Relapsed/Refractory Multiple Myeloma: A Review of Available Therapies and Clinical Scenarios Encountered in Myeloma Relapse

Abstract
1. Introduction
2. Overview of Available Therapies in RRMM
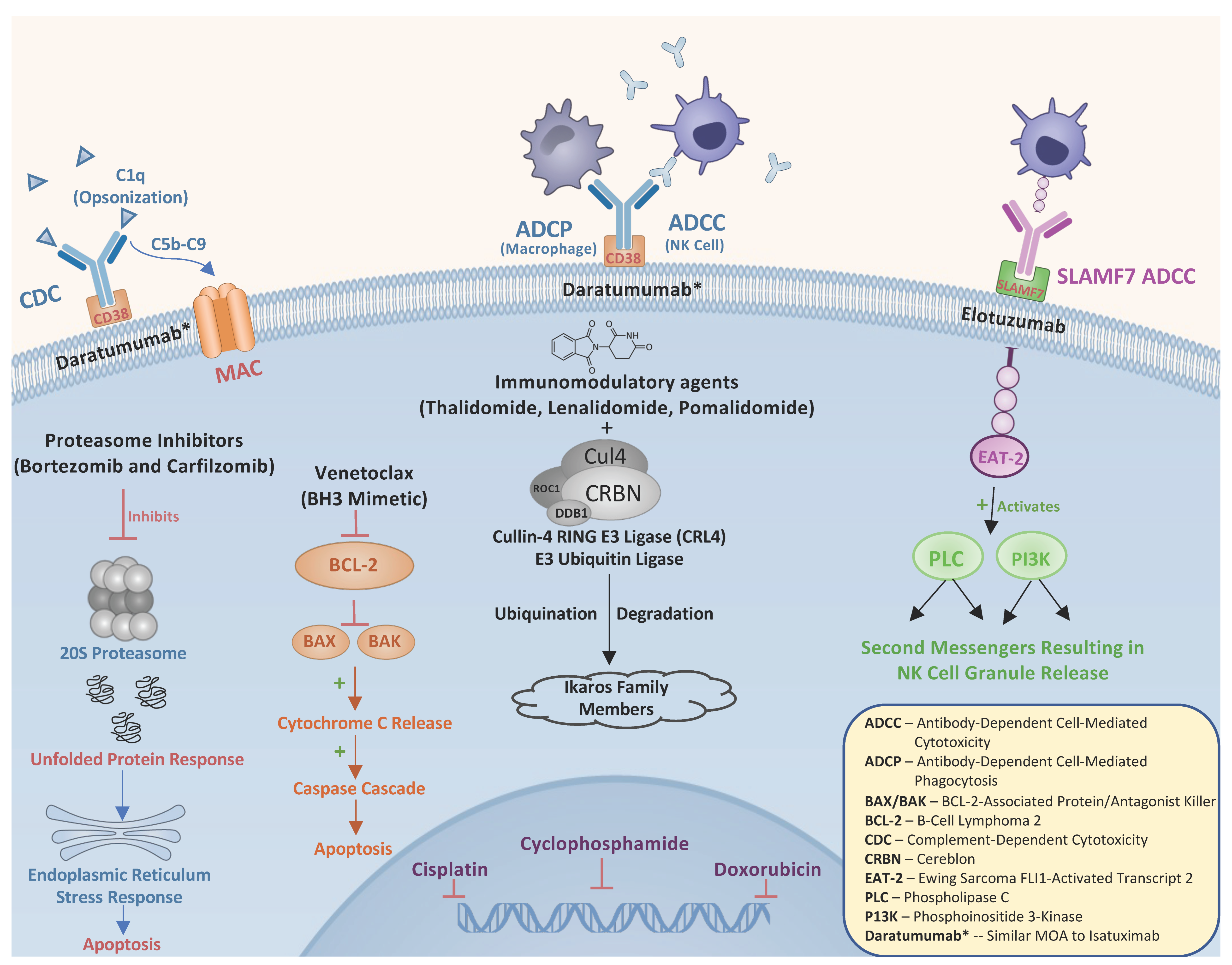
2.1. Immunomodulatory Drugs (IMiDs)
2.2. Proteasome Inhibitors
2.3. Monoclonal Antibodies
2.4. Chemotherapy
2.5. Venetoclax
2.6. Selinexor
2.7. CAR-T/BITE Therapy
3. Clinical Scenarios
3.1. Relapse Due to Lenalidomide Resistance/Refractoriness
3.1.1. Immunomodulation with Pomalidomide
3.1.2. Use of Monoclonal Antibody therapy
3.1.3. Switching to a Different Class of Medication
3.2. Relapse Due to Bortezomib Resistance/Refractoriness
3.3. Relapse Due to Daratumumab Resistance/Refractoriness
3.4. Autologous Stem Cell Transplant
3.5. Managing Myeloma Relapse with Renal Impairment
3.6. Managing Myeloma Relapse with Extramedullary Disease
4. Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ozen, S.; Aktay, N.; Lainka, E.; Duzova, A.; Bakkaloglu, A.; Kallinich, T. Disease severity in children and adolescents with Familial Mediterranean Fever: A comparative study to explore environmental effects on a monogenic disease. Ann. Rheum. Dis. 2008, 68, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Harousseau, J.L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Lonial, S.; Bladé, J.; Mateos, M.-V.; Dimopoulos, M.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Zervas, K.; Kouvatseas, G.; Galani, E.; Grigoraki, V.; Kiamouris, C.; Vervessou, E.; Samantas, E.; Papadimitriou, C.; Economou, O.; et al. Thalidomide and dexamethasone combination for refractory multiple myeloma. Ann. Oncol. 2001, 12, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kasserra, C.; Reyes, J.; Schafer, P.; Kosek, J.; Capone, L.; Parton, A.; Kim-Kang, H.; Surapaneni, S.; Kumar, G. Absorption, metabolism and excretion of [14C]pomalidomide in humans following oral administration. Cancer Chemother. Pharmacol. 2013, 71, 489–501. [Google Scholar] [CrossRef]
- Chen, N.; Wen, L.; Lau, H.; Surapaneni, S.; Kumar, G. Pharmacokinetics, metabolism and excretion of [(14)C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother. Pharmacol. 2012, 69, 789–797. [Google Scholar] [CrossRef]
- Gupta, D.; Treon, S.P.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.T.; Lin, B.; Lentzsch, S.; Davies, F.E.; Chauhan, D.; et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef]
- Li, S.; Pal, R.; Monaghan, S.A.; Schafer, P.; Ouyang, H.; Mapara, M.; Galson, D.L.; Lentzsch, S. IMiD immunomodulatory compounds block C/EBPbeta translation through eIF4E down-regulation resulting in inhibition of MM. Blood 2011, 117, 5157–5165. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.C.; Treon, S.P.; Anderson, K.C. Apoptotic signaling induced by immunomodulatory thalidomide analogs in human multiple myeloma cells: Therapeutic implications. Blood 2002, 99, 4525–4530. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Corral, L.G.; Haslett, P.A.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J. Immunol. 1999, 163, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.-T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, X.; He, X.; Zhou, Y.; Jiang, X.; Chen-Kiang, S.; Jaffrey, S.R.; Xu, G. A novel effect of thalidomide and its analogs: Suppression of cereblon ubiquitination enhances ubiquitin ligase function. FASEB J. 2015, 29, 4829–4839. [Google Scholar] [CrossRef]
- Morgan, B.; Sun, L.; Avitahl, N.; Andrikopoulos, K.; Ikeda, T.; Gonzales, E.; Wu, P.; Neben, S.; Georgopoulos, K. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J. 1997, 16, 2004–2013. [Google Scholar] [CrossRef]
- Cortés, M.; Georgopoulos, K. Aiolos Is Required for the Generation of High Affinity Bone Marrow Plasma Cells Responsible for Long-Term Immunity. J. Exp. Med. 2004, 199, 209–219. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.-X.; Kortuem, K.M.; Bruins, L.A.; Schmidt, J.E.; Chang, X.-B.; Langlais, P.; Luo, M.; Jedlowski, P.; et al. Identification of cereblon-binding proteins and relationship with response and survival after IMiDs in multiple myeloma. Blood 2014, 124, 536–545. [Google Scholar] [CrossRef]
- Shen, C.; Nayak, A.; Neitzel, L.R.; Adams, A.A.; Silver-Isenstadt, M.; Sawyer, L.M.; Benchabane, H.; Wang, H.; Bunnag, N.; Li, B.; et al. The E3 ubiquitin ligase component, Cereblon, is an evolutionarily conserved regulator of Wnt signaling. Nat. Commun. 2021, 12, 5263. [Google Scholar] [CrossRef] [PubMed]
- Manni, S.; Carrino, M.; Piazza, F. Role of protein kinases CK1α and CK2 in multiple myeloma: Regulation of pivotal survival and stress-managing pathways. J. Hematol. Oncol. 2017, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, C.; Prince, H.M.; Neeson, P.J. Understanding the Role of T-Cells in the Antimyeloma Effect of Immunomodulatory Drugs. Front. Immunol. 2021, 12, 632399. [Google Scholar] [CrossRef] [PubMed]
- Danhof, S.; Schreder, M.; Knop, S.; Rasche, L.; Strifler, S.; Loffler, C.; Gogishvili, T.; Einsele, H.; Hudecek, M. Expression of programmed death-1 on lymphocytes in myeloma patients is lowered during lenalidomide maintenance. Haematologica 2018, 103, e126–e129. [Google Scholar] [CrossRef] [PubMed]
