin. Med. 2021, 10, 2200. [Google Scholar] [CrossRef]
- Vos, N.; Oyen, R. Renal Angiomyolipoma: The Good, the Bad, and the Ugly. J. Belg. Soc. Radiol. 2018, 102, 41. [Google Scholar] [CrossRef]
- Woo, S.; Kim, S.Y.; Cho, J.Y.; Kim, S.H.; Lee, M.S. Exophytic Renal Angiomyolipoma and Perirenal Liposarcoma: Revisiting the Role of CT for Differential Diagnosis. Acta Radiol. 2016, 57, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Wildgruber, M.; Becker, K.; Feith, M.; Gaa, J. Perivascular Epitheloid Cell Tumor (PEComa) Mimicking Retroperitoneal Liposarcoma. World J. Surg. Oncol. 2014, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Tang, Y.L.; Tandon, A.A. Multifocal Retroperitoneal and Pelvic PEComas Mimicking Liposarcoma: A Case Report and Review of Literature. Radiol. Case Rep. 2021, 16, 2624–2629. [Google Scholar] [CrossRef]
- Weaver, J.; Downs-Kelly, E.; Goldblum, J.R.; Turner, S.; Kulkarni, S.; Tubbs, R.R.; Rubin, B.P.; Skacel, M. Fluorescence in Situ Hybridization for MDM2 Gene Amplification as a Diagnostic Tool in Lipomatous Neoplasms. Mod. Pathol. 2008, 21, 943–949. [Google Scholar] [CrossRef]
- Ryan, J.W.; Farrelly, C.; Geoghegan, T. What Are the Indications for Prophylactic Embolization of Renal Angiomyolipomas? A Review of the Current Evidence in the Literature. Can. Assoc. Radiol. J. 2018, 69, 236–239. [Google Scholar] [CrossRef]
- Agaram, N.P.; Sung, Y.-S.; Zhang, L.; Chen, C.-L.; Chen, H.-W.; Singer, S.; Dickson, M.A.; Berger, M.F.; Antonescu, C.R. Dichotomy of Genetic Abnormalities in PEComas with Therapeutic Implications. Am. J. Surg. Pathol. 2015, 39, 813–825. [Google Scholar] [CrossRef]
- Kenerson, H.; Folpe, A.L.; Takayama, T.K.; Yeung, R.S. Activation of the MTOR Pathway in Sporadic Angiomyolipomas and Other Perivascular Epithelioid Cell Neoplasms. Hum. Pathol. 2007, 38, 1361–1371. [Google Scholar] [CrossRef]
- Argani, P.; Aulmann, S.; Illei, P.B.; Netto, G.J.; Ro, J.; Cho, H.; Dogan, S.; Ladanyi, M.; Martignoni, G.; Goldblum, J.R.; et al. A Distinctive Subset of PEComas Harbors TFE3 Gene Fusions. Am. J. Surg. Pathol. 2010, 34, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.R.; Halpern, E.F.; Thiele, E.A. Renal Manifestations of Tuberous Sclerosis Complex: Incidence, Prognosis, and Predictive Factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous Sclerosis Complex. Nat. Rev. Dis. Prim. 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, R.; Jones, R.L.; Blay, J.-Y.; Le Cesne, A.; Provenzano, S.; Antoniou, G.; Mir, O.; Fucà, G.; Fumagalli, E.; Bertulli, R.; et al. Role of Chemotherapy, VEGFR Inhibitors, and MTOR Inhibitors in Advanced Perivascular Epithelioid Cell Tumors (PEComas). Clin. Cancer Res. 2019, 25, 5295–5300. [Google Scholar] [CrossRef]
- Jia, R.; Jiang, L.; Zhou, Y.; Wang, Y.; Guo, X.; Ji, Y.; Ni, X.; Yang, X. Clinical Features of 18 Perivascular Epithelioid Cell Tumor Cases. Medicine 2020, 99, e21659. [Google Scholar] [CrossRef]
- Flechter, E.; Zohar, Y.; Guralnik, L.; Passhak, M.; Sela, G.B. Long-Lasting Stable Disease with MTOR Inhibitor Treatment in a Patient with a Perivascular Epithelioid Cell Tumor: A Case Report and Literature Review. Oncol. Lett. 2016, 12, 4739–4743. [Google Scholar] [CrossRef]
