Targeting Pro-Survival Autophagy Enhanced GSK-3β Inhibition-Induced Apoptosis and Retarded Proliferation in Bladder Cancer Cells

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Cell Viability Assay
2.3. Protein Extraction and Western Blot Analysis
2.4. Estimation of Autophagic Flux
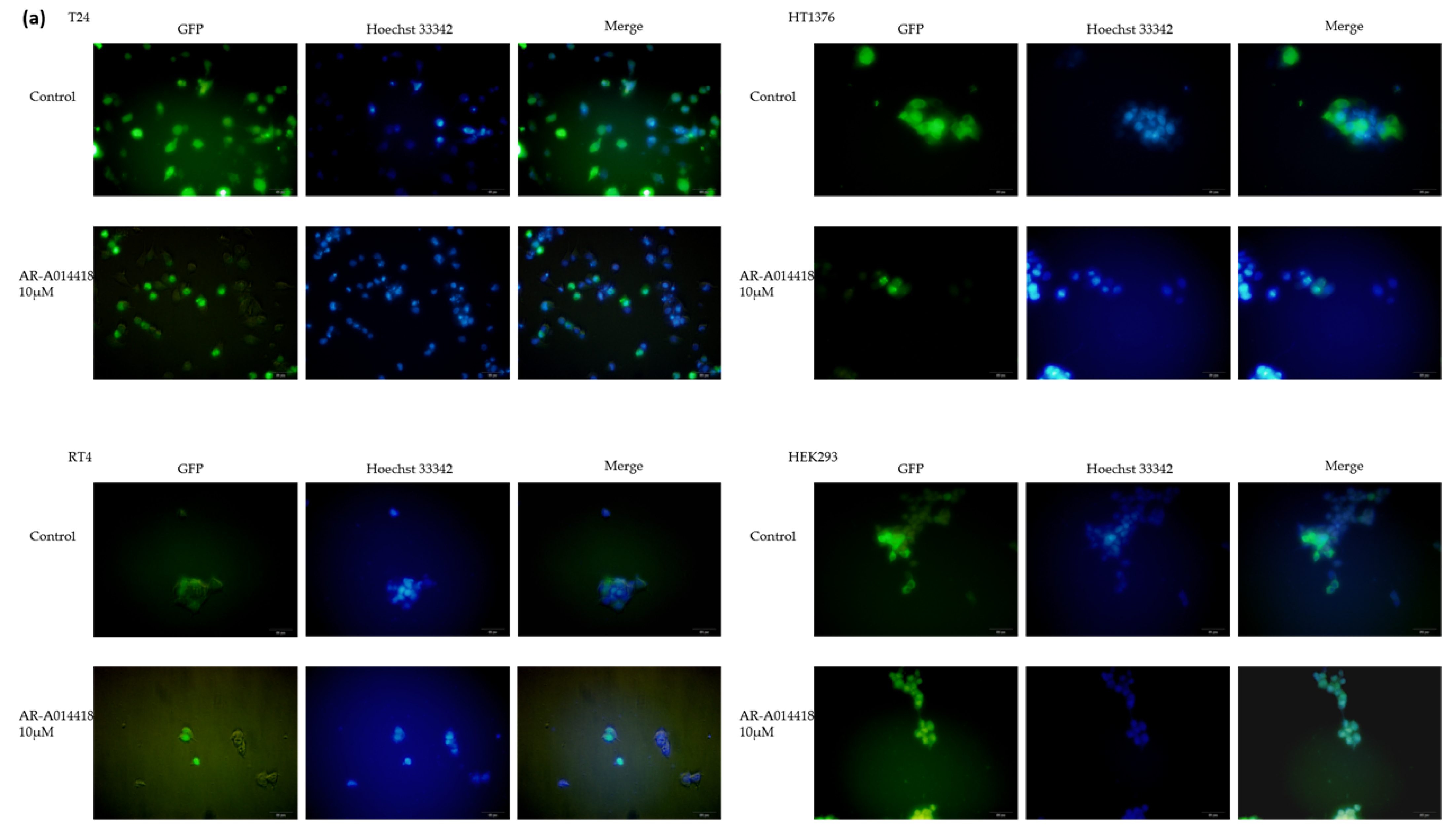
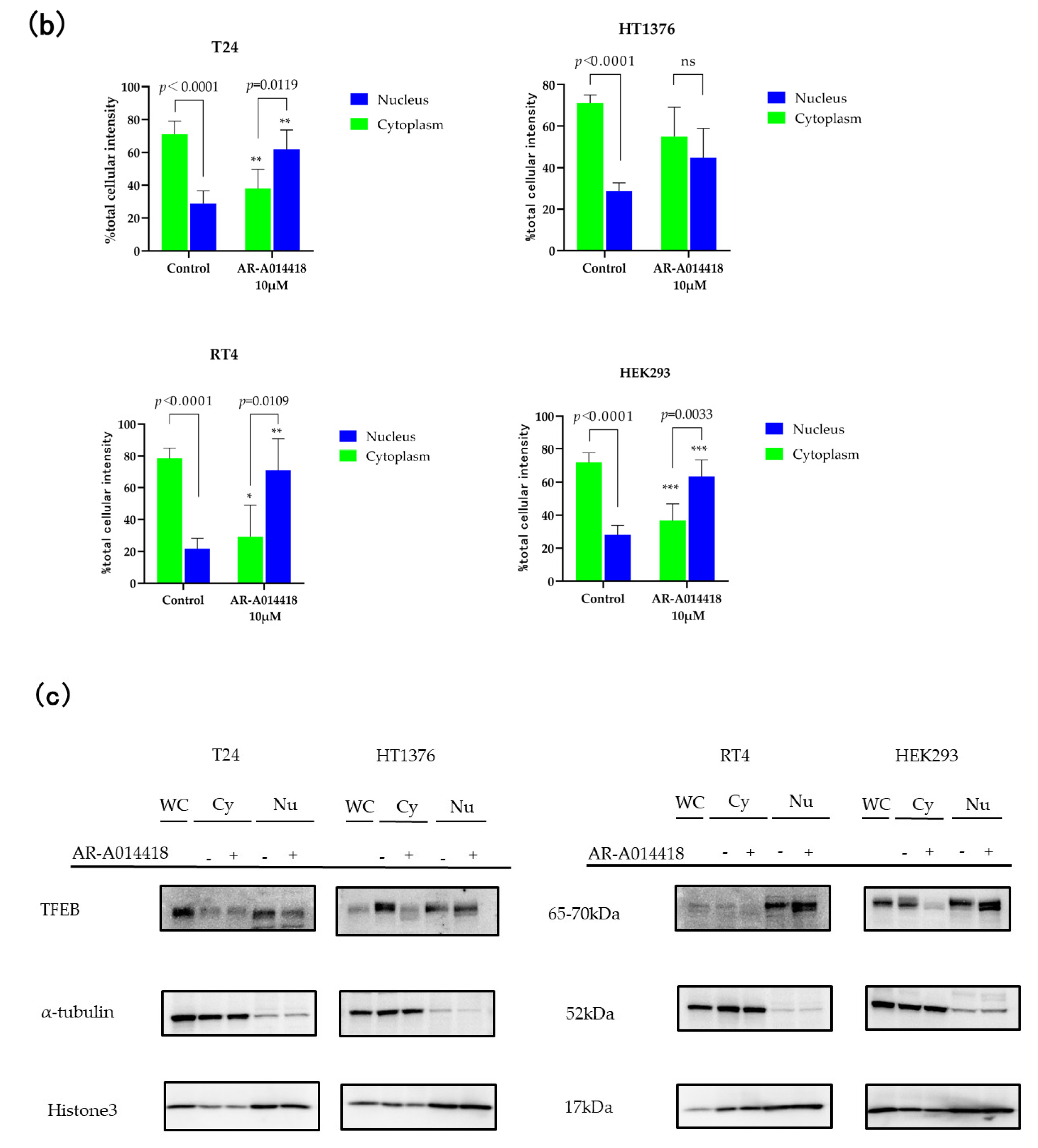
2.5. Nuclear Translocation of TFEB-EGFP
2.6. siRNA Transfection
2.7. Statistical Analysis
3. Results