- Luptakova, K.; Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Stroopinsky, D.; Kufe, T.; Vasir, B.; Arnason, J.; Tzachanis, D.; Zwicker, J.I.; et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol. Immunother. 2013, 62, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Zeh, D.; Janzen, V.; Mugge, L.O.; Wolf, D.; Fingerhut, L.; Hahn-Ast, C.; Maurer, O.; Brossart, P.; von Lilienfeld-Toal, M. Treatment with lenalidomide induces immunoactivating and counter-regulatory immunosuppressive changes in myeloma patients. Clin. Exp. Immunol. 2014, 177, 439–453. [Google Scholar] [CrossRef]
- Orlowski, R.Z.; Stinchcombe, T.E.; Mitchell, B.S.; Shea, T.C.; Baldwin, A.S.; Stahl, S.; Adams, J.; Esseltine, D.-L.; Elliott, P.J.; Pien, C.S.; et al. Phase I Trial of the Proteasome Inhibitor PS-341 in Patients With Refractory Hematologic Malignancies. J. Clin. Oncol. 2002, 20, 4420–4427. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.H.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Anderson, K.C. Extended follow-up of a phase II trial in relapsed, refractory multiple myeloma:: Final time-to-event results from the SUMMIT trial. Cancer 2006, 106, 1316–1319. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Matthews, G.M.; de Matos Simoes, R.; Dhimolea, E.; Sheffer, M.; Gandolfi, S.; Dashevsky, O.; Sorrell, J.D.; Mitsiades, C.S. NF-kappaB dysregulation in multiple myeloma. Semin. Cancer Biol. 2016, 39, 68–76. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Richardson, P.G.; Hideshima, T.; Munshi, N.; Treon, S.P.; Anderson, K.C. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: Therapeutic applications. Blood 2002, 99, 4079–4086. [Google Scholar] [CrossRef]
- Hideshima, T.; Richardson, P.G.; Anderson, K.C. Mechanism of Action of Proteasome Inhibitors and Deacetylase Inhibitors and the Biological Basis of Synergy in Multiple Myeloma. Mol. Cancer Ther. 2011, 10, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, V.; Gormley, N.J.; Luo, L.; Shen, Y.L.; Sridhara, R.; Subramaniam, S.; Shen, G.; Ma, L.; Shord, S.; Goldberg, K.B.; et al. FDA Approval Summary: Daratumumab for Treatment of Multiple Myeloma After One Prior Therapy. Oncologist 2017, 22, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Isatuximab: First Approval. Drugs 2020, 80, 905–912. [Google Scholar] [CrossRef]
- Sanchez, L.; Wang, Y.; Siegel, D.S.; Wang, M.L. Daratumumab: A first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J. Hematol. Oncol. 2016, 9, 51. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Wong, S.W.; Comenzo, R.L. CD38 Monoclonal Antibody Therapies for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2015, 15, 635–645. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef]
- Meyer, S.; Leusen, J.H.; Boross, P. Regulation of complement and modulation of its activity in monoclonal antibody therapy of cancer. mAbs 2014, 6, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Verploegen, S.; Bogels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef]
- D’Agostino, M.; Innorcia, S.; Boccadoro, M.; Bringhen, S. Monoclonal Antibodies to Treat Multiple Myeloma: A Dream Come True. Int. J. Mol. Sci. 2020, 21, 8192. [Google Scholar] [CrossRef] [PubMed]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef]
- Bouchon, A.; Cella, M.; Grierson, H.L.; Cohen, J.I.; Colonna, M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J. Immunol. 2001, 167, 5517–5521. [Google Scholar] [CrossRef]
- Ritchie, D.; Colonna, M. Mechanisms of Action and Clinical Development of Elotuzumab. Clin. Transl. Sci. 2018, 11, 261–266. [Google Scholar] [CrossRef]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef]
- Tassi, I.; Colonna, M. The Cytotoxicity Receptor CRACC (CS-1) Recruits EAT-2 and Activates the PI3K and Phospholipase Cγ Signaling Pathways in Human NK Cells1. J. Immunol. 2005, 175, 7996–8002. [Google Scholar] [CrossRef]
- Thompson, A.D.; Braun, B.S.; Arvand, A.; Stewart, S.D.; May, W.A.; Chen, E.; Korenberg, J.; Denny, C. EAT-2 is a novel SH2 domain containing protein that is up regulated by Ewing’s sarcoma EWS/FLI1 fusion gene. Oncogene 1996, 13, 2649–2658. [Google Scholar]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Yimer, H.; Melear, J.; Faber, E.; Bensinger, W.I.; Burke, J.M.; Narang, M.; Stevens, D.; Gray, K.S.; Lutska, Y.; Bobba, P.; et al. Daratumumab, cyclophosphamide, bortezomib, and dexamethasone for multiple myeloma: Final results of the LYRA study. Leuk. Lymphoma 2022, 63, 2383–2392. [Google Scholar] [CrossRef]
- Gomez-Arteaga, A.; Mark, T.M.; Guarneri, D.; Christos, P.J.; Gergis, U.; Greenberg, J.D.; Hsu, J.; Mayer, S.A.; Niesvizky, R.; Pearse, R.N.; et al. High-dose bendamustine and melphalan conditioning for autologous stem cell transplantation for patients with multiple myeloma. Bone Marrow Transpl. 2019, 54, 2027–2038. [Google Scholar] [CrossRef]
