HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature

 , , ,
, , ,  and
and
Abstract
1. Introduction
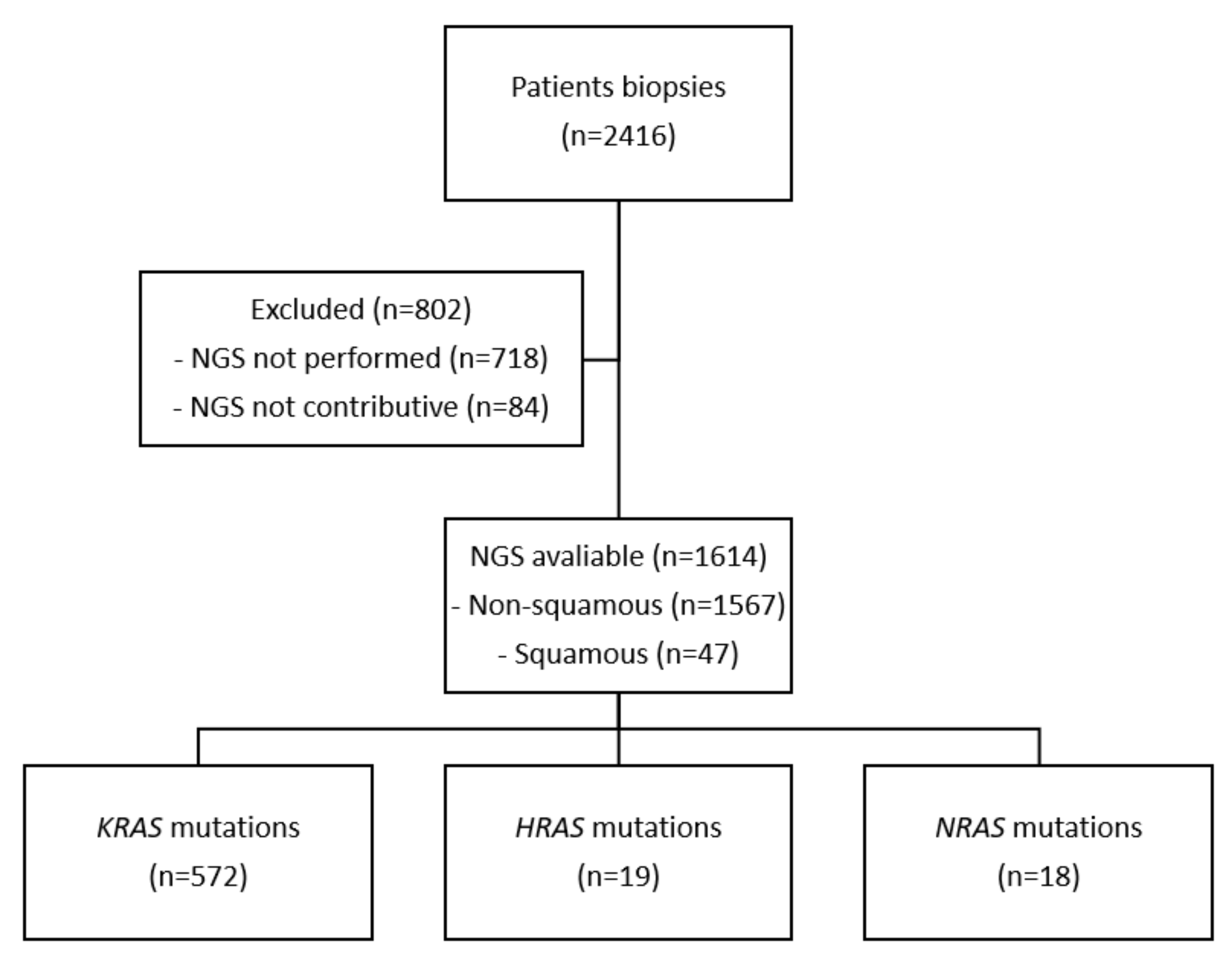
2. Patients and Methods
3. Results
3.1. Cases Presentation
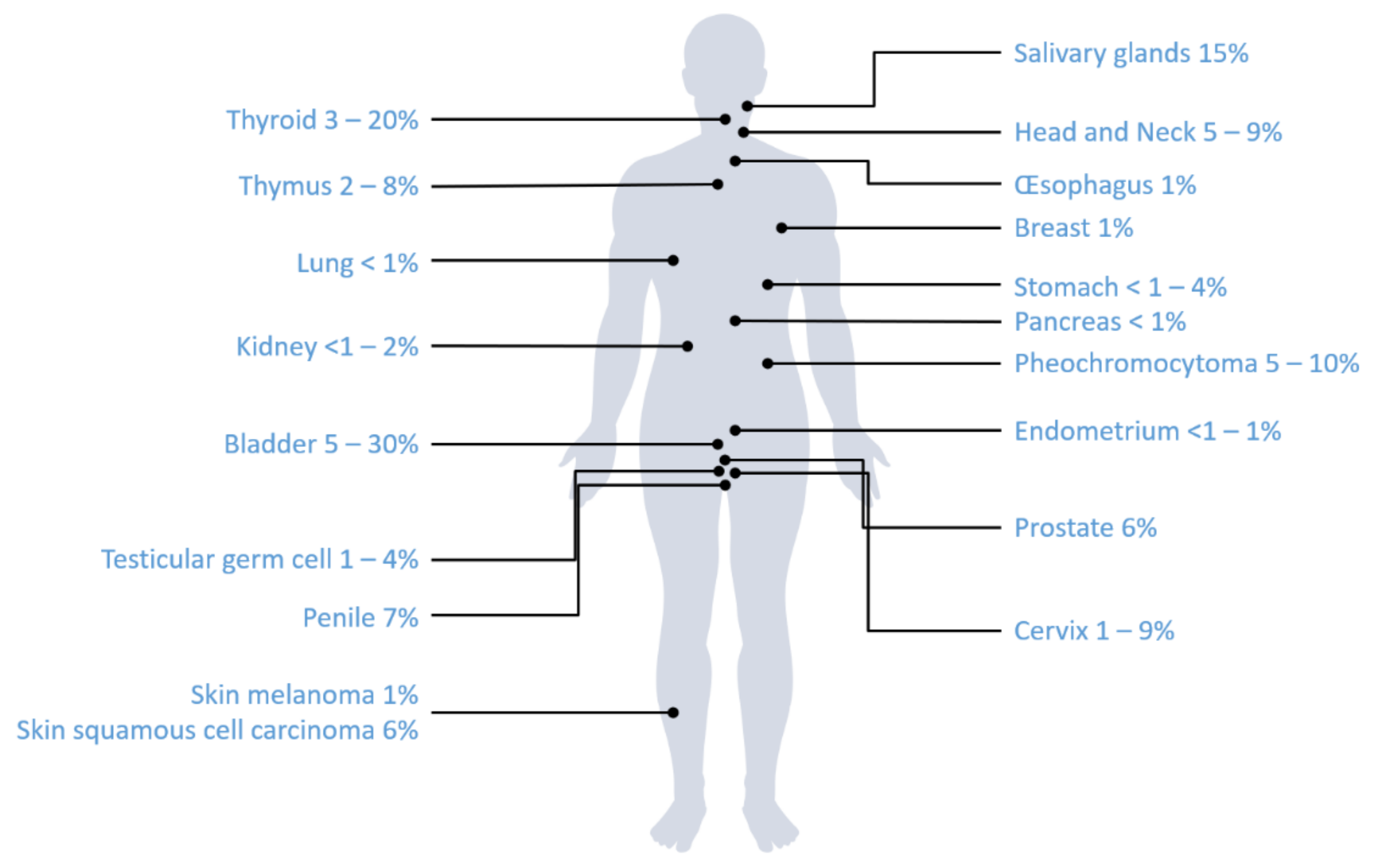
3.2. Review of Literature
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Sequenced Regions |
|---|
| AKT1 exon 3 (NM_001014431.1) |
| ALK exons 22 to 25 (NM_004304.1) |
| BRAF exons 11 and 15 (NM_004333.4) |
| CTNNB1 exon 3 (NM_001904.4) |
| DDR2 exons 5 to 10 and 14 to 19 (NM_001014796.2) |
| EGFR exons 18 to 21 (NM_005228.3) |
| ERBB2 (HER2) exons 19 to 22 (NM_004448.2) |
| ERBB4 codons 393 and 452 (NM_005235.2) |
| FGFR2 codons 252, 549 and 659 (NM_000141.4) |
| FGFR3 exons 6, 8 and 13 (NM_000142.4) |
| HRAS exons 2, 3 and 4 (NM_005343.2) |
| IDH1 codons 100 and 132 (NM_005896.3) |
| IDH2 codon 172 (NM_002168.3) |
| KIT exons 8, 9, 11, 13, 14, 17 and 18 (NM_000222.2) |
| KRAS exons 2, 3 and 4 (NM_033360.2) |
| MAP2K1 (MEK1) exon 2 (NM_002755.3) |
| MET exon 2, intron 13 and exons 14 to 20 (NM_001127500.1) |
| NRAS exons 2, 3 and 4 (NM_002524.3) |
| PDGFRA exons 12, 14 and 18 (NM_006206.4) |
| PIK3CA exons 10 and 21 (NM_006218.2) |
| RET exons 11 and 16 (NM_020975.6) |
| TP53 exons 2 to 11 (NM_000546.4) |
| Microsatellites: BAT25, BAT26, NR21, NR24, MONO27 |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Vansteenkiste, J.; Planchard, D.; Cho, B.C.; Gray, J.E.; Ohe, Y.; Zhou, C.; Reungwetwattana, T.; Cheng, Y.; Chewaskulyong, B.; et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Ryan, M.B.; Corcoran, R.B. Therapeutic Strategies to Target RAS-Mutant Cancers. Nat. Rev. Clin. Oncol. 2018, 15, 709–720. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS Oncogenes: Weaving a Tumorigenic Web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Cox, A.D.; Der, C.J. Ras History: The Saga Continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras Oncogenes: Split Personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational Landscape of Metastatic Cancer Revealed from Prospective Clinical Sequencing of 10,000 Patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Taniguchi-Tamura, H.; Araki, M.; Kawamura, T.; Miyamoto, R.; Tsuda, C.; Shima, F.; Kumasaka, T.; Okuno, Y.; Kataoka, T. Oncogenic Mutations Q61L and Q61H Confer Active Form-Like Structural Features to the Inactive State (State 1) Conformation of H-Ras Protein. Biochem. Biophys. Res. Commun. 