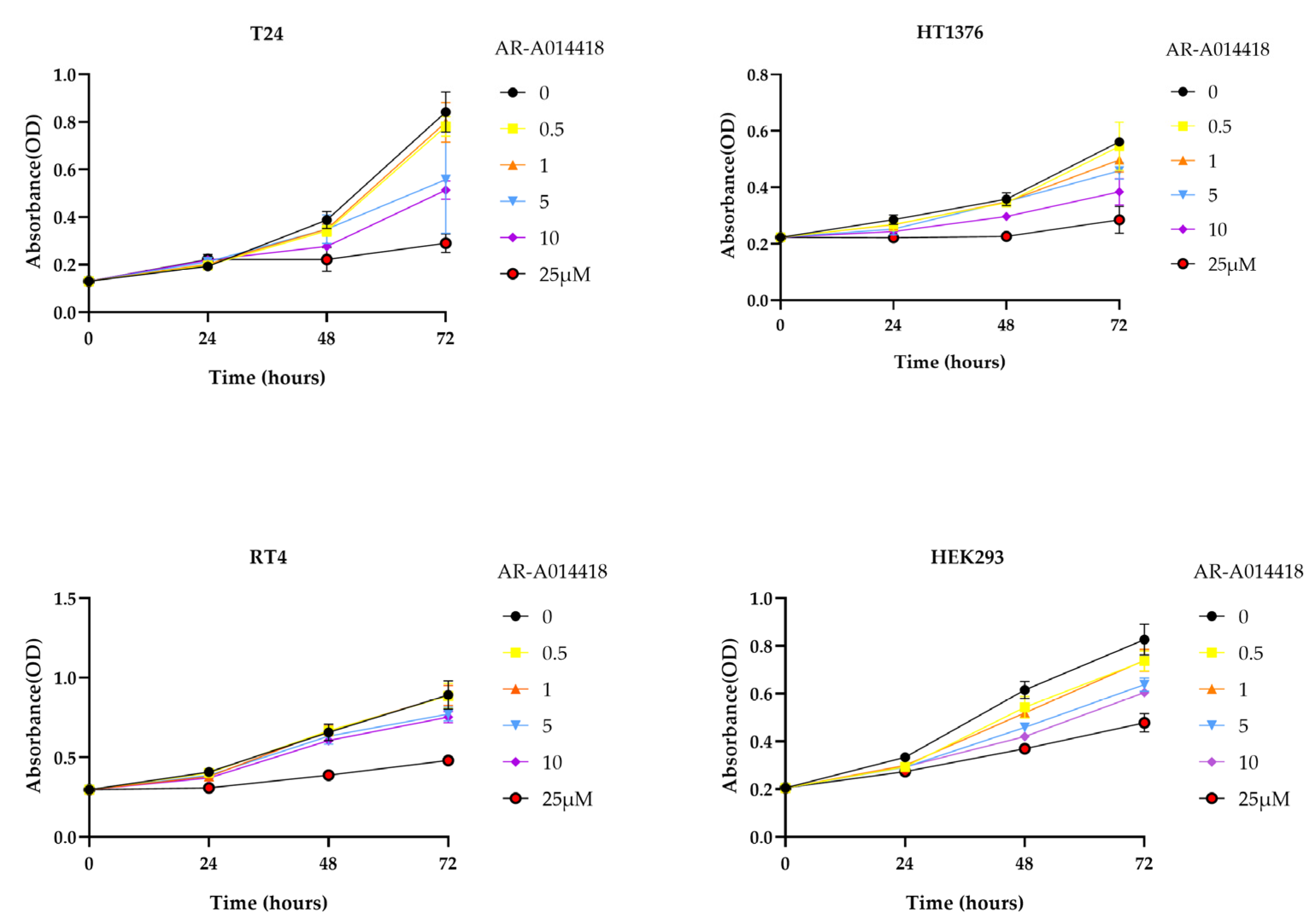
3.1. GSK-3 Inhibits the Proliferation of Bladder Cancer Cells and Has Low Cytotoxicity in Normal Cells
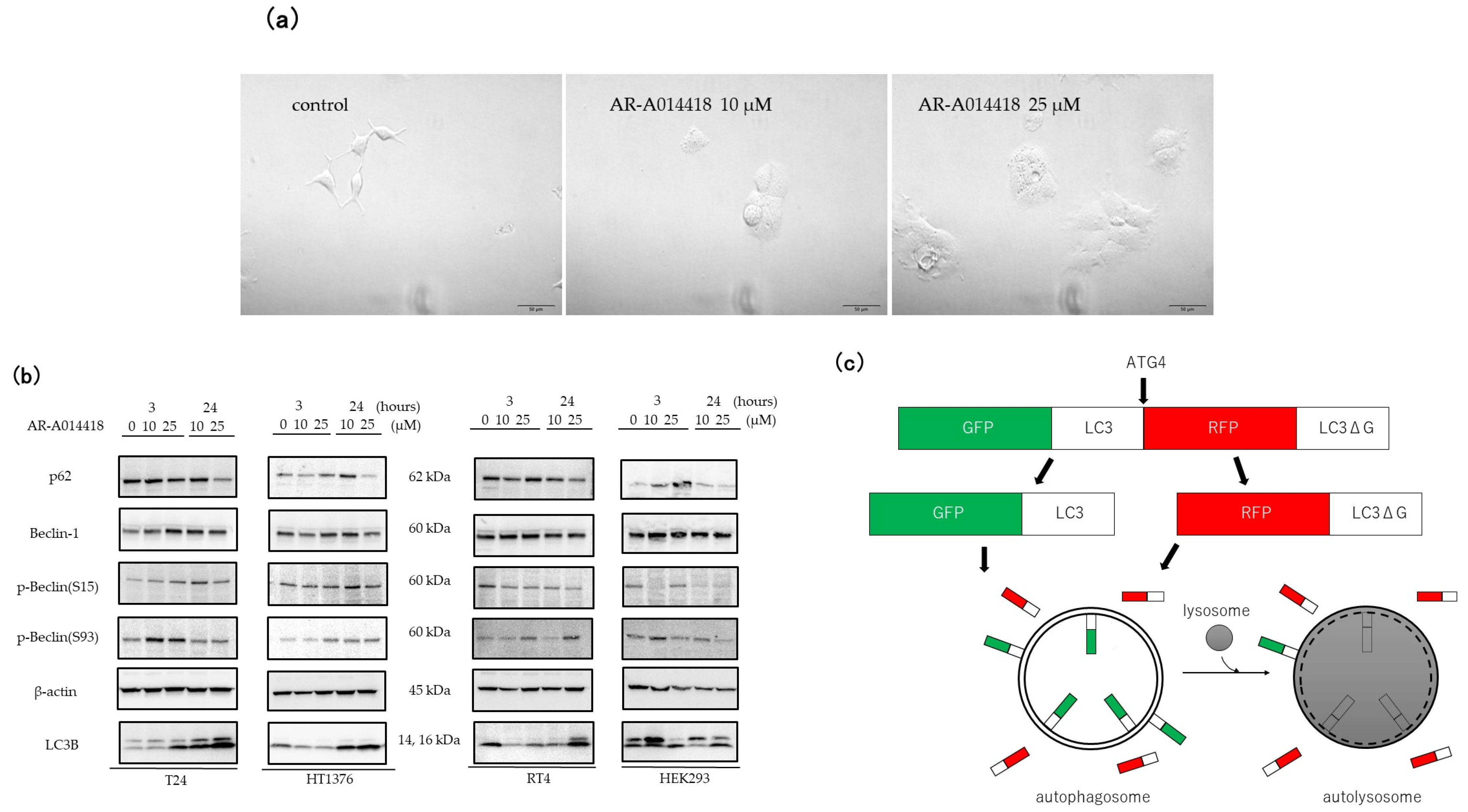
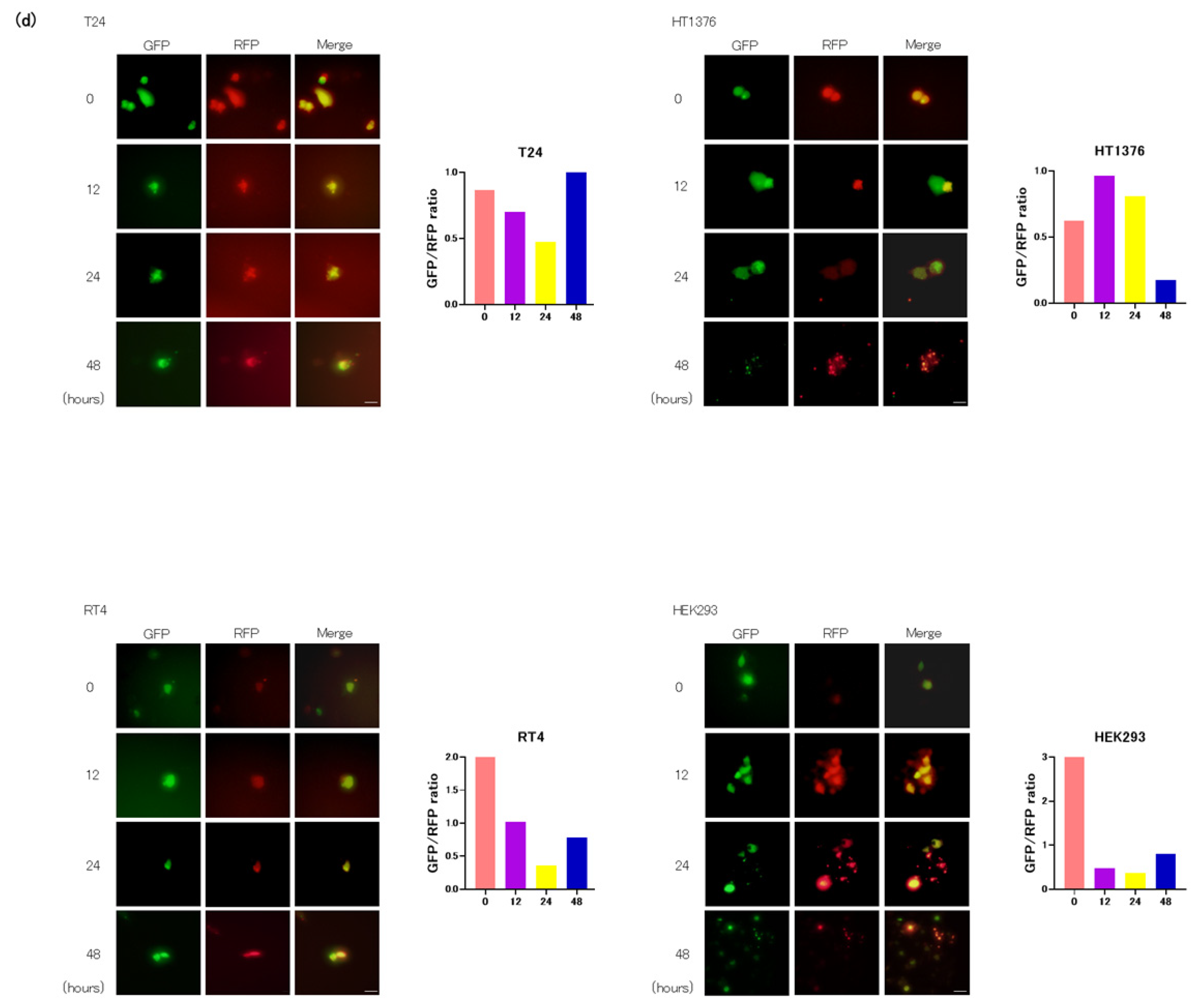
3.2. GSK-3 Inhibition Induces Autophagy in Cultured Bladder Cancer Cells
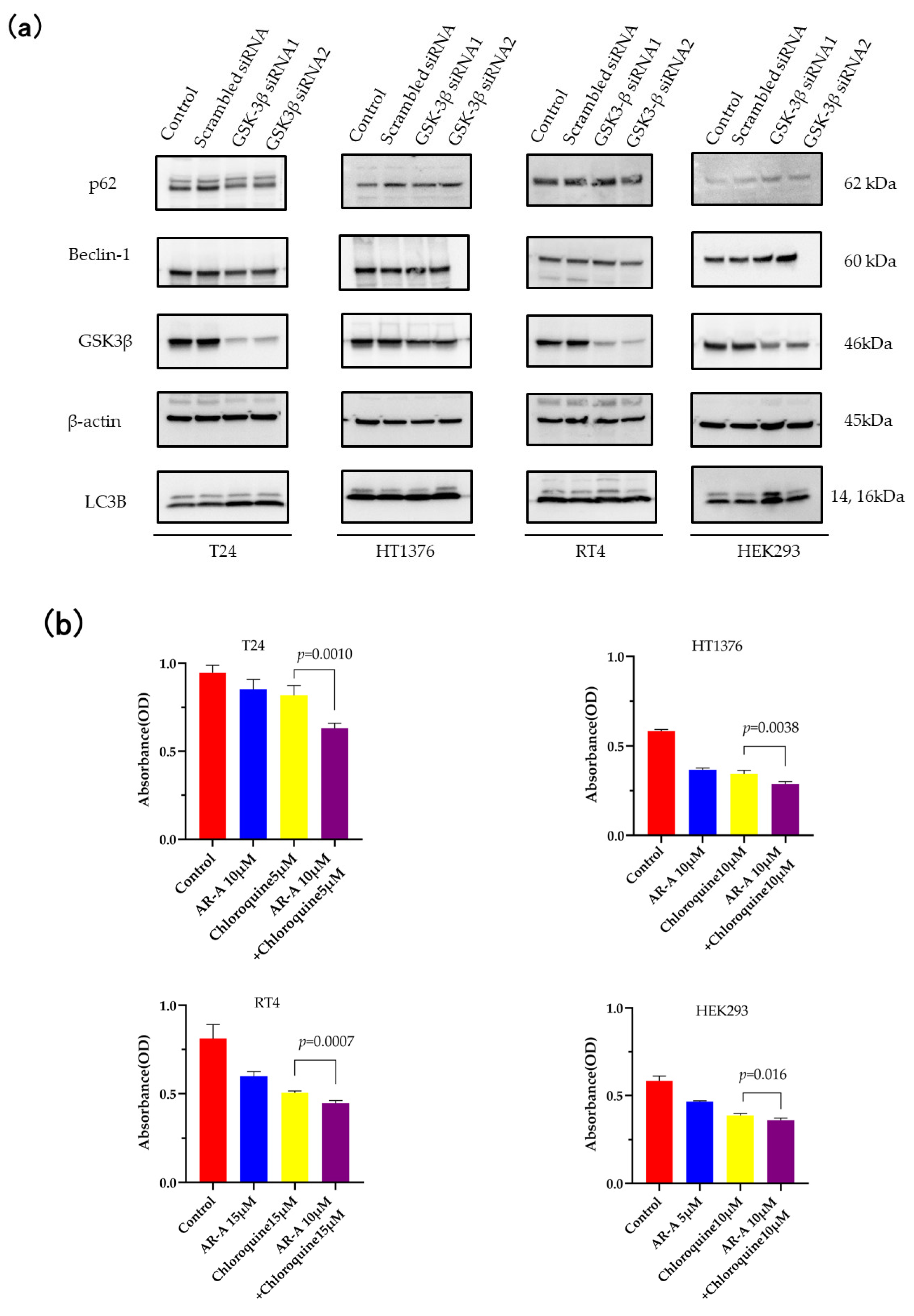
3.3. Inhibition of Autophagy Sensitizes the Cells to GSK-3 Inhibition-Induced Apoptosis
3.4. TFEB Nuclear Translocation Is Governed by GSK-3 Inhibition