- Dickson, M.A.; Schwartz, G.K.; Antonescu, C.R.; Kwiatkowski, D.J.; Malinowska, I.A. Extrarenal Perivascular Epithelioid Cell Tumors (PEComas) Respond to MTOR Inhibition: Clinical and Molecular Correlates. Int. J. Cancer 2013, 132, 1711–1717. [Google Scholar] [CrossRef]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.M.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical Activity of MTOR Inhibition with Sirolimus in Malignant Perivascular Epithelioid Cell Tumors: Targeting the Pathogenic Activation of MTORC1 in Tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef]
- Wagner, A.J.; Ravi, V.; Riedel, R.F.; Ganjoo, K.; Van Tine, B.A.; Chugh, R.; Cranmer, L.; Gordon, E.M.; Hornick, J.L.; Du, H.; et al. Nab-Sirolimus for Patients with Malignant Perivascular Epithelioid Cell Tumors. J. Clin. Oncol. 2021, 39, 3660–3670. [Google Scholar] [CrossRef]
- Pinto, A.; Dickman, P.; Parham, D. Pathobiologic Markers of the Ewing Sarcoma Family of Tumors: State of the Art and Prediction of Behaviour. Sarcoma 2011, 2011, 856190. [Google Scholar] [CrossRef]
- de Alava, E.; Gerald, W.L. Molecular Biology of the Ewing’s Sarcoma/Primitive Neuroectodermal Tumor Family. J. Clin. Oncol. 2000, 18, 204–213. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G. Gene Fusion with an ETS DNA-Binding Domain Caused by Chromosome Translocation in Human Tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Parham, D.M.; Hijazi, Y.; Steinberg, S.M.; Meyer, W.H.; Horowitz, M.; Tzen, C.Y.; Wexler, L.H.; Tsokos, M. Neuroectodermal Differentiation in Ewing’s Sarcoma Family of Tumors Does Not Predict Tumor Behavior. Hum. Pathol. 1999, 30, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Jürgens, H.; Bier, V.; Harms, D.; Beck, J.; Brandeis, W.; Etspüler, G.; Gadner, H.; Schmidt, D.; Treuner, J.; Winkler, K. Malignant Peripheral Neuroectodermal Tumors. A Retrospective Analysis of 42 Patients. Cancer 1988, 61, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Carpentieri, D.F.; Qualman, S.J.; Bowen, J.; Krausz, T.; Marchevsky, A.; Dickman, P.S. Protocol for the Examination of Specimens From Pediatric and Adult Patients with Osseous and Extraosseous Ewing Sarcoma Family of Tumors, Including Peripheral Primitive Neuroectodermal Tumor and Ewing Sarcoma. Arch. Pathol. Lab. Med. 2005, 129, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Herrmann, C.; Jürgens, H.; Harms, D. Malignant Peripheral Neuroectodermal Tumor and Its Necessary Distinction from Ewing’s Sarcoma. A Report from the Kiel Pediatric Tumor Registry. Cancer 1991, 68, 2251–2259. [Google Scholar] [CrossRef]
- Jaffe, R.; Santamaria, M.; Yunis, E.J.; Tannery, N.H.; Agostini, R.M.; Medina, J.; Goodman, M. The Neuroectodermal Tumor of Bone. Am. J. Surg. Pathol. 1984, 8, 885–898. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Cote, G.M.; Hornick, J.L. Contemporary Sarcoma Diagnosis, Genetics, and Genomics. J. Clin. Oncol. 2018, 36, 101–110. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A Second Ewing’s Sarcoma Translocation, t(21;22), Fuses the EWS Gene to Another ETS-Family Transcription Factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Tsuda, Y.; Dickson, B.C.; Swanson, D.; Sung, Y.-S.; Zhang, L.; Meyers, P.; Healey, J.H.; Antonescu, C.R. Ewing Sarcoma with FEV Gene Rearrangements Is a Rare Subset with Predilection for Extra-Skeletal Locations and Aggressive Behavior. Genes Chromosom. Cancer 2020, 59, 286–294. [Google Scholar] [CrossRef]