2021, 565, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Blockade of Mutant RAS Oncogenic Signaling with a Special Emphasis on KRAS. Pharmacol. Res. 2021, 172, 105806. [Google Scholar] [CrossRef] [PubMed]
- Cathcart-Rake, E.; Corless, C.; Sauer, D.; Lopez-Chavez, A. Elderly Former Smoker with HRAS Mutant Non-Small-Cell Lung Cancer. J. Thorac. Oncol. 2014, 9, e75–e78. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; He, M.; Chen, X.; Cui, X.; Qin, T.; Niu, X.; Zhao, L. Poor Prognosis of Pulmonary Adenosquamous Carcinoma with NRAS and HRAS Double Mutation. Onco Targets Ther. 2021, 14, 1113–1116. [Google Scholar] [CrossRef]
- Long, Y.; Tang, Y.; Cai, C.; Yu, M.; Zhang, M.; Chen, R.; Huang, M. The Influence of STK11 Mutation on Acquired Resistance to Immunotherapy in Advanced Non-Small Cell Lung Cancer with Lynch Syndrome: A Case Report and Literature Review. Ann. Palliat. Med. 2021, 10, 7088–7094. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Mendiratta, G.; Ke, E.; Aziz, M.; Liarakos, D.; Tong, M.; Stites, E.C. Cancer Gene Mutation Frequencies for the U.S. Population. Nat. Commun. 2021, 12, 5961. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; Gabriel, S.; et al. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Bal-asundaram, M.; Birol, I.; et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Naemi, H.A.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-Mediated Oncogenic Signaling Pathways in Human Malignancies. Semin. Cancer Biol. 2019, 54, 1–13. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556. [Google Scholar] [CrossRef]
- Stenman, A.; Welander, J.; Gustavsson, I.; Brunaud, L.; Bäckdahl, M.; Söderkvist, P.; Gimm, O.; Juhlin, C.C.; Larsson, C. HRAS Mutation Prevalence and Associated Expression Patterns in Pheochromocytoma. Genes Chromosomes Cancer 2016, 55, 452–459. [Google Scholar] [CrossRef]