- Gold, J.M.; Raja, A. Cisplatin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ogino, M.H.; Tadi, P. Cyclophosphamide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Johnson-Arbor, K.; Dubey, R. Doxorubicin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Reyhanoglu, G.; Tadi, P. Etoposide. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sidiqi, M.H.; Al Saleh, A.S.; Kumar, S.K.; Leung, N.; Jevremovic, D.; Muchtar, E.; Gonsalves, W.I.; Kourelis, T.V.; Warsame, R.; Buadi, F.K.; et al. Venetoclax for the treatment of multiple myeloma: Outcomes outside of clinical trials. Am. J. Hematol. 2021, 96, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Czabotar, P.E. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb. Perspect. Biol. 2020, 12, a036400. [Google Scholar] [CrossRef] [PubMed]
- FDA. Selinexor. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selinexor-refractory-or-relapsed-multiple-myeloma (accessed on 1 February 2023).
- Mo, C.C.; Jagannath, S.; Chari, A.; Nooka, A.K.; Lonial, S.; Siegel, D.; Biran, N.; Gasparetto, C.; Bahlis, N.J.; Richardson, P. Selinexor for the treatment of patients with previously treated multiple myeloma. Expert Rev. Hematol. 2021, 14, 697–706. [Google Scholar] [CrossRef]
- Xu, D.; Grishin, N.V.; Chook, Y.M. NESdb: A database of NES-containing CRM1 cargoes. Mol. Biol. Cell 2012, 23, 3673–3676. [Google Scholar] [CrossRef]
- Argueta, C.; Kashyap, T.; Klebanov, B.; Unger, T.J.; Guo, C.; Harrington, S.; Baloglu, E.; Lee, M.; Senapedis, W.; Shacham, S.; et al. Selinexor synergizes with dexamethasone to repress mTORC1 signaling and induce multiple myeloma cell death. Oncotarget 2018, 9, 25529–25544. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Landesman, Y.; Acharya, C.; Calle, Y.; Zhong, M.Y.; Cea, M.; Tannenbaum, D.; Cagnetta, A.; Reagan, M.; Munshi, A.A.; et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Senapedis, W.; McCauley, D.; Baloglu, E.; Shacham, S.; Festuccia, C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 2014, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, T.; Argueta, C.; Aboukameel, A.; Unger, T.J.; Klebanov, B.; Mohammad, R.M.; Muqbil, I.; Azmi, A.S.; Drolen, C.; Senapedis, W.; et al. Selinexor, a Selective Inhibitor of Nuclear Export (SINE) compound, acts through NF-kappaB deactivation and combines with proteasome inhibitors to synergistically induce tumor cell death. Oncotarget 2016, 7, 78883–78895. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef]
- Rodriguez-Lobato, L.G.; Ganzetti, M.; Fernandez de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front. Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef]
- Cho, S.F.; Yeh, T.J.; Anderson, K.C.; Tai, Y.T. Bispecific antibodies in multiple myeloma treatment: A journey in progress. Front. Oncol. 2022, 12, 1032775. [Google Scholar] [CrossRef]
- Martinez-Hoyer, S.; Karsan, A. Mechanisms of lenalidomide sensitivity and resistance. Exp. Hematol. 2020, 91, 22–31. [Google Scholar] [CrossRef]
- Miguel, J.S.; Weisel, K.; Moreau, P.; Lacy, M.; Song, K.; Delforge, M.; Karlin, L.; Goldschmidt, H.; Banos, A.; Oriol, A.; et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013, 14, 1055–1066. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Palumbo, A.; Corradini, P.; Cavo, M.; Delforge, M.; Di Raimondo, F.; Weisel, K.C.; Oriol, A.; Hansson, M.; Vacca, A.; et al. Safety and efficacy of pomalidomide plus low-dose dexamethasone in STRATUS (MM-010): A phase 3b study in refractory multiple myeloma. Blood 2016, 128, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Schjesvold, F.; Weisel, K.; Moreau, P.; Anderson, L.D., Jr.; White, D.; Rodriguez-Otero, P.; Sonneveld, P.; Engelhardt, M.; Jenner, M.; et al. Pomalidomide, bortezomib, and dexamethasone at first relapse in lenalidomide-pretreated myeloma: A subanalysis of OPTIMISMM by clinical characteristics. Eur. J. Haematol. 2022, 108, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Overall Survival With Daratumumab, Bortezomib, and Dexamethasone in Previously Treated Multiple Myeloma (CASTOR): A Randomized, Open-Label, Phase III Trial. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Gavriatopoulou, M.; Oriol, A.; Rabin, N.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022, 23, 65–76. [Google Scholar] [CrossRef]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Spicka, I.; Baker, R.; Kim, K.; et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): A multicentre, open-label, randomised phase 3 trial. Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Jakubowiak, A.; Offidani, M.; Pegourie, B.; De La Rubia, J.; Garderet, L.; Laribi, K.; Bosi, A.; Marasca, R.; Laubach, J.; Mohrbacher, A.; et al. Randomized phase 2 study: Elotuzumab plus bortezomib/dexamethasone vs. bortezomib/dexamethasone for relapsed/refractory MM. Blood 2016, 127, 2833–2840. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Terpos, E.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Baldini, L.; Symeonidis, A.; Bila, J.; et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): An open-label, randomised, phase 3 trial. Lancet Oncol. 2021, 22, 801–812. [Google Scholar] [CrossRef]