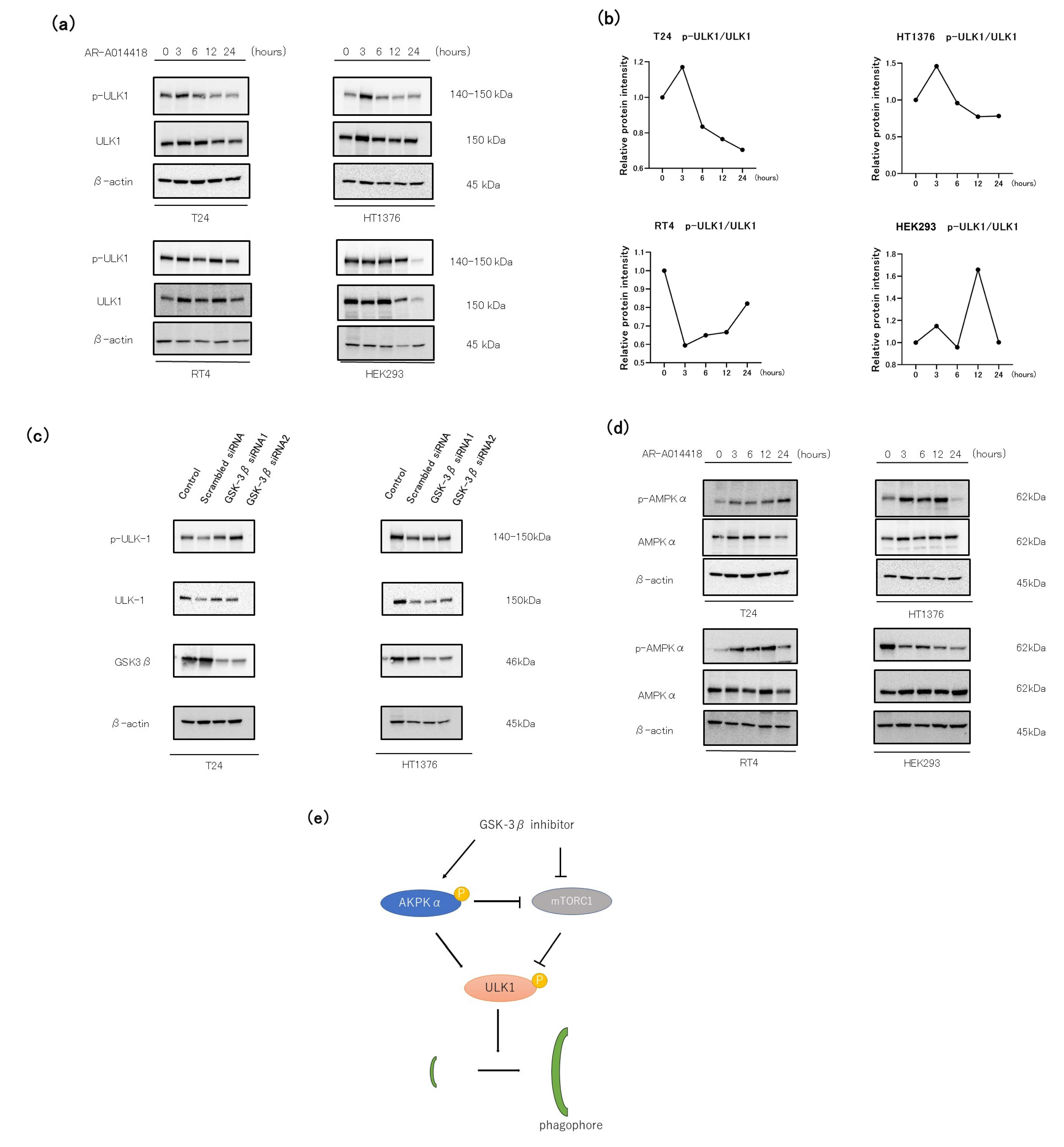
3.5. The Role of the AMP-AMPK-ULK1 Pathway in GSK-3 Inhibition-Induced Autophagy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Douchet, G. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. Oncologie 2017, 19, 205–206. [Google Scholar]
- Bladder Cancer: Statistics. Available online: https://www.cancer.net/cancer-types/bladder-cancer/statistics (accessed on 23 February 2023).
