Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
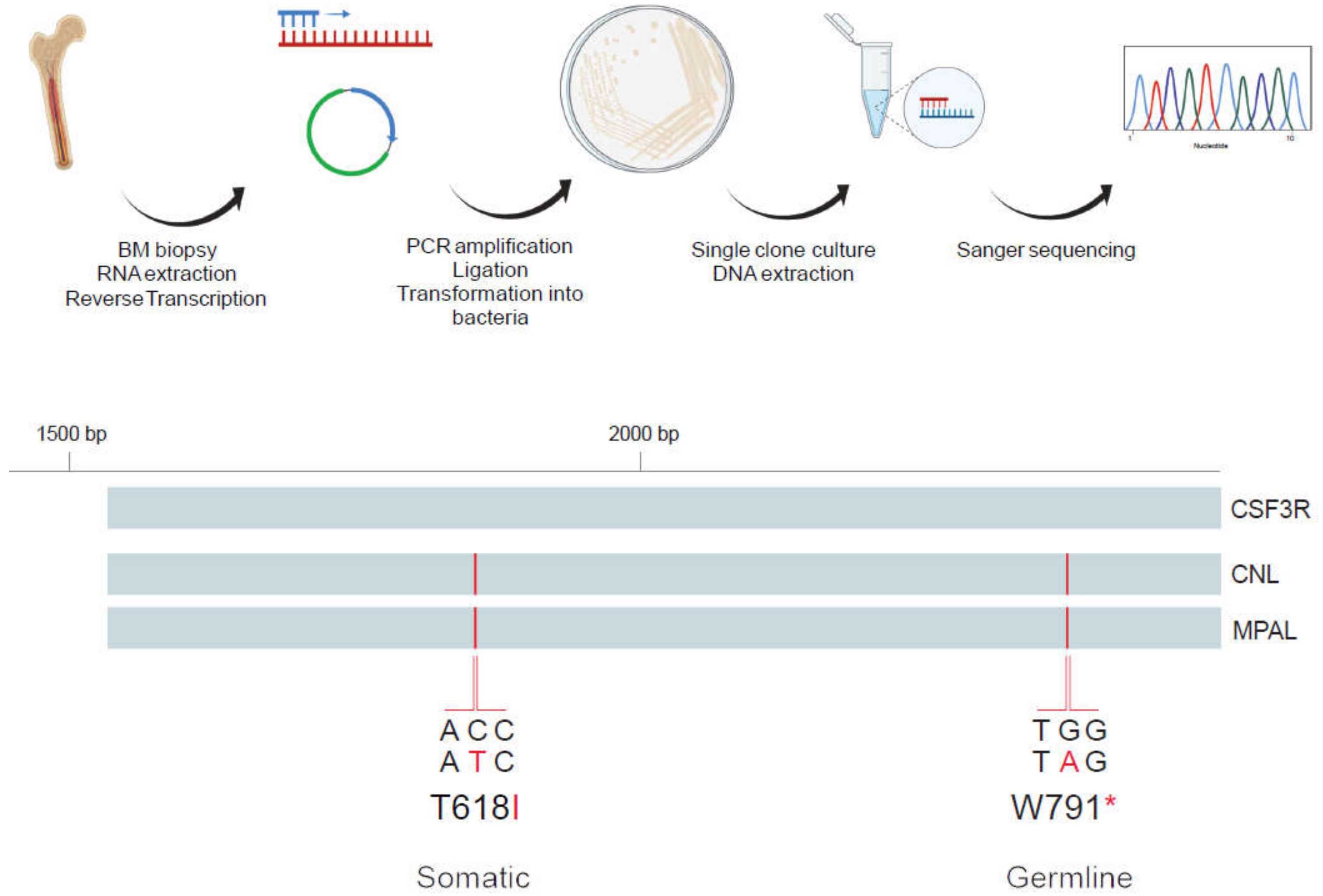
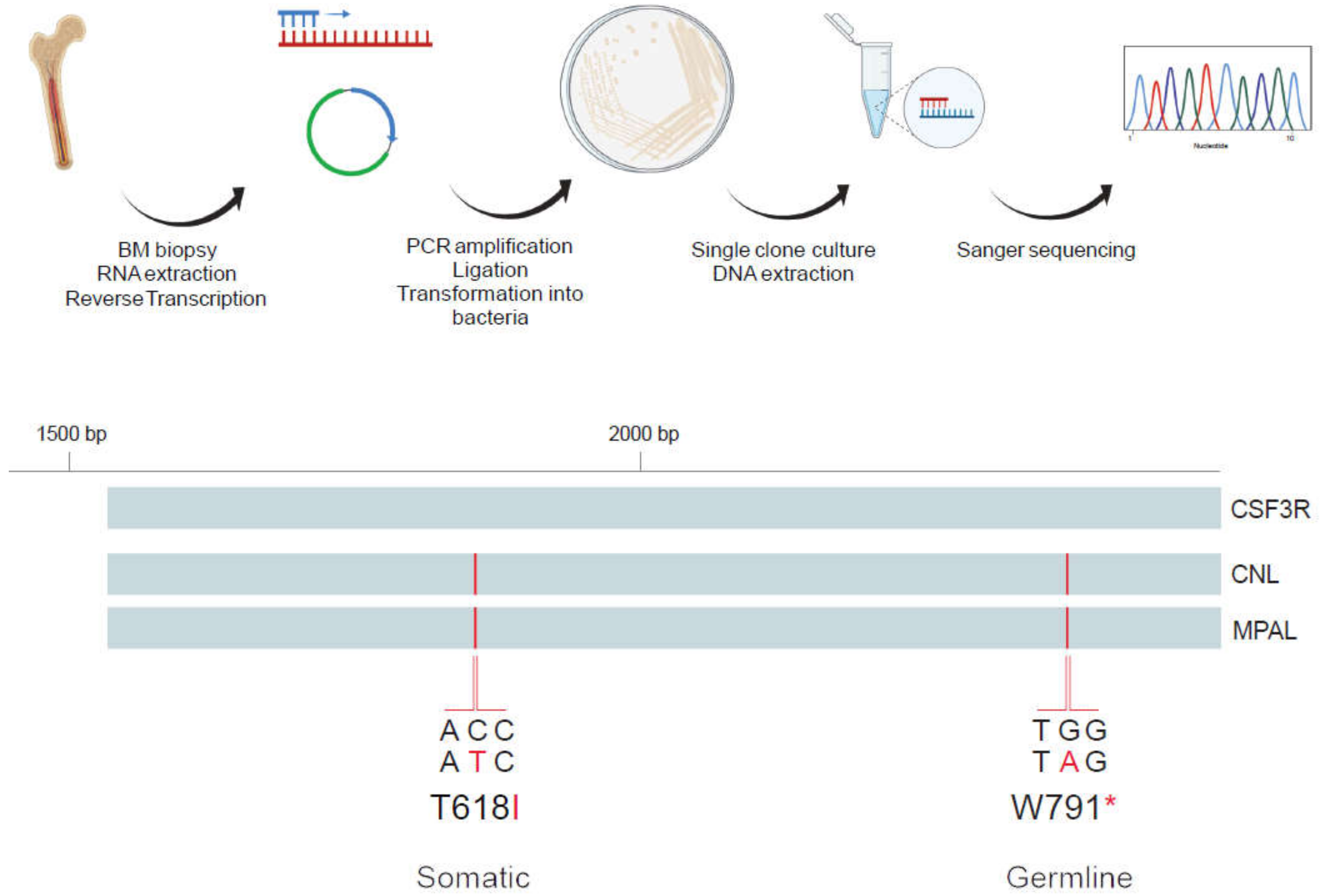
2.1. Mutational Analysis by Next Generation Sequencing
2.2. Analysis of Allelic Localization of CSF3R Mutations by Sequencing of Plasmid DNA Clones
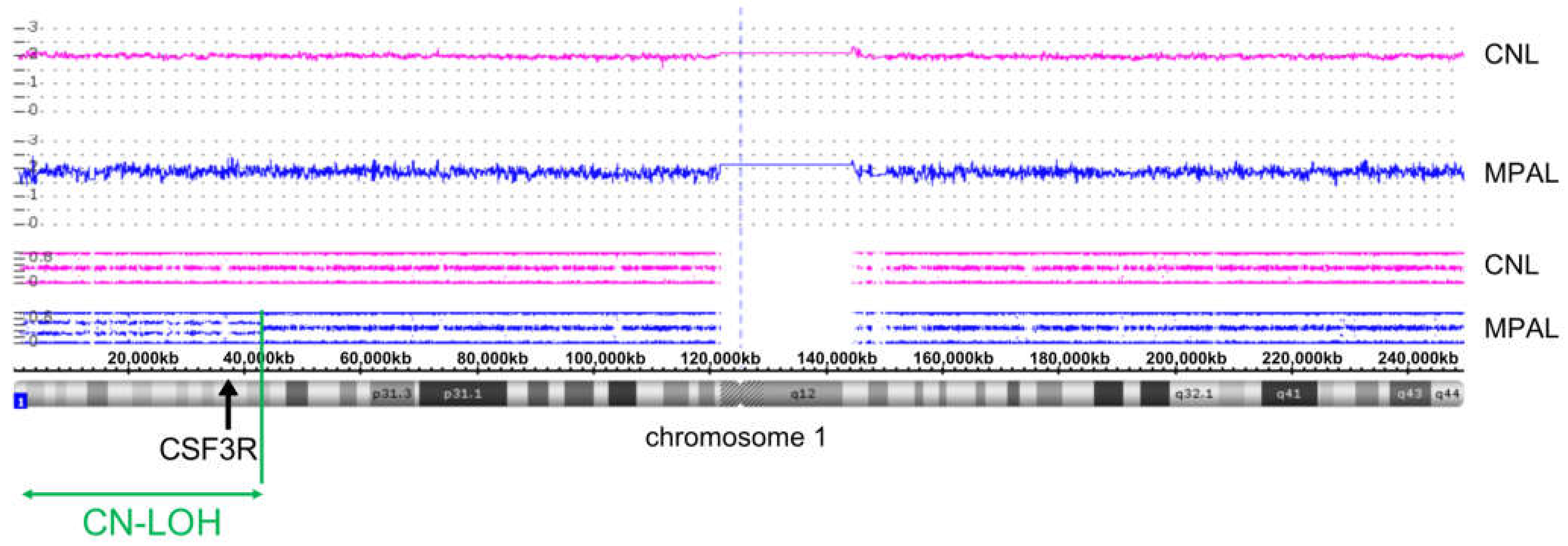
2.3. SNP Array Karyotyping
3. Results
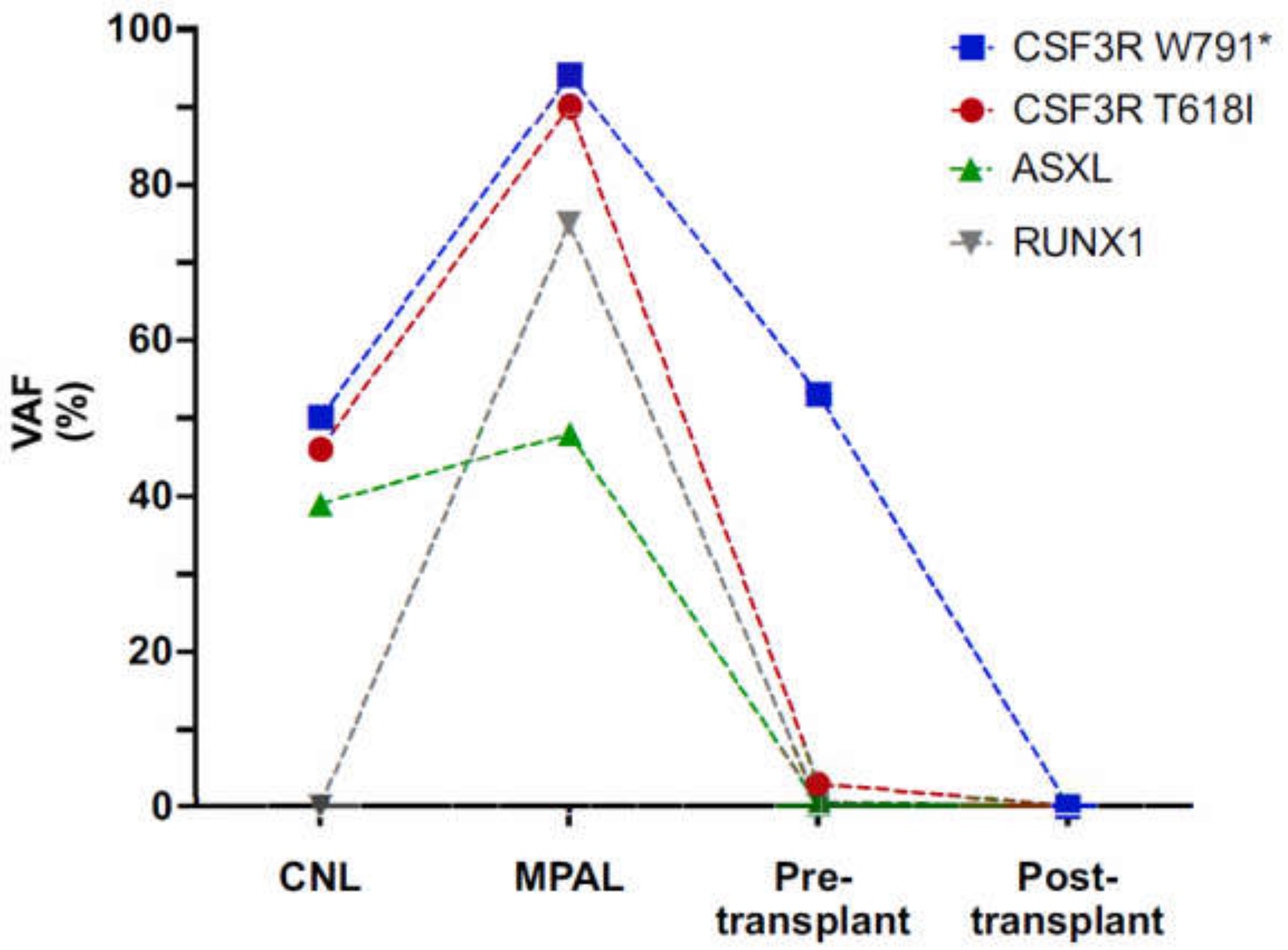
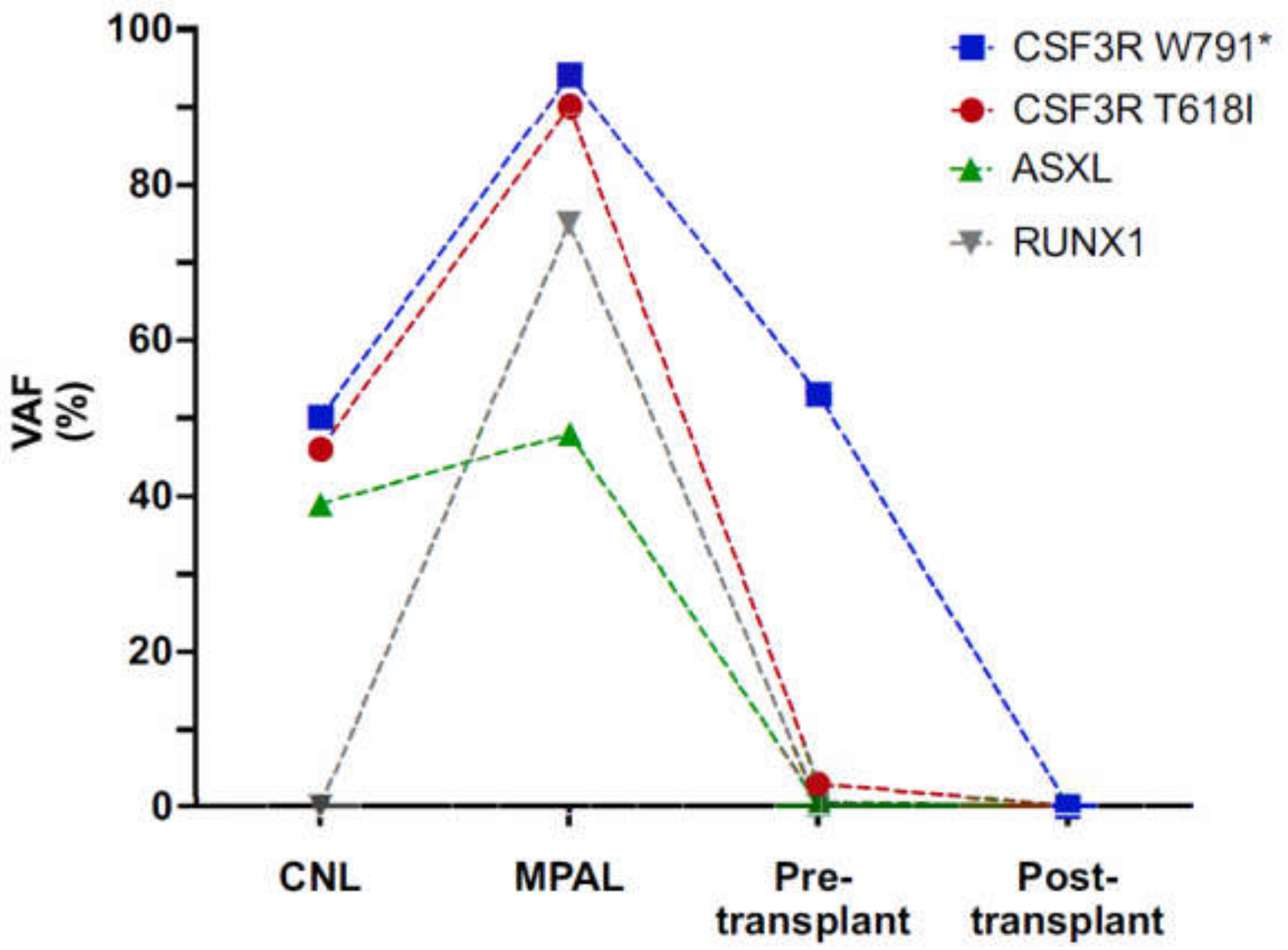
3.1. Early Onset CNL Demonstrates Progression to Mixed Phenotype Acute Leukemia
3.2. Characterization of CSF3R Mutations Reveals W791* in Germline and in Cis with Somatic T618I
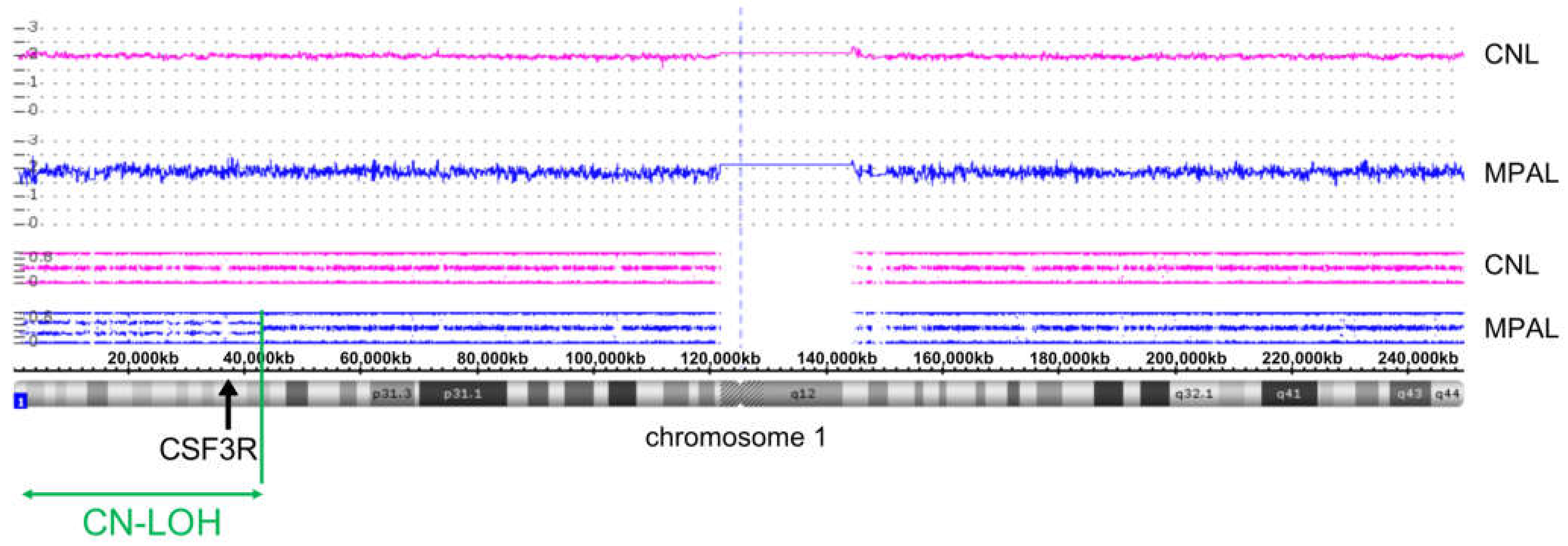
3.3. Clonal Progression Driven by CSF3R LOH and RUNX1 Mutation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Szuber, N.; Elliott, M.; Tefferi, A. Chronic neutrophilic leukemia: 2020 update on diagnosis, molecular genetics, prognosis, and management. Am. J. Hematol. 2020, 95, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beekman, R.; Valkhof, M.; Van Strien, P.; Valk, P.J.; Touw, I.P. Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica 2013, 98, e62–e63. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. Am. Soc. Hematol. 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.; Maxson, J.E.; Luty, S.B.; Agarwal, A.; Royer, L.R.; Abel, M.L.; MacManiman, J.D.; Loriaux, M.M.; Druker, B.; Tyner, J.W. The CSF3R T618I mutation causes a lethal neutrophilic neoplasia in mice that is responsive to therapeutic JAK inhibition. Blood 2013, 122, 3628–3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dao, K.-H.T.; Gotlib, J.; Deininger, M.M.N.; Oh, S.T.; Cortes, J.E.; Collins, R.H.; Winton, E.F.; Parker, D.R.; Lee, H.; Reister, A.; et al. Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia. J. Clin. Oncol. 2020, 38, 1006–1018. [Google Scholar] [CrossRef]
- Plo, I.; Zhang, Y.; Le Couédic, J.-P.; Nakatake, M.; Boulet, J.-M.; Itaya, M.; Smith, S.O.; Debili, N.; Constantinescu, S.; Vainchenker, W.; et al. An activating mutation in the CSF3R gene induces a hereditary chronic neutrophilia. J. Exp. Med. 2009, 206, 1701–1707. [Google Scholar] [CrossRef]
- Spiciarich, D.R.; Oh, S.T.; Foley, A.; Hughes, S.B.; Mauro, M.J.; Abdel-Wahab, O.; Press, R.D.; Viner, R.; Thompson, S.L.; Chen, Q.; et al. A Novel Germline Variant in CSF3R Reduces N-Glycosylation and Exerts Potent Oncogenic Effects in Leukemia. Cancer Res. 2018, 78, 6762–6770. [Google Scholar] [CrossRef] [Green Version]
- Druhan, L.J.; McMahon, D.P.; Steuerwald, N.; Price, A.E.; Lance, A.; Gerber, J.M.; Avalos, B.R. Chronic neutrophilic leukemia in a child with a CSF3R T618I germ line mutation. Blood 2016, 128, 2097–2099. [Google Scholar] [CrossRef] [Green Version]
- Maxson, J.E.; Tyner, J.W. Genomics of chronic neutrophilic leukemia. Blood 2017, 129, 715–722. [Google Scholar] [CrossRef] [Green Version]
- Maxson, J.E.; Luty, S.B.; MacManiman, J.D.; Abel, M.L.; Druker, B.; Tyner, J.W. Ligand Independence of the T618I Mutation in the Colony-stimulating Factor 3 Receptor (CSF3R) Protein Results from Loss of O-Linked Glycosylation and Increased Receptor Dimerization. J. Biol. Chem. 2014, 289, 5820–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, M.A.; Pardanani, A.; Hanson, C.A.; Lasho, T.L.; Finke, C.M.; Belachew, A.A.; Tefferi, A. ASXL1mutations are frequent and prognostically detrimental inCSF3R-mutated chronic neutrophilic leukemia. Am. J. Hematol. 