- Andersson, P.; Kolaric, A.; Windahl, T.; Kirrander, P.; Söderkvist, P.; Karlsson, M.G. PIK3CA, HRAS and KRAS Gene Mutations in Human Penile Cancer. J. Urol. 2008, 179, 2030–2034. [Google Scholar] [CrossRef]
- Kiessling, M.K.; Curioni-Fontecedro, A.; Samaras, P.; Atrott, K.; Cosin-Roger, J.; Lang, S.; Scharl, M.; Rogler, G. Mutant HRAS as Novel Target for MEK and MTOR Inhibitors. Oncotarget 2015, 6, 42183–42196. [Google Scholar] [CrossRef]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- End, D.W.; Smets, G.; Todd, A.V.; Applegate, T.L.; Fuery, C.J.; Angibaud, P.; Venet, M.; Sanz, G.; Poignet, H.; Skrzat, S.; et al. Characterization of the Antitumor Effects of the Selective Farnesyl Protein Transferase Inhibitor R115777 In Vivo and In Vitro. Cancer Res. 2001, 61, 131–137. [Google Scholar] [PubMed]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C Inhibitor: Activity and Resistance. Cancer Gene Ther. 2021, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma With HRAS Mutations. J. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef]
- Hanna, G.J.; Guenette, J.P.; Chau, N.G.; Sayehli, C.M.; Wilhelm, C.; Metcalf, R.; Wong, D.J.; Brose, M.; Razaq, M.; Pérez-Ruiz, E.; et al. Tipifarnib in Recurrent, Metastatic HRAS-Mutant Salivary Gland Cancer. Cancer 2020, 126, 3972–3981. [Google Scholar] [CrossRef]
- Lee, H.W.; Sa, J.K.; Gualberto, A.; Scholz, C.; Sung, H.H.; Jeong, B.C.; Choi, H.Y.; Kwon, G.Y.; Park, S.H. A Phase II Trial of Tipifarnib for Patients with Previously Treated, Metastatic Urothelial Carcinoma Harboring HRAS Mutations. Clin. Cancer Res. 2020, 26, 5113–5119. [Google Scholar] [CrossRef]
- Jazieh, K.; Molina, J.; Allred, J.; Yin, J.; Reid, J.; Goetz, M.; Lim, V.-S.; Kaufmann, S.H.; Adjei, A. A Phase I Study of the Farnesyltransferase Inhibitor Tipifarnib in Combination with the Epidermal Growth Factor Tyrosine Kinase Inhibitor Erlotinib in Patients with Advanced Solid Tumors. Investig. New Drugs 2019, 37, 307–314. [Google Scholar] [CrossRef]