- Murthy, S.S.; Challa, S.; Raju, K.; Rajappa, S.J.; Fonseca, D.; Gundimeda, S.D.; Rao, B.V.; Ahmed, F.; Kodandapani, S.; Nambaru, L.; et al. Ewing Sarcoma with Emphasis on Extra-Skeletal Ewing Sarcoma: A Decade’s Experience From a Single Centre in India. Clin. Pathol. 2020, 13. [Google Scholar] [CrossRef]
- Applebaum, M.A.; Worch, J.; Matthay, K.K.; Goldsby, R.; Neuhaus, J.; West, D.C.; Dubois, S.G. Clinical Features and Outcomes in Patients with Extraskeletal Ewing Sarcoma. Cancer 2011, 117, 3027–3032. [Google Scholar] [CrossRef]
- Thyavihally, Y.B.; Tongaonkar, H.B.; Gupta, S.; Kurkure, P.A.; Amare, P.; Muckaden, M.A.; Desai, S.B. Primitive Neuroectodermal Tumor of the Kidney: A Single Institute Series of 16 Patients. Urology 2008, 71, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Marinova, L. Retroperitoneal Primitive Neuroectodermal Tumour (PNET). A Case Report and Review of the Literature. Rep. Pract. Oncol. Radiother. 2009, 14, 221–224. [Google Scholar] [CrossRef]
- Li, J.; Nie, F.; Li, Y. Extraosseous Ewing’s Sarcoma/Peripheral Primitive Neuroectodermal Tumour of the Kidney: A Case Report and Literature Review. BMC Urol. 2022, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Frigo, S.; Lecointre, L.; Hummel, M.; Akladios, C.Y.; Bergerat, J.P.; Noël, G.; Wattiez, A. Retroperitoneal Primitive Neuroectodermal Tumor (PNET): Case Report and Review of Literature. Eur. J. Gynaecol. Oncol. 2017, 38, 314–318. [Google Scholar] [PubMed]
- Javalgi, A.P. Blue Cell Tumour at Unusual Site: Retropritoneal Ewings Sarcoma. J. Clin. Diagn. Res. 2016, 10, ED19–ED20. [Google Scholar] [CrossRef]
- Genov, P.; Serbezova, I.; Georgieva, D.; Koleva, G.; Hristova, I. A Rare Case of Retroperitoneal Primitive Neuroectodermal Tumor (PNET). Urol. Case Rep. 2021, 35, 101554. [Google Scholar] [CrossRef]
- Das, S.; Aggarwal, G.; Gupta, S.; Midha, D. Primary Renal Ewing’s Sarcoma in an Adult: An Enigma. Innov. Surg. Sci. 2021, 6, 20200022. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Patkar, S.; Purandare, N.; Mokal, S.; Goel, M. Management of Abdominal Ewing’s Sarcoma: A Single Institute Experience. Indian. J. Surg. Oncol. 2021, 12, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.; Grimer, R.J.; Spooner, D.; Peake, D.; Carter, S.R.; Tillman, R.M.; Abudu, A.; Jeys, L. Oncological Outcomes of Patients with Ewing’s Sarcoma: Is There a Difference between Skeletal and Extra-Skeletal Ewing’s Sarcoma? J. Bone Jt. Surg. Br. 2011, 93, 531–536. [Google Scholar] [CrossRef]
- Lynch, A.D.; Gani, F.; Meyer, C.F.; Morris, C.D.; Ahuja, N.; Johnston, F.M. Extraskeletal versus Skeletal Ewing Sarcoma in the Adult Population: Controversies in Care. Surg. Oncol. 2018, 27, 373–379. [Google Scholar] [CrossRef]
- Cuesta Alcalá, J.A.; Solchaga Martínez, A.; Caballero Martínez, M.C.; Gómez Dorronsoro, M.; Pascual Piédrola, I.; Ripa Saldías, L.; Aldave Villanueva, J.; Arrondo Arrondo, J.L.; Grasa Lanau, V.; Ponz González, M.; et al. Primary neuroectodermal tumor (PNET) of the kidney: 26 cases. Current status of its diagnosis and treatment. Arch. Esp. Urol. 2001, 54, 1081–1093. [Google Scholar]