- Woodgett, J.R.; Cohen, P. Multisite phosphorylation of glycogen synthase: Molecular basis for the substrate specificity of glycogen synthease kinase-3 and casein kinase-II (glycogen synthase kinase-5). Biochim. Biophys. Acta 1984, 788, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H. Glycogen synthase kinase 3: An emerging therapeutic target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef]
- Naito, S.; Bilim, V.; Yuuki, K.; Ugolkov, A.; Motoyama, T.; Nagaoka, A.; Kato, T.; Tomita, Y. Glycogen Synthase Kinase-3β: A Prognostic Marker and a Potential Therapeutic Target in Human Bladder Cancer. Clin. Cancer Res. 2010, 16, 5124–5132. [Google Scholar] [CrossRef]
- Kuroki, H.; Anraku, T.; Kazama, A.; Bilim, V.; Tasaki, M.; Schmitt, D.; Mazar, A.P.; Giles, F.J.; Ugolkov, A.; Tomita, Y. 9-ING-41, a small molecule inhibitor of GSK-3beta, potentiates the effects of anticancer therapeutics in bladder cancer. Sci. Rep. 2019, 9, 19977. [Google Scholar] [CrossRef]
- Schlütermann, D.; Skowron, M.A.; Berleth, N.; Böhler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J.; et al. Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 160.e1–160.e13. [Google Scholar] [CrossRef]
- Lin, J.-F.; Lin, Y.-C.; Tsai, T.-F.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Dev. Ther. 2017, 11, 1517–1533. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03678883 (accessed on 23 February 2023).