2015, 90, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wilmot, B.; Bottomly, D.; Dao, K.-H.T.; Stevens, S.S.; Eide, C.A.; Khanna, V.; Rofelty, A.; Savage, S.; Schultz, A.R.; et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 2019, 134, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Dale, D.C.; Link, D.C. The Many Causes of Severe Congenital Neutropenia. N. Engl. J. Med. 2009, 360, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Prim. 2017, 3, 17032. [Google Scholar] [CrossRef]
- Dong, F.; Brynes, R.K.; Tidow, N.; Welte, K.; Löwenberg, B.; Touw, I. Mutations in the Gene for the Granulocyte Colony-Stimulating–Factor Receptor in Patients with Acute Myeloid Leukemia Preceded by Severe Congenital Neutropenia. N. Engl. J. Med. 1995, 333, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Skokowa, J.; Steinemann, D.; Katsman-Kuipers, J.E.; Zeidler, C.; Klimenkova, O.; Klimiankou, M.; Ünalan, M.; Kandabarau, S.; Makaryan, V.; Beekman, R.; et al. Cooperativity of RUNX1 and CSF3R mutations in severe congenital neutropenia: A unique pathway in myeloid leukemogenesis. Blood 2014, 123, 2229–2237. [Google Scholar] [CrossRef]