- Li, T.; Zhang, F.; Cao, Y.; Ning, S.; Bi, Y.; Xue, W.; Ren, L. Primary Ewing’s Sarcoma/Primitive Neuroectodermal Tumor of the Ileum: Case Report of a 16-Year-Old Chinese Female and Literature Review. Diagn. Pathol. 2017, 12, 37. [Google Scholar] [CrossRef]
- Mir, A.; Lashkari, M.; Jafari, F.; Molavi, B. Primitive Neuroectodermal Tumour Invading the Inferior Vena Cava. Eur. J. Case Rep. Intern. Med. 2020, 7. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, Y.; Zhang, F.; Hu, K.; Qiu, J.; Hou, X.; Yan, J.; Lian, X.; Sun, S.; Liu, Z.; et al. Outcome of Multidisciplinary Treatment of Peripheral Primitive Neuroectodermal Tumor. Sci. Rep. 2020, 10, 15656. [Google Scholar] [CrossRef] [PubMed]
- Rud, N.P.; Reiman, H.M.; Pritchard, D.J.; Frassica, F.J.; Smithson, W.A. Extraosseous Ewing’s Sarcoma. A Study of 42 Cases. Cancer 1989, 64, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Brennan, B.; Kirton, L.; Marec-Bérard, P.; Gaspar, N.; Laurence, V.; Martín-Broto, J.; Sastre, A.; Gelderblom, H.; Owens, C.; Fenwick, N.; et al. Comparison of Two Chemotherapy Regimens in Patients with Newly Diagnosed Ewing Sarcoma (EE2012): An Open-Label, Randomised, Phase 3 Trial. Lancet 2022, 400, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Huang, W.; Wang, H.; Zheng, C.; Jiang, J. Risk Factors for Lung Metastasis at Presentation with Malignant Primary Osseous Neoplasms: A Population-Based Study. J. Orthop. Surg. Res. 2020, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Perotti, D.; Corletto, V.; Giardini, R.; Parafioriti, A.; Fossati-Bellani, F.; Luksch, R. Retrospective Analysis of Ploidy in Primary Osseous and Extraosseous Ewing Family Tumors in Children. Tumori 1998, 84, 493–498. [Google Scholar] [CrossRef]
- Orr, W.S.; Denbo, J.W.; Billups, C.A.; Wu, J.; Navid, F.; Rao, B.N.; Davidoff, A.M.; Krasin, M.J. Analysis of Prognostic Factors in Extraosseous Ewing Sarcoma Family of Tumors: Review of St. Jude Children’s Research Hospital Experience. Ann. Surg. Oncol. 2012, 19, 3816–3822. [Google Scholar] [CrossRef]
- Kolb, E.A.; Kushner, B.H.; Gorlick, R.; Laverdiere, C.; Healey, J.H.; LaQuaglia, M.P.; Huvos, A.G.; Qin, J.; Vu, H.T.; Wexler, L.; et al. Long-Term Event-Free Survival after Intensive Chemotherapy for Ewing’s Family of Tumors in Children and Young Adults. J. Clin. Oncol. 2003, 21, 3423–3430. [Google Scholar] [CrossRef]
- Strauss, S.J.; Frezza, A.M.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; Bonvalot, S.; et al. Bone Sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical Practice Guideline for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 1520–1536. [Google Scholar] [CrossRef]
- El Weshi, A.; Allam, A.; Ajarim, D.; Al Dayel, F.; Pant, R.; Bazarbashi, S.; Memon, M. Extraskeletal Ewing’s Sarcoma Family of Tumours in Adults: Analysis of 57 Patients from a Single Institution. Clin. Oncol. (R. Coll. Radiol.) 2010, 22, 374–381. [Google Scholar] [CrossRef]
- Rijswijk, C.S.P.V.; Lieng, J.G.S.T.A.; Kroon, H.M.; Hogendoorn, P.C.W. Retroperitoneal Extraskeletal Osteosarcoma. J. Clin. Pathol. 2001, 54, 77–78. [Google Scholar] [CrossRef]