- Whitehead, R.P.; McCoy, S.; Macdonald, J.S.; Rivkin, S.E.; Neubauer, M.A.; Dakhil, S.R.; Lenz, H.-J.; Tanaka, M.S.; Abbruzzese, J.L.; Southwest Oncology Group. Phase II Trial of R115777 (NSC #70818) in Patients with Advanced Colorectal Cancer: A Southwest Oncology Group Study. Investig. New Drugs 2006, 24, 335–341. [Google Scholar] [CrossRef]
- Lara, P.N.J.; Law, L.Y.; Wright, J.J.; Frankel, P.; Twardowski, P.; Lenz, H.J.; Lau, D.H.M.; Kawaguchi, T.; Gumerlock, P.H.; Doroshow, J.H.; et al. Intermittent Dosing of the Farnesyl Transferase Inhibitor Tipifarnib (R115777) in Advanced Malignant Solid Tumors: A Phase I California Cancer Consortium Trial. Anti-Cancer Drugs 2005, 16, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Heymach, J.V.; Johnson, D.H.; Khuri, F.R.; Safran, H.; Schlabach, L.L.; Yunus, F.; DeVore, R.F.; De Porre, P.M.; Richards, H.M.; Jia, X.; et al. Phase II Study of the Farnesyl Transferase Inhibitor R115777 in Patients with Sensitive Relapse Small-Cell Lung Cancer. Ann. Oncol. 2004, 15, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II Study of the Farnesyl Transferase Inhibitor R115777 in Patients with Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.M.; Bernhard, E.J.; Regine, W.; Mohiuddin, M.; Haller, D.G.; Stevenson, J.P.; Smith, D.; Pramanik, B.; Tepper, J.; DeLaney, T.F.; et al. A Phase I Trial of the Farnesyltransferase Inhibitor L-778,123 and Radiotherapy for Locally Advanced Lung and Head and Neck Cancer. Clin. Cancer Res. 2002, 8, 1065–1072. [Google Scholar] [PubMed]
- Storck, E.M.; Morales-Sanfrutos, J.; Serwa, R.A.; Panyain, N.; Lanyon-Hogg, T.; Tolmachova, T.; Ventimiglia, L.N.; Martin-Serrano, J.; Seabra, M.C.; Wojciak-Stothard, B.; et al. Dual Chemical Probes Enable Quantitative System-Wide Analysis of Protein Prenylation and Prenylation Dynamics. Nat. Chem. 2019, 11, 552–561. [Google Scholar] [CrossRef]
- Gualberto, A.; Scholz, C.; Mishra, V.; Janes, M.R.; Kessler, L.; Cutsem, E.V.; Ho, A.L.; Witzig, T. Abstract CT191: Mechanism of Action of the Farnesyltransferase Inhibitor, Tipifarnib and Its Clinical Applications. Cancer Res. 2019, 79, CT191. [Google Scholar] [CrossRef]


| HRAS Mutations | Numbers of Patients |
|---|---|
| p.Q61L (p.Gln61Leu; C.182A>T) | 4 |
| p.G13V (p.Gly13Val; c.38G>T) | 2 |