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.-M. Therapeutic Targeting of Autophagy in Disease: Biology and Pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed]
- Bishop, E.; Bradshaw, T.D. Autophagy modulation: A prudent approach in cancer treatment? Cancer Chemother. Pharmacol. 2018, 82, 913–922. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy–Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Chao, X.; Wang, S.; Zhao, K.; Li, Y.; Williams, J.A.; Li, T.; Chavan, H.; Krishnamurthy, P.; He, X.C.; Li, L.; et al. Impaired TFEB-Mediated Lysosome Biogenesis and Autophagy Promote Chronic Ethanol-Induced Liver Injury and Steatosis in Mice. Gastroenterology 2018, 155, 865–879.e12. [Google Scholar] [CrossRef]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE 3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 9, 605–621. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H + -ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 Controls Phagophore Expansion during Autophagosome Formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733. [Google Scholar] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Morten, L.; Hijlkema, K.-J.; Coppes, R.P.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H.; et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell. Physiol. Biochem. 2017, 45, 54–66. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Konac, E.; Kurman, Y.; Baltaci, S. Contrast effects of autophagy in the treatment of bladder cancer. Exp. Biol. Med. 2021, 246, 354–367. [Google Scholar] [CrossRef]
- Yang, C.-F.K.; Sathiyaseelan, P.; Ho, C.; Gorski, S.M. Evolution of tools and methods for monitoring autophagic flux in mammalian cells. Biochem. Soc. Trans. 2018, 46, 97–110. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Sun, A.; Li, C.; Chen, R.; Huang, Y.; Chen, Q.; Cui, X.; Liu, H.; Thrasher, J.B.; Li, B. GSK-3β controls autophagy by modulating LKB1-AMPK pathway in prostate cancer cells. Prostate 2015, 76, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Cao, Y.; Gaisina, I.N.; Bhattacharya, S.; Dutta, S.K.; Wang, E.; Gunosewoyo, H.; Kozikowski, A.P.; Billadeau, D.D.; Mukhopadhyay, D. Inhibition of GSK-3 Induces Differentiation and Impaired Glucose Metabolism in Renal Cancer. Mol. Cancer Ther. 2014, 13, 285–296. [Google Scholar] [CrossRef]
- Kawamoto, M.; Horibe, T.; Kohno, M.; Kawakami, K. HER2-Targeted Hybrid Peptide That Blocks HER2 Tyrosine Kinase Disintegrates Cancer Cell Membrane and Inhibits Tumor Growth In Vivo. Mol. Cancer Ther. 2013, 12, 384–393. [Google Scholar] [CrossRef]
- Cao, Q.; You, X.; Xu, L.; Wang, L.; Chen, Y. PAQR3 suppresses the growth of non-small cell lung cancer cells via modulation of EGFR-mediated autophagy. Autophagy 2020, 16, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kim, S.O.; Li, Y.; Han, J. Autophagy Contributes to Caspase-independent Macrophage Cell Death. J. Biol. Chem. 2006, 281, 19179–19187. [Google Scholar] [CrossRef]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.-M.; Boucher, M.-J. Glycogen Synthase Kinase-3 (GSK3) Inhibition Induces Prosurvival Autophagic Signals in Human Pancreatic Cancer Cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef]
- Russi, S.; Sgambato, A.; Bochicchio, A.M.; Zoppoli, P.; Aieta, M.; Capobianco, A.M.L.; Ruggieri, V.; Zifarone, E.; Falco, G.; Laurino, S. CHIR99021, trough GSK-3β Targeting, Reduces Epithelioid Sarcoma Cell Proliferation by Activating Mitotic Catastrophe and Autophagy. Int. J. Mol. Sci. 2021, 22, 11147. [Google Scholar] [CrossRef]
- Piya, S.; Andreeff, M.; Borthakur, G. Targeting autophagy to overcome chemoresistance in acute myleogenous leukemia. Autophagy 2017, 13, 214–215. [Google Scholar] [CrossRef]