- Richardson, P.G.; Attal, M.; Campana, F.; Le-Guennec, S.; Hui, A.M.; Risse, M.L.; Corzo, K.; Anderson, K.C. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Future Oncol. 2018, 14, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; LeBlanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab Plus Pomalidomide and Dexamethasone for Relapsed/Refractory Multiple Myeloma: Final Overall Survival Analysis From the Randomized Phase II ELOQUENT-3 Trial. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef] [PubMed]
- Bringhen, S.; Pour, L.; Vorobyev, V.; Vural, F.; Warzocha, K.; Benboubker, L.; Koh, Y.; Maisnar, V.; Karlin, L.; Pavic, M.; et al. Isatuximab plus pomalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma according to prior lines of treatment and refractory status: ICARIA-MM subgroup analysis. Leuk. Res. 2021, 104, 106576. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Perrot, A.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; Huang, J.S.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): Follow-up analysis of a randomised, phase 3 study. Lancet Oncol. 2022, 23, 416–427. [Google Scholar] [CrossRef]
- Terpos, E.; Dimopoulos, M.A.; Boccadoro, M.; Delimpasi, S.; Beksac, M.; Katodritou, E.; Moreau, P.; Pompa, A.; Symeonidis, A.; Bila, J.; et al. Health-related quality of life in patients with relapsed/refractory multiple myeloma treated with pomalidomide and dexamethasone +/− subcutaneous daratumumab: Patient-reported outcomes from the APOLLO trial. Am. J. Hematol. 2022, 97, 481–490. [Google Scholar] [CrossRef]
- Grosicki, S.; Simonova, M.; Spicka, I.; Pour, L.; Kriachok, I.; Gavriatopoulou, M.; Pylypenko, H.; Auner, H.W.; Leleu, X.; Doronin, V.; et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): A randomised, open-label, phase 3 trial. Lancet 2020, 396, 1563–1573. [Google Scholar] [CrossRef]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Stewart, A.K.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.; Mihaylov, G.G.; Goranova-Marinova, V.; et al. Carfilzomib, lenalidomide, and dexamethasone in patients with relapsed multiple myeloma categorised by age: Secondary analysis from the phase 3 ASPIRE study. Br. J. Haematol. 2017, 177, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hajek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in Overall Survival With Carfilzomib, Lenalidomide, and Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Moreau, P.; Niesvizky, R.; Ludwig, H.; Oriol, A.; Chng, W.J.; Goldschmidt, H.; Yang, Z.; Kimball, A.S.; Dimopoulos, M. Carfilzomib-Dexamethasone Versus Bortezomib-Dexamethasone in Relapsed or Refractory Multiple Myeloma: Updated Overall Survival, Safety, and Subgroups. Clin. Lymphoma Myeloma Leuk. 2019, 19, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef]
- Richardson, P.G.; Kumar, S.K.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; et al. Final Overall Survival Analysis of the TOURMALINE-MM1 Phase III Trial of Ixazomib, Lenalidomide, and Dexamethasone in Patients With Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2021, 39, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, P.; Zweegman, S.; Cavo, M.; Kazem, N.; Broyl, A.; Troia, R.; Pour, L.; Croockewit; Corradini, P.; Bos, G.; et al. Carfilzomib, Pomalidomide and Dexamethasone (KPd) in Patients with First Progression of Multiple Myeloma Refractory to Bortezomib and Lenalidomide. Final Report of the EMN011/HOVON114 Trial. Blood 2021, 138, 1664. [Google Scholar] [CrossRef]
- Mateos, M.V.; Ocio, E.M.; Balari, A.; Oriol, A.; Garcia, E.; Moreno, M.; Granell, M.; Escalante, F.; Gonzalez De La Calle, E.; Lopez, J.; et al. Randomized Phase 2 Study of Weekly Carfilzomib 70 Mg/m2 and Dexamethasone Plus/Minus Cyclophosphamide in Relapsed and/or Refractory Multiple Myeloma (RRMM) Patients (GEM-KyCyDex). Blood 2021, 136, 8–9. [Google Scholar] [CrossRef]
- Garderet, L.; Kuhnowski, F.; Berge, B.; Roussel, M.; Escoffre-Barbe, M.; Lafon, I.; Facon, T.; Leleu, X.; Karlin, L.; Perrot, A.; et al. Pomalidomide, cyclophosphamide, and dexamethasone for relapsed multiple myeloma. Blood 2018, 132, 2555–2563. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Franssen, L.E.; Levin, M.D.; Bos, G.M.J.; Broijl, A.; Klein, S.K.; Koene, H.R.; Bloem, A.C.; Beeker, A.; Faber, L.M.; et al. Phase 1/2 study of lenalidomide combined with low-dose cyclophosphamide and prednisone in lenalidomide-refractory multiple myeloma. Blood 2016, 128, 2297–2306. [Google Scholar] [CrossRef]
- Nooka, A.K.; Joseph, N.S.; Kaufman, J.L.; Heffner, L.T.; Gupta, V.A.; Gleason, C.; Boise, L.H.; Lonial, S. Clinical efficacy of daratumumab, pomalidomide, and dexamethasone in patients with relapsed or refractory myeloma: Utility of re-treatment with daratumumab among refractory patients. Cancer 2019, 125, 2991–3000. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef] [PubMed]
- GSK. Available online: https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-dreamm-3-phase-iii-trial-for-blenrep/ (accessed on 1 February 2023).