- Touw, I.P.; Beekman, R. Severe congenital neutropenia and chronic neutrophilic leukemia: An intriguing molecular connection unveiled by oncogenic mutations in CSF3R. Haematologica 2013, 98, 1490–1492. [Google Scholar] [CrossRef]
- Mitsui, T.; Watanabe, S.; Taniguchi, Y.; Hanada, S.; Ebihara, Y.; Sato, T.; Heike, T.; Mitsuyama, M.; Nakahata, T.; Tsuji, K. Impaired neutrophil maturation in truncated murine G-CSF receptor–transgenic mice. Blood 2003, 101, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.A.; Hanson, C.A.; Dewald, G.W.; Smoley, S.A.; Lasho, T.L.; Tefferi, A. WHO-defined chronic neutrophilic leukemia: A long-term analysis of 12 cases and a critical review of the literature. Leukemia 2004, 19, 313–317. [Google Scholar] [CrossRef]
- Ruan, G.J.; Smith, C.J.; Day, C.; Harmsen, W.S.; Zblewski, D.L.; Alkhateeb, H.; Begna, K.; Al-Kali, A.; Litzow, M.R.; Hogan, W.; et al. A population-based study of chronic neutrophilic leukemia in the United States. Blood Cancer J. 2020, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Boddy, C.S.; Tan, B.T.; Aoki, J. B-lymphoblastic leukemia arising in a patient with chronic neutrophilic leukemia. Blood Adv. 2020, 4, 5389–5392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Schultz, A.R.; Luty, S.; Rofelty, A.; Su, Y.; Means, S.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tyner, J.W. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukmia 2017, 31, 2752–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoner, R.C.; Press, R.D.; Maxson, J.E.; Tyner, J.W.; Dao, K.-H.T. Insights on mechanisms of clonal evolution in chronic neutrophilic leukemia on ruxolitinib therapy. Leukemia 2019, 34, 1684–1688. [Google Scholar] [CrossRef] [PubMed]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Harada, Y.; Niimi, H.; Kyo, T.; Kimura, A.; Inaba, T. High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 2004, 103, 2316–2324. [Google Scholar] [CrossRef]
- Takahashi, K.; Wang, F.; Morita, K.; Yan, Y.; Hu, P.; Zhao, P.; Zhar, A.A.; Wu, C.J.; Gumbs, C.; Little, L.; et al. Integrative genomic analysis of adult mixed phenotype acute leukemia delineates lineage associated molecular subtypes. Nat. Commun. 2018, 9, 2670. [Google Scholar] [CrossRef] [Green Version]