- Ratnagiri, R.; Garg, V.; Chaturvedi, R. Extra Osseous Osteosarcoma of the Retroperitoneum: An Unusual Entity. J. Cancer Res. 2012, 8, 424–426. [Google Scholar] [CrossRef]
- Nagpal, A.P.; Chandra, S.; Goel, S. Large Retroperitoneal Extraosseous Osteosarcoma Invading into the Spine: A Case Report. Indian J. Surg. Oncol. 2016, 7, 464–466. [Google Scholar] [CrossRef]
- D’Andrea, G.; Caroli, E.; Capponi, M.G.; Scicchitano, F.; Osti, M.F.; Bellotti, C.; Ferrante, L. Retroperitoneal Mesenchymal Chondrosarcoma Mimicking a Large Retroperitoneal Sacral Schwannoma. Neurosurg. Rev. 2008, 31, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Koc, M.; Ozercan, M.R. Retroperitoneal Extraskeletal Mesenchymal Chondrosarcoma: A Very Rare Case Report. Erciyes Med. J. 2016, 38, 115–118. [Google Scholar] [CrossRef]
- Taori, K.; Patil, P.; Attarde, V.; Chandanshive, S.; Rangankar, V.; Rewatkar, N. Primary Retroperitoneal Extraskeletal Mesenchymal Chondrosarcoma: A Computed Tomography Diagnosis. BJR 2007, 80, e268–e270. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.L.; Khurana, K.K.; Kemp, B.L.; Ayala, A.G. Heterologous Elements in the Dedifferentiated Component of Dedifferentiated Liposarcoma. Am. J. Surg. Pathol. 1994, 18, 1150–1157. [Google Scholar] [CrossRef]
- Henricks, W.H.; Chu, Y.C.; Goldblum, J.R.; Weiss, S.W. Dedifferentiated Liposarcoma: A Clinicopathological Analysis of 155 Cases with a Proposal for an Expanded Definition of Dedifferentiation. Am. J. Surg. Pathol. 1997, 21, 271–281. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Sobczuk, P.; Zdzienicki, M.; Spałek, M.; Rutkowski, P. Malignant peripheral nerve sheath tumour (MPNST). Oncol. Clin. Pract. 2018, 14, 364–376. [Google Scholar] [CrossRef]
- Yamashita, K.; Kohashi, K.; Yamada, Y.; Nishida, Y.; Urakawa, H.; Oda, Y.; Toyokuni, S. Primary Extraskeletal Osteosarcoma: A Clinicopathological Study of 18 Cases Focusing on MDM2 Amplification Status. Hum. Pathol. 2017, 63, 63–69. [Google Scholar] [CrossRef]
- Crombé, A.; Spinnato, P.; Righi, A.; Leopardi, M.P.; Carpenzano, M.; Izzo, F.; Parmeggiani, A.; Linck, P.-A.; Perret, R.; Cesari, M.; et al. Imaging Presentation of Extraskeletal Osteosarcomas on CT and MRI and Correlation with Patients Outcome: A Two-Center Retrospective Study of 54 Patients. Diagn. Interv. Imaging 2023, 104, 297–306. [Google Scholar] [CrossRef]
- O’Regan, K.N.; Jagannathan, J.; Krajewski, K.; Zukotynski, K.; Souza, F.; Wagner, A.J.; Ramaiya, N. Imaging of Liposarcoma: Classification, Patterns of Tumor Recurrence, and Response to Treatment. AJR Am. J. Roentgenol. 2011, 197, W37–W43. [Google Scholar] [CrossRef]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and Prognosis with Osteosarcoma: Outcomes in More than 2000 Patients in the EURAMOS-1 (European and American Osteosarcoma Study) Cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef]
- Heng, M.; Gupta, A.; Chung, P.W.; Healey, J.H.; Vaynrub, M.; Rose, P.S.; Houdek, M.T.; Lin, P.P.; Bishop, A.J.; Hornicek, F.J.; et al. The Role of Chemotherapy and Radiotherapy in Localized Extraskeletal Osteosarcoma. Eur. J. Cancer 2020, 125, 130–141. [Google Scholar] [CrossRef]