- Chiao, M.-T.; Cheng, W.-Y.; Yang, Y.-C.; Shen, C.-C.; Ko, J.-L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Amantini, C.; Morelli, M.B.; Liberati, S.; Farfariello, V.; Nabissi, M.; Bonfili, L.; Eleuteri, A.M.; Mozzicafreddo, M.; Burattini, L.; et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br. J. Cancer 2013, 109, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Xu, J.; Deng, X.; Xu, J.; Li, J.; Zhu, D.Q.; Zhu, J.; Jin, H.; Tian, Z.; Huang, H.; et al. New compound ChlA-F induces autophagy-dependent anti-cancer effect via upregulating Sestrin-2 in human bladder cancer. Cancer Lett. 2018, 436, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xu, K.; Liu, Y. Anticancer effect of salidroside reduces viability through autophagy/PI3K/Akt and MMP-9 signaling pathways in human bladder cancer cells. Oncol. Lett. 2018, 16, 3162–3168. [Google Scholar] [CrossRef]
- Kou, B.; Liu, W.; Xu, X.; Yang, Y.; Yi, Q.; Guo, F.; Li, J.; Zhou, J.; Kou, Q. Autophagy induction enhances tetrandrine-induced apoptosis via the AMPK/mTOR pathway in human bladder cancer cells. Oncol. Rep. 2017, 38, 3137–3143. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition. Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nature 2016, 18, 1065–1077. [Google Scholar] [CrossRef]
- Chen, W.R.; Bin Liu, H.; Chen, Y.D.; Sha, Y.; Ma, Q.; Zhu, P.J.; Mu, Y. Melatonin Attenuates Myocardial Ischemia/Reperfusion Injury by Inhibiting Autophagy Via an AMPK/mTOR Signaling Pathway. Cell. Physiol. Biochem. 2018, 47, 2067–2076. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Grunwald, D.S.; Otto, N.M.; Park, J.-M.; Song, D.; Kim, D.-H. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy 2020, 16, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Azoulay-Alfaguter, I.; Elya, R.; Avrahami, L.; Katz, A.; Eldar-Finkelman, H. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cell growth. Oncogene 2014, 34, 4613–4623. [Google Scholar] [CrossRef] [PubMed]
- Nave, O.; Hareli, S.; Elbaz, M.; Iluz, I.H.; Bunimovich-Mendrazitsky, S. BCG and IL—2 model for bladder cancer treatment with fast and slow dynamics based on SPVF method—Stability analysis. Math. Biosci. Eng. 2019, 16, 5346–5379. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Suo, H.; Zhou, S.; Hou, Z.; Bu, M.; Liu, X.; Xu, W. Afatinib induces pro-survival autophagy and increases sensitivity to apoptosis in stem-like HNSCC cells. Cell Death Dis. 2021, 12, 728. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Song, Y.; Huang, X.; Wang, L.; Luo, S.; Zhang, H.; Pan, H.; Jiang, W.; Qian, J.; Yao, G.; et al. mTORC1-dependent TFEB nucleus translocation and pro-survival autophagy induced by zeolitic imidazolate framework-8. Biomater. Sci. 2020, 8, 4358–4369. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Q.; Song, D.; Zen, R.; Zhang, L.; Wang, Y.; Yang, H.; Zhang, D.; Jia, J.; Zhang, J.; et al. Lysosomal dysfunction and autophagy blockade contribute to autophagy-related cancer suppressing peptide-induced cytotoxic death of cervical cancer cells through the AMPK/mTOR pathway. J. Exp. Clin. Cancer Res. 2020, 39, 197. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shirono, Y.; Bilim, V.; Anraku, T.; Kuroki, H.; Kazama, A.; Murata, M.; Hiruma, K.; Tomita, Y. Targeting Pro-Survival Autophagy Enhanced GSK-3β Inhibition-Induced Apoptosis and Retarded Proliferation in Bladder Cancer Cells. Curr. Oncol. 2023, 30, 5350-5365. https://doi.org/10.3390/curroncol30060406
Shirono Y, Bilim V, Anraku T, Kuroki H, Kazama A, Murata M, Hiruma K, Tomita Y. Targeting Pro-Survival Autophagy Enhanced GSK-3β Inhibition-Induced Apoptosis and Retarded Proliferation in Bladder Cancer Cells. Current Oncology. 2023; 30(6):5350-5365. https://doi.org/10.3390/curroncol30060406
Chicago/Turabian StyleShirono, Yuko, Vladimir Bilim, Tsutomu Anraku, Hiroo Kuroki, Akira Kazama, Masaki Murata, Kaede Hiruma, and Yoshihiko Tomita. 2023. "Targeting Pro-Survival Autophagy Enhanced GSK-3β Inhibition-Induced Apoptosis and Retarded Proliferation in Bladder Cancer Cells" Current Oncology 30, no. 6: 5350-5365. https://doi.org/10.3390/curroncol30060406
APA StyleShirono, Y., Bilim, V., Anraku, T., Kuroki, H., Kazama, A., Murata, M., Hiruma, K., & Tomita, Y. (2023). Targeting Pro-Survival Autophagy Enhanced GSK-3β Inhibition-Induced Apoptosis and Retarded Proliferation in Bladder Cancer Cells. Current Oncology, 30(6), 5350-5365. https://doi.org/10.3390/curroncol30060406