| p.E98K (p.Glu98Lys; c.292G>A) | 1 |
| p.S89F (p.Ser89Phe; c.266C>T) | 1 |
| p.A11P (p.Ala11Pro; c.31G>C) | 1 |
| p.K117N (p.Lys117Asn; c.351G>T) | 1 |
| p.R102L (p.Arg102Leu; c.305G>T) | 1 |
| p.D107fs (p.Asp107fs; c.319del) | 1 |
| p.V109L (p.Val109Leu; c.325G>T) | 1 |
| p.T58I (p.Thr58Ile; c.173C>T) | 1 |
| p.T148P (p.Thr148Pro; c.442A>C) | 1 |
| p.R41W (p.Arg41Trp; c.121C>T) | 1 |
| p.R135Q (p.Arg135Gln; c.404G>A) | 1 |
| p.M72I (p.Met72Ile; c.216G>T) | 1 |
| p.E76D (p.Glu76Asp; c.228G>T) | 1 |
| Reference | Sex (Female/Male) | Age at Diagnosis (Years) | Smoking Status | Pathology | PD-L1 (%) | Other Alterations | Metastatic Site | Treatment | PFS (Weeks) | OS (Weeks) |
|---|---|---|---|---|---|---|---|---|---|---|
| Current | F | 50 | Active | ADC | <1 | None | Lung/Liver/Pericardial effusion (4 years after adjuvant chemotherapy) | Carboplatin—Pemetrexed | 11 | 15 |
| Current | M | 55 | Former | NOS | <1 | KRAS p.Gly12Cys | Locally-advanced disease | Carboplatin—Pemetrexed | 22 | 30 |
| Current | M | 63 | Active | NOS | 60 | KRAS p.Gly12Ser TP53 c.784_809del | Brain/Pericardial effusion | Carboplatin—Pemetrexed—Pembrolizumab WBRT | On treatment | On treatment |
| Current | F | 61 | Active | ADC | 60 | TP53 p.Ile195Thr | Pleural effusion | Not treated | NA | 3 |
| Cathcart-Rake E., 2014 [17] | M | 79 | Former | ADC | NA | None | Brain/Bone/Liver/Adrenal (10 months after adjuvant chemotherapy) | Adjuvant Carboplatin—Pemetrexed Brain surgery—Stereotactic radiosurgery | NA | 64 |
| Zhao J., 2021 [18] | M | 58 | Active | ADSQ | NA | EGFR p.Leu858Arg and p.Thr790Met (only on pleural effusion), NRAS p.Gln61Lys | Pleural effusion | Cisplatin—Osimertinib | 2 | 4 |
| Long Y., 2021 [19] | M | 76 | Active | SCC | 50 | TP53 p.Arg158Leu LRP1B p.Val3711Phe LRP1B p.Leu4013Met DNMT3A p.Wrp601 * DDR2 p.Val336Leu NTM p.Thr240Ile TCF7L2 p.Arg420Pro POLE p.Arg579Leu | Locally-advanced disease | Pembrolizumab | 24 | NA |