- Liao, Z.; Qiu, M.; Yang, J.; Yang, Y.; Zhu, L.; Yang, B.; Bai, X.; Xing, P.; Zhang, J.; Xing, R.; et al. Outcomes of Surgery and/or Combination Chemotherapy for Extraskeletal Osteosarcoma: A Single-Center Retrospective Study from China. Sci. Rep. 2019, 9, 4816. [Google Scholar] [CrossRef]
- Fan, Z.; Patel, S.; Lewis, V.O.; Guadagnolo, B.A.; Lin, P.P. Should High-Grade Extraosseous Osteosarcoma Be Treated with Multimodality Therapy Like Other Soft Tissue Sarcomas? Clin. Orthop. Relat. Res. 2015, 473, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Miao, R.; Jacobson, A.; Harmon, D.; Choy, E.; Hornicek, F.; Raskin, K.; Chebib, I.; DeLaney, T.F.; Chen, Y.-L.E. Extraskeletal Osteosarcoma: A Large Series Treated at a Single Institution. Rare Tumors 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Mavrogenis, A.F.; Angelelli, L.; Righi, A.; Filardo, G.; Kido, A.; Honoki, K.; Tanaka, Y.; Tanaka, Y.; Errani, C. The Effect of Adjuvant Chemotherapy on Localized Extraskeletal Osteosarcoma: A Systematic Review. Cancers 2022, 14, 2559. [Google Scholar] [CrossRef]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.P.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft Tissue and Visceral Sarcomas: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up☆. Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, R.; Fuchs, J.; Rodeberg, D. Rhabdomyosarcoma. Semin. Pediatr. Surg. 2016, 25, 276–283. [Google Scholar] [CrossRef]
- Benjelloun, E.G.; Hatim, G.; Meryem, Z.; Razzouki, I.; Guérisse, N.B.; Jouhadi, H.; Chekrine, T.; Bouchbika, Z.; Benchekroun, N.; Tawfiq, N.; et al. Case Report: Adult Retroperitoneal Rhabdomyosarcoma. Eur. J. Med. Health Sci. 2022, 4, 1–4. [Google Scholar] [CrossRef]
- Yadav, S.K.; Sinha, D.K.; Ahmed, A.; Azhar, T.; Sinha, M. Primary Intra-Abdominal Rhabdomyosarcoma in an Adult: An Unusual Presentation and Review of Literature. Indian J. Surg. Oncol. 2015, 6, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yang, S.J. Spindle Cell Rhabdomyosarcoma of the Retroperitoneum: An Unusual Case Developed in a Pregnant Woman but Obscured by Pregnancy. Int. J. Clin. Exp. Pathol. 2014, 7, 4904–4912. [Google Scholar] [PubMed]
- Improta, L.; Tzanis, D.; Bouhadiba, T.; Abdelhafidh, K.; Bonvalot, S. Overview of Primary Adult Retroperitoneal Tumours. Eur. J. Surg. Oncol. 2020, 46, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Wang, C.-W.; Hong, R.-L.; Kuo, S.-H. Prognostic Factors and Treatment Outcomes of Adult Patients with Rhabdomyosarcoma After Multimodality Treatment. Anticancer Res. 2019, 39, 1355–1364. [Google Scholar] [CrossRef]
- Perez, E.A.; Kassira, N.; Cheung, M.C.; Koniaris, L.G.; Neville, H.L.; Sola, J.E. Rhabdomyosarcoma in Children: A SEER Population Based Study. J. Surg. Res. 2011, 170, e243–e251. [Google Scholar] [CrossRef]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing Adult and Pediatric Rhabdomyosarcoma in the Surveillance, Epidemiology and End Results Program, 1973 to 2005: An Analysis of 2600 Patients. JCO 2009, 27, 3391–3397. [Google Scholar] [CrossRef]
- Mäkinen, V.-N.; Safwat, A.; Aggerholm-Pedersen, N. Rhabdomyosarcoma in Adults: A Retrospective Analysis of Case Records Diagnosed between 1979 and 2018 in Western Denmark. Sarcoma 2021, 2021, e9948885. [Google Scholar] [CrossRef]