- Andrews, C.; Tierens, A.; Minden, M. The genomic and biological complexity of mixed phenotype acute leukemia. Crit. Rev. Clin. Lab. Sci. 2021, 58, 153–166. [Google Scholar] [CrossRef]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Bhatt, A.; Pan, L.; Pedamallu, C.S.; Severson, E.; Gutierrez, A.; Dorfman, D.M.; Kuo, F.C.; Kluk, M.; Kung, A.L.; et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: Genetic and epigenetic findings in an outlier case. Mol. Case Stud. 2015, 1, a000539. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Variation | Phase | Treatment |
|---|---|---|
| Pre-diagnosis | Germline CSF3R truncation mutation W791* |
|
| Diagnosis | CNL |
|
| Transformation | Acute leukemia of MPAL type |
|
| Last follow-up | Complete molecular remission |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adam, F.C.; Szybinski, J.; Halter, J.P.; Cantoni, N.; Wenzel, F.; Leonards, K.; Brkic, S.; Passweg, J.R.; Touw, I.; Maxson, J.E.; et al. Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia. Curr. Oncol. 2022, 29, 805-815. https://doi.org/10.3390/curroncol29020068
Adam FC, Szybinski J, Halter JP, Cantoni N, Wenzel F, Leonards K, Brkic S, Passweg JR, Touw I, Maxson JE, et al. Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia. Current Oncology. 2022; 29(2):805-815. https://doi.org/10.3390/curroncol29020068
Chicago/Turabian StyleAdam, Franziska C., Jakub Szybinski, Jörg P. Halter, Nathan Cantoni, Friedel Wenzel, Katharina Leonards, Sime Brkic, Jakob R. Passweg, Ivo Touw, Julia E. Maxson, and et al. 2022. "Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia" Current Oncology 29, no. 2: 805-815. https://doi.org/10.3390/curroncol29020068
APA StyleAdam, F. C., Szybinski, J., Halter, J. P., Cantoni, N., Wenzel, F., Leonards, K., Brkic, S., Passweg, J. R., Touw, I., Maxson, J. E., & Meyer, S. C. (2022). Co-Occurring CSF3R W791* Germline and Somatic T618I Driver Mutations Induce Early CNL and Clonal Progression to Mixed Phenotype Acute Leukemia. Current Oncology, 29(2), 805-815. https://doi.org/10.3390/curroncol29020068