| Reference | Phase | Tumour Site | Number of Patients | Setting | Biomarker | Tipifarnib Dose & Schedule | Primary Endpoint | ORR (%) (SD) | Median PFS (Months) | Median OS (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| Ho A.L., 2021 [36] | 2 | HNSCC | 22 | Relapsed | Missense HRAS mutation/VAF > 20% either in blood, primary tumour tissue, recurrent or metastatic disease | 800 or 900 mg PO twice daily on days 1–7 and 15–21 of 28-day cycles | ORR | 50 (41) | 5.6 | 15.4 |
| Haddad R., 2021 | 2 (Ongoing) | HNSCC | NA | Relapsed | R/M mHRAS VAF ≥ 20% (tumour tissue) detected by NGS | 600 mg PO with a meal twice a day for 7 days in alternating weeks (Days 1–7 and 15–21) of 28-day cycles | ORR in High VAF population | 55 (NA) | NA | 15.4 |
| Hanna G.J., 2020 [35] | 2 | Salivary gland carcinoma | 13 | Relapsed | Missense HRAS mutation with a VAF > 20%: 54% p.Gln61Arg (tumour tissue) | 900 mg PO twice daily on days 1 to 7 and days 15 to 21 of a 28-day | ORR | 8 (54) | 7.0 | 18.0 |
| Lee H.W., 2020 [36] | 2 | Urothelial carcinoma | 21 | Relapsed | Missense, nonsynonymous HRAS mutations (p.Gly13Arg, n = 7; p.Gln61Arg, n = 4; p.Gly12Ser, n = 3; p.Gly12Cys, n = 2) (tumour tissue) | 900 mg PO twice daily on days 1–7 and 15–21 of 28-day | 6-month PFS | 24 (62) | 4.7 | 6.1 |
| Jazieh K., 2019 [37] | 1 | Advanced, recurrent or metastatic solid tumours | 27 | Relapsed | No selection on HRAS status Tumour tissue (diagnostic) | 4 dose levels, ranging from tipifarnib 200 mg PO twice daily plus erlotinib 75 mg PO once daily to tipifarnib 300 mg PO twice daily plus erlotinib 150 mg PO once daily | Safety, tolerability, maximum tolerated dose | 7.4 (37) | NA | NA |
| Whitehead R.P., 2006 [38] | 2 | Metastatic colorectal adenocarcinoma | 62 | No prior chemo: 33/55 Prior chemo: 22/55 | No selection on HRAS status Tumour tissue (diagnostic) | Fixed dose of 300 mg PO, twice daily, immediately after a meal, days 1–21, every 28 days, until tumour progression or toxicity | Confirmed response probability | 2 (20) | 1.7 | 8.1 |
| Lara Jr P.N., 2005 [39] | 1 | Advanced, recurrent or metastatic malignant tumours: (8) NSCLC, (6) colorectal, (3) prostate, (1) oesophagial, (1) pancreatic, (1) parotid, (1) renal | 21 | Relapsed | No selection on HRAS status Tumour tissue (diagnostic) | Starting dose was 300 mg PO twice daily with escalation by 300 mg. Increments over six dose levels to a maximum of 1800 mg PO twice daily, on days 1–7 and 15–21 of 28-day treatment cycles | Not mentioned | 0 (32) | NA | NA |