- Esnaola, N.F.; Rubin, B.P.; Baldini, E.H.; Vasudevan, N.; Demetri, G.D.; Fletcher, C.D.M.; Singer, S. Response to Chemotherapy and Predictors of Survival in Adult Rhabdomyosarcoma. Ann. Surg. 2001, 234, 215–223. [Google Scholar] [CrossRef]
- Ferrari, A.; Dileo, P.; Casanova, M.; Bertulli, R.; Meazza, C.; Gandola, L.; Navarria, P.; Collini, P.; Gronchi, A.; Olmi, P.; et al. Rhabdomyosarcoma in Adults. A Retrospective Analysis of 171 Patients Treated at a Single Institution. Cancer 2003, 98, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Gerber, N.K.; Wexler, L.H.; Singer, S.; Alektiar, K.M.; Keohan, M.L.; Shi, W.; Zhang, Z.; Wolden, S. Adult Rhabdomyosarcoma Survival Improved with Treatment on Multimodality Protocols. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Elsebaie, M.A.T.; Amgad, M.; Elkashash, A.; Elgebaly, A.S.; Ashal, G.G.E.I.; Shash, E.; Elsayed, Z. Management of Low and Intermediate Risk Adult Rhabdomyosarcoma: A Pooled Survival Analysis of 553 Patients. Sci. Rep. 2018, 8, 9337. [Google Scholar] [CrossRef] [PubMed]
- Little, D.J.; Ballo, M.T.; Zagars, G.K.; Pisters, P.W.T.; Patel, S.R.; El-Naggar, A.K.; Garden, A.S.; Benjamin, R.S. Adult Rhabdomyosarcoma: Outcome Following Multimodality Treatment. Cancer 2002, 95, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Bernasconi, A.; Bergamaschi, L.; Botta, L.; Andreano, A.; Castaing, M.; Rugge, M.; Bisogno, G.; Falcini, F.; Sacerdote, C.; et al. Impact of Rhabdomyosarcoma Treatment Modalities by Age in a Population-Based Setting. J. Adolesc. Young Adult Oncol. 2021, 10, 309–315. [Google Scholar] [CrossRef]
- von Mehren, M.; Kane, J.M.; Bui, M.M.; Choy, E.; Connelly, M.; Dry, S.; Ganjoo, K.N.; George, S.; Gonzalez, R.J.; Heslin, M.J.; et al. NCCN Guidelines Insights: Soft Tissue Sarcoma, Version 1.2021: Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2020, 18, 1604–1612. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Common Genetic or Molecular Findings | * Special Management Considerations | |
|---|---|---|
| PEComa-family tumours | ||
| Angiomyolipoma | TSC1/2 or FLCN deletions, SFPQ/DVL2/NONO::TFE3 | Embolization, marginal resection, or surveillance |
| PEComa-NOS/CCMMT/CCST | TSC1/2 or FLCN deletions, SFPQ/DVL2/NONO::TFE3 | * mTOR inhibitors may have role in TSC mutated patients |
| Extraosseus and classic Orthopaedic sarcoma | ||
| Ewing sarcoma | EWSR1::FLI1/ERG/FUS/FEV/ETV/E1AF | Neo- or adjuvant VDC/IE |
| Osteosarcoma | FISH amplification of MDM2, but not CDK4 (unlike LPS) | * Treat similar to RPS, unlike intraosseous osteosarcoma |
| Myxoid chondrosarcoma [6] | NR4A3::EWSR1/TAF1 | * sunitinib/pazopanib may have role (phase II trials) |
| Synovial sarcoma [7] | SS18::SSX1/2 | * |
| Rhabdomyosarcoma [8] | PAX3/7 fusions. Alveolar: FKHR/NCOA1, spindle-cell: (NCOA2), embryonal: (complex alterations) | Neo- or adjuvant chemo (paediatric protocols). Pleomorphic subtype: chemoresistant |
| Other non-Ewing round cell RPS [9] | ||
| EWSR1 non-ETS sarcoma | EWSR1::PATZ1 | * |
| CIC-rearranged sarcoma | CIC::DUX4 | * |
| BCOR-altered sarcoma | BCOR::CCNB3 | * |
| Desmoplastic small round cell tumour | EWSR1::WT1 | * Potential role for CRS-HIPEC |