- FDA. Teclistamab. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-teclistamab-cqyv-relapsed-or-refractory-multiple-myeloma (accessed on 1 February 2023).
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-cell maturation antigen x CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MajesTEC-1): A multicentre, open-label, single-arm, phase 1 study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, A.; Singh, P.P.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; Dingli, D.; Hwa, Y.L.; Fonder, A.L.; et al. Efficacy of VDT PACE-like regimens in treatment of relapsed/refractory multiple myeloma. Am. J. Hematol. 2018, 93, 179–186. [Google Scholar] [CrossRef]
- Lazzarino, M.; Corso, A.; Barbarano, L.; Alessandrino, E.P.; Cairoli, R.; Pinotti, G.; Ucci, G.; Uziel, L.; Rodeghiero, F.; Fava, S.; et al. DCEP (dexamethasone, cyclophosphamide, etoposide, and cisplatin) is an effective regimen for peripheral blood stem cell collection in multiple myeloma. Bone Marrow Transpl. 2001, 28, 835–839. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; Maracci, L.; Liberati, A.M.; Ballanti, S.; Attolico, I.; Caraffa, P.; Alesiani, F.; Caravita di Toritto, T.; Gentili, S.; et al. Efficacy and tolerability of bendamustine, bortezomib and dexamethasone in patients with relapsed-refractory multiple myeloma: A phase II study. Blood Cancer J. 2013, 3, e162. [Google Scholar] [CrossRef]
- Lentzsch, S.; O’Sullivan, A.; Kennedy, R.C.; Abbas, M.; Dai, L.; Pregja, S.L.; Burt, S.; Boyiadzis, M.; Roodman, G.D.; Mapara, M.Y.; et al. Combination of bendamustine, lenalidomide, and dexamethasone (BLD) in patients with relapsed or refractory multiple myeloma is feasible and highly effective: Results of phase 1/2 open-label, dose escalation study. Blood 2012, 119, 4608–4613. [Google Scholar] [CrossRef]
- Richardson, P.G.; Jacobus, S.J.; Weller, E.A.; Hassoun, H.; Lonial, S.; Raje, N.S.; Medvedova, E.; McCarthy, P.L.; Libby, E.N.; Voorhees, P.M.; et al. Triplet Therapy, Transplantation, and Maintenance until Progression in Myeloma. N. Engl. J. Med. 2022, 387, 132–147. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Cazaubiel, T.; Facon, T.; Caillot, D.; Clement-Filliatre, L.; Macro, M.; Decaux, O.; Belhadj, K.; Mohty, M.; et al. Early Versus Late Autologous Stem Cell Transplant in Newly Diagnosed Multiple Myeloma: Long-Term Follow-up Analysis of the IFM 2009 Trial. Blood 2020, 136, 39. [Google Scholar] [CrossRef]
- Cook, G.; Ashcroft, A.J.; Cairns, D.A.; Williams, C.D.; Brown, J.M.; Cavenagh, J.D.; Snowden, J.A.; Parrish, C.; Yong, K.; Cavet, J.; et al. The effect of salvage autologous stem-cell transplantation on overall survival in patients with relapsed multiple myeloma (final results from BSBMT/UKMF Myeloma X Relapse [Intensive]): A randomised, open-label, phase 3 trial. Lancet Haematol. 2016, 3, e340–e351. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Baertsch, M.-A.; Schlenzka, J.; Becker, N.; Christina, H.; Hielscher, T.; Raab, M.; Hillengass, J.; Müller-Tidow, C.; Luntz, S.; et al. Salvage Autologous Transplant and Lenalidomide Maintenance Versus Continuous Lenalidomide/Dexamethasone for Relapsed Multiple Myeloma: Results of the Randomized GMMG Phase III Multicenter Trial Relapse. Blood 2018, 132, 253. [Google Scholar] [CrossRef]
- Cook, G.; Royle, K.L.; O’Connor, S.; Cairns, D.A.; Ashcroft, A.J.; Williams, C.D.; Hockaday, A.; Cavenagh, J.D.; Snowden, J.A.; Ademokun, D.; et al. The impact of cytogenetics on duration of response and overall survival in patients with relapsed multiple myeloma (long-term follow-up results from BSBMT/UKMF Myeloma X Relapse [Intensive]): A randomised, open-label, phase 3 trial. Br. J. Haematol. 2019, 185, 450–467. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, M.-A.; Schlenzka, J.; Christina, H.; Hielscher, T.; Raab, M.; Hillengass, J.; Müller-Tidow, C.; Luntz, S.; Jauch, A.; Brossart, P.; et al. Subgroup Analyses of the Randomized GMMG Phase III Multicenter Trial Relapse Suggest Survival Benefit of Salvage Autologous Transplant Primarily in Low Risk Multiple Myeloma. Blood 2018, 132, 254. [Google Scholar] [CrossRef]
- Michaelis, L.C.; Saad, A.; Zhong, X.; Le-Rademacher, J.; Freytes, C.O.; Marks, D.I.; Lazarus, H.M.; Bird, J.M.; Holmberg, L.; Kamble, R.T.; et al. Salvage second hematopoietic cell transplantation in myeloma. Biol. Blood Marrow Transpl. 2013, 19, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.; D’Souza, A.; Kleman, A.; Chhabra, S.; Mohan, M.; Hari, P. Salvage second transplantation in relapsed multiple myeloma. Leukemia 2021, 35, 1214–1217. [Google Scholar] [CrossRef]
- Vakiti, A.; Padala, S.A.; Mewawalla, P. Myeloma Kidney. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Abbott, K.C.; Agodoa, L.Y. Multiple myeloma and light chain-associated nephropathy at end-stage renal disease in the United States: Patient characteristics and survival. Clin. Nephrol. 2001, 56, 207–210. [Google Scholar]