| Heymach J.V., 2004 [42] | 2 | SCLC | 22 | Relapsed | Missense HRAS mutation | 3-week cycles at a dose of 400 mg PO twice daily for 14 consecutive days followed by 7 days off treatment | ORR | 0 (5) | 1.4 | 6.8 |
| Adjei A., 2003 [43] | 2 | NSCLC | 44 | 100% No prior chemotherapy (eligibility criteria) 9/44 had radiotherapy | No selection on HRAS status | 300 mg PO twice daily for 21 of every 28 days | ORR | 0 (16) | 2.7 | 7.7 |
| Hahn S., 2002 [44] | 1 | NSCLC | 9 | No prior therapy: 7/9 Prior chemotherapy: 2/9 | No selection on HRAS status Tumour tissue (diagnostic) | Dose-escalation study of tipifarnib Dose level, 280 mg/m2 daily PO during weeks 1, 2, 4, and 5 of radiotherapy Dose level, 560 mg/m2 daily during weeks 1, 2, 4, 5, and 7 of radiotherapy | Maximum tolerated dose, dose limiting toxicity | NA | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathiot, L.; Herbreteau, G.; Robin, S.; Fenat, C.; Bennouna, J.; Blanquart, C.; Denis, M.; Pons-Tostivint, E. HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature. Curr. Oncol. 2022, 29, 3748-3758. https://doi.org/10.3390/curroncol29050300
Mathiot L, Herbreteau G, Robin S, Fenat C, Bennouna J, Blanquart C, Denis M, Pons-Tostivint E. HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature. Current Oncology. 2022; 29(5):3748-3758. https://doi.org/10.3390/curroncol29050300
Chicago/Turabian StyleMathiot, Laurent, Guillaume Herbreteau, Siméon Robin, Charlotte Fenat, Jaafar Bennouna, Christophe Blanquart, Marc Denis, and Elvire Pons-Tostivint. 2022. "HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature" Current Oncology 29, no. 5: 3748-3758. https://doi.org/10.3390/curroncol29050300
APA StyleMathiot, L., Herbreteau, G., Robin, S., Fenat, C., Bennouna, J., Blanquart, C., Denis, M., & Pons-Tostivint, E. (2022). HRAS Q61L Mutation as a Possible Target for Non-Small Cell Lung Cancer: Case Series and Review of Literature. Current Oncology, 29(5), 3748-3758. https://doi.org/10.3390/curroncol29050300