| Other ultra-rare RPS histologies | ||
| Epithelioid sarcoma [10] | SMARCB1/INI1 mutations | * Consider resecting associated lymph node basins |
| Clear cell sarcoma [11] | EWSR1::ATF1/CREB1 | * |
| Angiosarcoma [12] | Complex alterations (secondary angiosarcoma: MYC amplification) | * |
| Inflammatory myofibroblastic tumour [13] | TPM3/4/CLTC::ALK | * |
| NTRK-fusion sarcomas [14,15] | NTRK3::ETV6/EML4 | * Tropomyosin kinase-receptor therapy has a role |
| Myxoinflammatory fibroblastic cell tumour [16] | Complex alterations TGFBR3, VGLL3, BRAF | * |
| Myofibroblastic sarcoma [17] | Complex or unknown alterations | * |
| Fibrosarcoma [18] | Complex alterations, CDK2NA/B, TP53, EWSR1 | * |
| Myxofibrosarcoma [19,20] | Complex alterations, CDK2NA/B, TP53, RB1 | * |
| Low grade fibromyxoid sarcoma [21] | FUS::CREB3L2 | * |
| Alveolar soft part sarcoma [22] | ASPSCR1::TFE3 | * |
| Extrarenal malignant rhabdoid tumour [23] | SMARCB1 inactivation | * |
| Epithelioid hemangioendothelioma [24] | WWTR1::CAMTA1, YAP1::TFE3 | * |
| Angiomatoid Fibrous Histiocytoma [25] | EWSR1::CREB1/ATF1, FUS::ATF1 | * |
| Intimal Sarcoma [26] | Complex alterations, notably MDM2/CDK4 amplification. Additionally, PDGFRA/B, NOTCH2, CDKN2A/B | * |
| Malignant myoepithelioma [27,28] | Complex alterations, EWSR1 rearrangements | * |
| Benign | Malignant | Uncertain Malignant Potential | |
|---|---|---|---|
| High-risk features | |||
| Folpe [31] | |||
| Size > 5 cm Mitotic rate ≥ 1/50 HPF Infiltrative growth pattern High nuclear grade and cellularity Necrosis Vascular invasion | <2 high-risk features and size ≤ 5 cm | 2 or more of any high-risk features | (1) Size > 5 cm with no other high-risk features, or (2) only high nuclear grade/multi-nucleated giant cells |
| Bleeker [29] | |||
| Size > 5 cm Mitotic rate ≥ 1/50 HPF | No high-risk features | Both high-risk features | Only one high-risk feature |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apte, S.S.; Mor, E.; Mitchell, C.; Gyorki, D.E. Practical Management of Adult Ultra-Rare Primary Retroperitoneal Soft Tissue Sarcoma: A Focus on Perivascular Epithelioid Tumours and Extraosseous Ewing Sarcoma. Curr. Oncol. 2023, 30, 5953-5972. https://doi.org/10.3390/curroncol30070445
Apte SS, Mor E, Mitchell C, Gyorki DE. Practical Management of Adult Ultra-Rare Primary Retroperitoneal Soft Tissue Sarcoma: A Focus on Perivascular Epithelioid Tumours and Extraosseous Ewing Sarcoma. Current Oncology. 2023; 30(7):5953-5972. https://doi.org/10.3390/curroncol30070445
Chicago/Turabian StyleApte, Sameer S., Eyal Mor, Catherine Mitchell, and David E. Gyorki. 2023. "Practical Management of Adult Ultra-Rare Primary Retroperitoneal Soft Tissue Sarcoma: A Focus on Perivascular Epithelioid Tumours and Extraosseous Ewing Sarcoma" Current Oncology 30, no. 7: 5953-5972. https://doi.org/10.3390/curroncol30070445
APA StyleApte, S. S., Mor, E., Mitchell, C., & Gyorki, D. E. (2023). Practical Management of Adult Ultra-Rare Primary Retroperitoneal Soft Tissue Sarcoma: A Focus on Perivascular Epithelioid Tumours and Extraosseous Ewing Sarcoma. Current Oncology, 30(7), 5953-5972. https://doi.org/10.3390/curroncol30070445