- Tsakiris, D.J.; Stel, V.S.; Finne, P.; Fraser, E.; Heaf, J.; de Meester, J.; Schmaldienst, S.; Dekker, F.; Verrina, E.; Jager, K.J. Incidence and outcome of patients starting renal replacement therapy for end-stage renal disease due to multiple myeloma or light-chain deposit disease: An ERA-EDTA Registry study. Nephrol. Dial. Transpl. 2010, 25, 1200–1206. [Google Scholar] [CrossRef]
- Bozic, B.; Rutner, J.; Zheng, C.; Ruckser, R.; Selimi, F.; Racz, K.; Kocher, M.; Tatzreiter, G.; Sebesta, C. Advances in the Treatment of Relapsed and Refractory Multiple Myeloma in Patients with Renal Insufficiency: Novel Agents, Immunotherapies and Beyond. Cancers 2021, 13, 36. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Mikhael, J.; Terpos, E.; Leleu, X.; Moreau, P.; Blade, J.; Kim, J.S.; Stockerl-Goldstein, K.; Richardson, P.G. An overview of treatment options for patients with relapsed/refractory multiple myeloma and renal impairment. Adv. Hematol. 2022, 13, 20406207221088458. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Leung, N.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Dingli, D.; Kapoor, P.; Go, R.S.; et al. Improvement in renal function and its impact on survival in patients with newly diagnosed multiple myeloma. Blood Cancer J. 2015, 5, e296. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Leleu, X.; Moreau, P.; Richardson, P.G.; Liberati, A.M.; Harrison, S.J.; Miles Prince, H.; Ocio, E.M.; Assadourian, S.; Campana, F.; et al. Isatuximab plus pomalidomide and dexamethasone in relapsed/refractory multiple myeloma patients with renal impairment: ICARIA-MM subgroup analysis. Leukemia 2021, 35, 562–572. [Google Scholar] [CrossRef]
- Capra, M.; Martin, T.; Moreau, P.; Baker, R.; Pour, L.; Min, C.K.; Leleu, X.; Mohty, M.; Segura, M.R.; Turgut, M.; et al. Isatuximab plus carfilzomib and dexamethasone versus carfilzomib and dexamethasone in relapsed multiple myeloma patients with renal impairment: IKEMA subgroup analysis. Haematologica 2022, 107, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraman, P.; Bhasin, A.; Dayal, N.; Pathak, S.; Naithani, R. Daratumumab in dialysis-dependent multiple myeloma. Blood Res. 2020, 55, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.; Glavey, S.; Melotti, D.; Thornton, P.; Sargent, J.; Conlon, P.; Murphy, P.; Quinn, J. Dialysis independence following single-agent daratumumab in refractory myeloma with renal failure. Ir. J. Med. Sci. 2019, 188, 1079–1080. [Google Scholar] [CrossRef]
- Kastritis, E.; Terpos, E.; Symeonidis, A.; Delimpasi, S.; Cavo, M.; Zamagni, E.; Katodritou, E.; Rivolti, E.; Kyrtsonis, M.-C.; Gavriatopoulou, M.; et al. Daratumumab with Dexamethasone in Patients with Relapsed/Refractory Multiple Myeloma and Severe Renal Impairment: Results on Efficacy and Safety of the Phase 2 Dare Study. Blood 2020, 136, 48–49. [Google Scholar] [CrossRef]
- Antlanger, M.; Dust, T.; Reiter, T.; Bohm, A.; Lamm, W.W.; Gornicec, M.; Willenbacher, E.; Nachbaur, D.; Weger, R.; Rabitsch, W.; et al. Impact of renal impairment on outcomes after autologous stem cell transplantation in multiple myeloma: A multi-center, retrospective cohort study. BMC Cancer 2018, 18, 1008. [Google Scholar] [CrossRef]
- Lazana, I.; Floro, L.; Christmas, T.; Shah, S.; Bramham, K.; Cuthill, K.; Bassett, P.; Schey, S.; Kazmi, M.; Potter, V.; et al. Autologous stem cell transplantation for multiple myeloma patients with chronic kidney disease: A safe and effective option. Bone Marrow Transpl. 2022, 57, 959–965. [Google Scholar] [CrossRef]
- St Bernard, R.; Chodirker, L.; Masih-Khan, E.; Jiang, H.; Franke, N.; Kukreti, V.; Tiedemann, R.; Trudel, S.; Reece, D.; Chen, C.I. Efficacy, toxicity and mortality of autologous SCT in multiple myeloma patients with dialysis-dependent renal failure. Bone Marrow Transpl. 2015, 50, 95–99. [Google Scholar] [CrossRef]
- Lee, C.K.; Zangari, M.; Barlogie, B.; Fassas, A.; van Rhee, F.; Thertulien, R.; Talamo, G.; Muwalla, F.; Anaissie, E.; Hollmig, K.; et al. Dialysis-dependent renal failure in patients with myeloma can be reversed by high-dose myeloablative therapy and autotransplant. Bone Marrow Transpl. 2004, 33, 823–828. [Google Scholar] [CrossRef]
- Touzeau, C.; Moreau, P. How I treat extramedullary myeloma. Blood 2016, 127, 971–976. [Google Scholar] [CrossRef]
- Pour, L.; Sevcikova, S.; Greslikova, H.; Kupska, R.; Majkova, P.; Zahradova, L.; Sandecka, V.; Adam, Z.; Krejci, M.; Kuglik, P.; et al. Soft-tissue extramedullary multiple myeloma prognosis is significantly worse in comparison to bone-related extramedullary relapse. Haematologica 2014, 99, 360–364. [Google Scholar] [CrossRef]
- Bansal, R.; Rakshit, S.; Kumar, S. Extramedullary disease in multiple myeloma. Blood Cancer J. 2021, 11, 161. [Google Scholar] [CrossRef]
- Rasche, L.; Strifler, S.; Duell, J.; Rosenwald, A.; Buck, A.; Maeder, U.; Einsele, H.; Knop, S. The lymphoma-like polychemotherapy regimen "Dexa-BEAM" in advanced and extramedullary multiple myeloma. Ann. Hematol. 2014, 93, 1207–1214. [Google Scholar] [CrossRef]
- Zhou, X.; Fluchter, P.; Nickel, K.; Meckel, K.; Messerschmidt, J.; Bockle, D.; Knorz, S.; Steinhardt, M.J.; Krummenast, F.; Danhof, S.; et al. Carfilzomib Based Treatment Strategies in the Management of Relapsed/Refractory Multiple Myeloma with Extramedullary Disease. Cancers 2020, 12, 1035. [Google Scholar] [CrossRef]
- Short, K.D.; Rajkumar, S.V.; Larson, D.; Buadi, F.; Hayman, S.; Dispenzieri, A.; Gertz, M.; Kumar, S.; Mikhael, J.; Roy, V.; et al. Incidence of extramedullary disease in patients with multiple myeloma in the era of novel therapy, and the activity of pomalidomide on extramedullary myeloma. Leukemia 2011, 25, 906–908. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Weiss, B.M.; Plesner, T.; Bahlis, N.J.; Belch, A.; Lonial, S.; Lokhorst, H.M.; Voorhees, P.M.; Richardson, P.G.; Chari, A.; et al. Clinical efficacy of daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma. Blood 2016, 128, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qiu, Y.; Personett, D.; Huang, P.; Edenfield, B.; Katz, J.; Babusis, D.; Tang, Y.; Shirely, M.A.; Moghaddam, M.F.; et al. Pomalidomide shows significant therapeutic activity against CNS lymphoma with a major impact on the tumor microenvironment in murine models. PLoS ONE 2013, 8, e71754. [Google Scholar] [CrossRef] [PubMed]
- Muscal, J.A.; Sun, Y.; Nuchtern, J.G.; Dauser, R.C.; McGuffey, L.H.; Gibson, B.W.; Berg, S.L. Plasma and cerebrospinal fluid pharmacokinetics of thalidomide and lenalidomide in nonhuman primates. Cancer Chemother. Pharm. 2012, 69, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kalff, A.; Low, M.; Gangatharan, S.; Ho, P.; Bajel, A.; Ritchie, D.; Grigg, A.; Spencer, A. Central nervous system multiple myeloma--potential roles for intrathecal therapy and measurement of cerebrospinal fluid light chains. Br. J. Haematol. 2013, 162, 371–375. [Google Scholar] [CrossRef]
- Chen, C.I.; Masih-Khan, E.; Jiang, H.; Rabea, A.; Cserti-Gazdewich, C.; Jimenez-Zepeda, V.H.; Chu, C.M.; Kukreti, V.; Trudel, S.; Tiedemann, R.; et al. Central nervous system involvement with multiple myeloma: Long term survival can be achieved with radiation, intrathecal chemotherapy, and immunomodulatory agents. Br. J. Haematol. 2013, 162, 483–488. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kegyes, D.; Constantinescu, C.; Vrancken, L.; Rasche, L.; Gregoire, C.; Tigu, B.; Gulei, D.; Dima, D.; Tanase, A.; Einsele, H.; et al. Patient selection for CAR T or BiTE therapy in multiple myeloma: Which treatment for each patient? J. Hematol. Oncol. 2022, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Garfall, A.L.; van de Donk, N.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Nooka, A.K.; Martin, T.; Rosinol, L.; Chari, A.; et al. Teclistamab in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2022, 387, 495–505. [Google Scholar] [CrossRef]
- Thakurta, A.; Pierceall, W.E.; Amatangelo, M.D.; Flynt, E.; Agarwal, A. Developing next generation immunomodulatory drugs and their combinations in multiple myeloma. Oncotarget 2021, 12, 1555–1563. [Google Scholar] [CrossRef]
- Lonial, S.; Popat, R.; Hulin, C.; Jagannath, S.; Oriol, A.; Richardson, P.G.; Facon, T.; Weisel, K.; Larsen, J.T.; Minnema, M.C.; et al. Iberdomide plus dexamethasone in heavily pretreated late-line relapsed or refractory multiple myeloma (CC-220-MM-001): A multicentre, multicohort, open-label, phase 1/2 trial. Lancet Haematol. 2022, 9, e822–e832. [Google Scholar] [CrossRef]
- Richardson, P.G.; Vangsted, A.J.; Ramasamy, K.; Trudel, S.; Martínez, J.; Mateos, M.-V.; Otero, P.R.; Lonial, S.; Popat, R.; Oriol, A.; et al. First-in-human phase I study of the novel CELMoD agent CC-92480 combined with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J. Clin. Oncol. 2020, 38, 8500. [Google Scholar] [CrossRef]
- van de Donk, N.W.C.J.; Popat, R.; Larsen, J.; Minnema, M.C.; Jagannath, S.; Oriol, A.; Zonder, J.; Richardson, P.G.; Rodriguez-Otero, P.; Badros, A.Z.; et al. First Results of Iberdomide (IBER; CC-220) in Combination with Dexamethasone (DEX) and Daratumumab (DARA) or Bortezomib (BORT) in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 16–17. [Google Scholar] [CrossRef]
- Richardson, P.G.; Ocio, E.; Raje, N.S.; Gregory, T.; White, D.; Oriol, A.; Sandhu, I.; Raab, M.-S.; LeBlanc, R.; Rodriguez, C.; et al. CC-92480, a Potent, Novel Cereblon E3 Ligase Modulator (CELMoD) Agent, in Combination with Dexamethasone (DEX) and Bortezomib (BORT) in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Preliminary Results from the Phase 1/2 Study CC-92480-MM-002. Blood 2021, 138, 2731. [Google Scholar] [CrossRef]
- Mann, H.; Comenzo, R.L. Evaluating the Therapeutic Potential of Idecabtagene Vicleucel in the Treatment of Multiple Myeloma: Evidence to Date. Onco Targets Ther. 2022, 15, 799–813. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Response | Definition |
|---|---|
| Complete response (CR) | Negative immunofixation in the serum or urine and disappearance of any soft tissue plasmacytomas and <5% plasma cells on bone marrow aspirate |
| Stringent Complete Response (sCR) | Complete response as defined above plus a normal serum free light chain ratio (FLC) and absence of clonal plasma cells on bone marrow aspirate |
| Very Good Partial Response (VGPR) | Serum and urine M-protein detectable on immunofixation but not on electrophoresis -or- >90% reduction in serum M-protein plus urine M-protein < 100 mg on 24 h collection |
| Partial Response (PR) |
|
| Minimal Response (MR) |
|
| Stable Disease (SD) | Not meeting criteria for CR, VGPR, PR, MR, or PD. Generally, not recommended for use as an indicator of response |
| Progressive Disease (PD) | Increase of 25% from the lowest confirmed response value in one or more of the following criteria:
|
| Clinical Relapse | Any one or more of the following criteria:
|
| Relapse from CR | Any one or more of the following
|
| Trial | % LEN Refractory | ORR (%) | PFS | OS |
|---|---|---|---|---|
| NIMBUS (MM-003) POM-loDex vs. HiDex | 92 vs. 95 | 30 vs. 9 | 3.9 vs. 1.9 months (HR 0.50, p < 0.001) | 12.7 vs. 8.0 months (HR 0.73, p = 0.0234) |
| STRATUS (MM-010) POM-loDex | 95 | 32.1 | 4.6 months | 11.9 months |
| OPTIMISMM POM-BOR-dex vs. BOR-dex | 69 vs. 71 | 82.2 vs. 50.0 | 11.2 vs. 7.1 months (HR 0.65, p < 0.0001) | Data not yet mature |
| CASTOR DARA-BOR-dex vs. BOR-dex | 17.9 vs. 24 | 80.5 vs. 50.0 | 9.3 vs. 4.4 months (HR 0.36, p = 0.0002) | 28.9 vs. 32.6 months (HR 0.96, NS) |
| CANDOR DARA-Kd vs. Kd | 32 vs. 36 | 90 vs. 67 * and 78 vs. 71 ** | 28.1 vs. 11.1 months (HR 0.46, p < 0.001) | Data not yet mature |
| IKEMA ISA-Kd vs. Kd | 32 vs. 34 | NT | NR vs. 15.7 months (HR 0.60, p = 0.56) | Data not yet mature |
| APOLLO DARA-POM-dex vs. POM-dex | 79 vs. 80 | 69 vs. 46 ¶ | 9.9 vs. 6.6 months (HR 0.66, p = Sig, NT) | Data not yet mature |
| ICARIA ISA-POM-dex vs. POM-dex | 93.5 vs. 91.5 | 59.0 vs. 31.4 | 11.4 vs. 5.59 months (HR 0.593, p = NT) | Data not yet mature |
| ELOQUENT-3 (Phase 2) ELO-POM-dex vs. POM-dex | 90 vs. 84 | 53 vs. 26 ¶ | 10.2 vs. 4.7 months (HR 0.56, p = NT) ¥ | 28.3 vs. 16.0 months (HR 0.42, p = NT) ¥ |
| BOSTON SEL-BOR-dex vs. BOR-dex | 37 vs. 39 ¶¶ | 67.5 vs. 53.2 | HR 0.63, p = NT PFS duration not reported for LEN ref. subgroup | OS duration not reported for LEN ref. subgroup |
| BELLINI VEN-BOR-dex vs. BOR-dex | 20 vs. 28 | NT | NR vs. 14.8 months (HR 0.75, p = NT | HR 1.82, p = NT OS duration not reported for LEN ref. subgroup |
| Trial | % Previous BOR Exposure | ORR (%) | PFS | OS |
|---|---|---|---|---|
| ASPIRE * KRd vs. K-dex | 62–67 vs. 64–66 | NT for subgroup analysis | NT for subgroup analysis | 45.9 vs. 33.9 months, HR 0.82, p = NT |
| ENDEAVOR * Kd vs. BOR-dex | 54 vs. 54 | NT for subgroup analysis | 15.6 vs. 8.1 months (HR 0.56, p = NT) | 41.8 vs. 32.7 months (HR 0.851, p = NT) |
| TOURMALINE MM1 * IRd vs. Rd | 69 vs. 69 | NT for subgroup analysis | 18.4 vs. 13.6 months (HR 0.74, p = NT) | 53.0 vs. 55.8 months (HR 0.994, p = NT) |
| EMN011/HOVON114 (Phase 2) K-POM-dex | 100% | 92 | 26 months | 67 months |
| APOLLO DARA-POM-dex vs. POM-dex | 47 vs. 49 | 69 vs. 46 ¶ | 8.3 vs. 6.3 months (HR = 0.73, p = NS, NT) | Data not yet mature |
| ICARIA ISA-POM-dex vs. POM-dex | 76.6 vs. 75.2 | 60.2 vs. 32.2 | 11.4 vs. 5.59 months (HR 0.578, p = NT) | Data not yet mature |
| ELOQUENT-3 (Phase 2) ELO-POM-dex vs. POM-dex | 78 vs. 82 | 53 vs. 26 ¶ | 10.2 vs. 4.7 months (HR 0.56, p = NT) ¥ | 28.3 vs. 16.0 months (HR 0.42, p = NT) ¥ |
| Trial | % DARA Refractory | ORR | PFS | OS |
|---|---|---|---|---|
| DREAMM-2 Belantamab Mafodotin | 100% | 32% | 2.8 months (95% CI: 1.6–3.6 months) | 13.7 months (95% CI: 9.9–NR) |
| DREAMM-3 BEL vs. POM-dex | NT | 41% vs. 36% | 11.2 vs. 7 months (HR 1.03) | 21.2 vs. 21.2 months (HR 1.13) |
| MajesTEC-1 Teclistamab | 89.7% | 63% | 11.3 months (95% CI: 8.8–17.1 months) | 18.3 months (95% CI: 15.1-NE)* |
| Trial | ORR | PFS | OS |
|---|---|---|---|
| Myeloma X (ASCT vs. Cy maintenance) | sCR or CR: −39.3 vs. 22.4% VGPR or PR: −43.8 vs. 52.9% | 19 vs. 11 months, HR 0.45, p < 0.0001 | 67 vs. 52 months, HR 0.56, p = 0.0169 |
| ReLApsE (ASCT + LEN maintenance vs. LEN-dex) | 77.9% vs. 74.6% (p = 0.57) | 20.7 vs. 18.8 months, HR 0.87, p = 0.34 | NR vs. 62.7 months, HR 0.81, p = 0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatt, P.; Kloock, C.; Comenzo, R. Relapsed/Refractory Multiple Myeloma: A Review of Available Therapies and Clinical Scenarios Encountered in Myeloma Relapse. Curr. Oncol. 2023, 30, 2322-2347. https://doi.org/10.3390/curroncol30020179
Bhatt P, Kloock C, Comenzo R. Relapsed/Refractory Multiple Myeloma: A Review of Available Therapies and Clinical Scenarios Encountered in Myeloma Relapse. Current Oncology. 2023; 30(2):2322-2347. https://doi.org/10.3390/curroncol30020179
Chicago/Turabian StyleBhatt, Parva, Colin Kloock, and Raymond Comenzo. 2023. "Relapsed/Refractory Multiple Myeloma: A Review of Available Therapies and Clinical Scenarios Encountered in Myeloma Relapse" Current Oncology 30, no. 2: 2322-2347. https://doi.org/10.3390/curroncol30020179
APA StyleBhatt, P., Kloock, C., & Comenzo, R. (2023). Relapsed/Refractory Multiple Myeloma: A Review of Available Therapies and Clinical Scenarios Encountered in Myeloma Relapse. Current Oncology, 30(2), 2322-2347. https://doi.org/10.3390/curroncol30020179