Value of Combined Diagnosis for Choroidal Lymphoma: A Case Report

Abstract
:1. Introduction
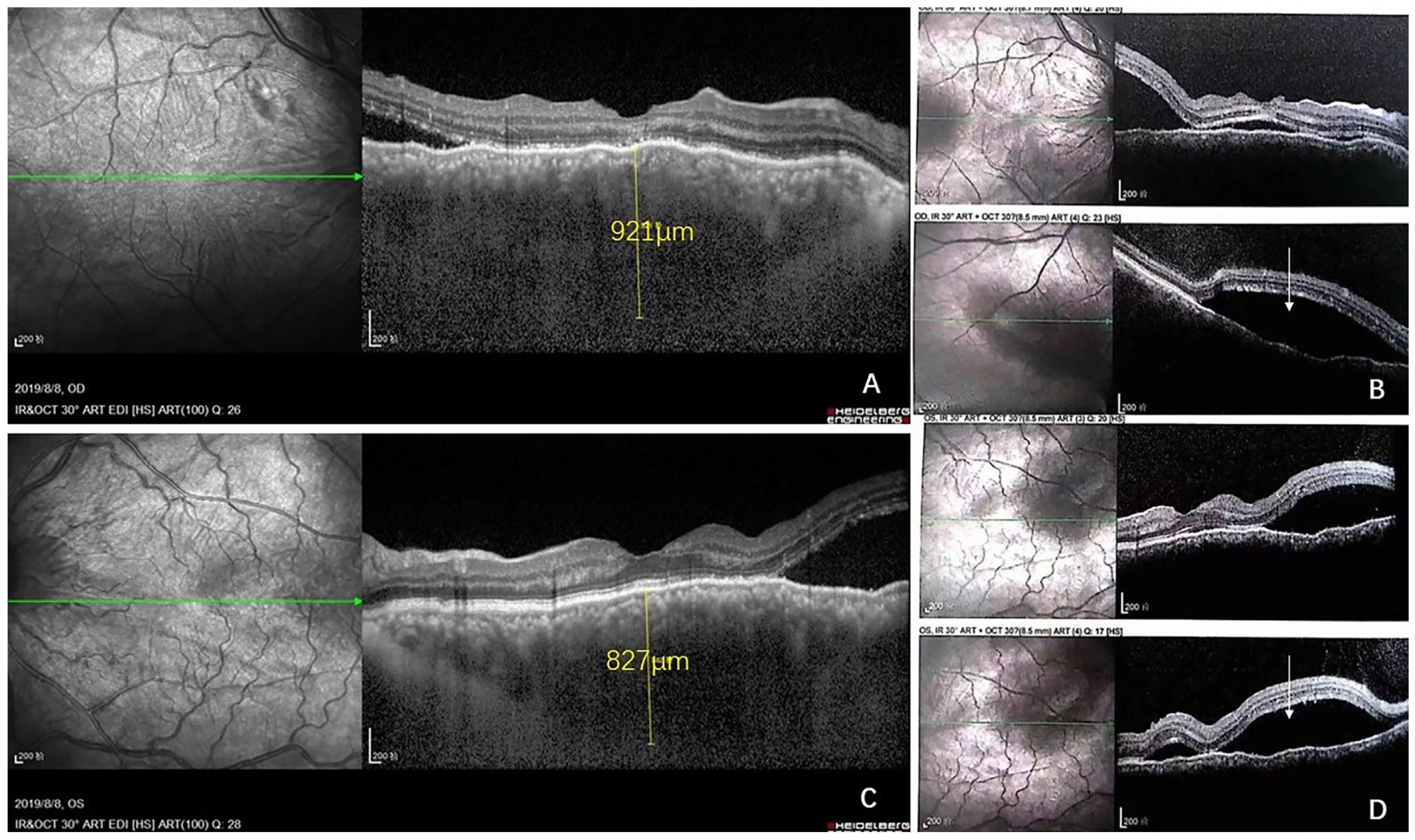
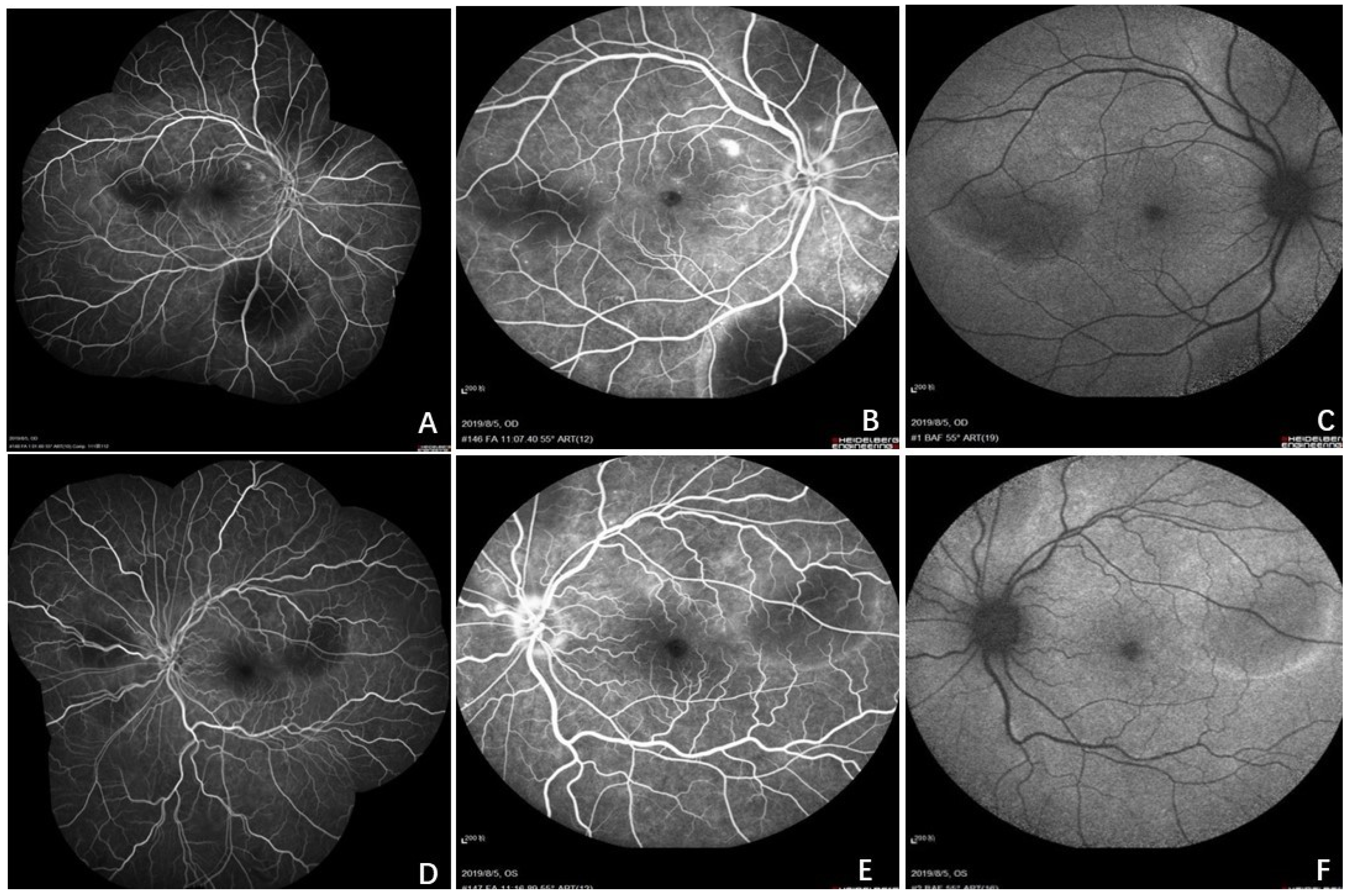
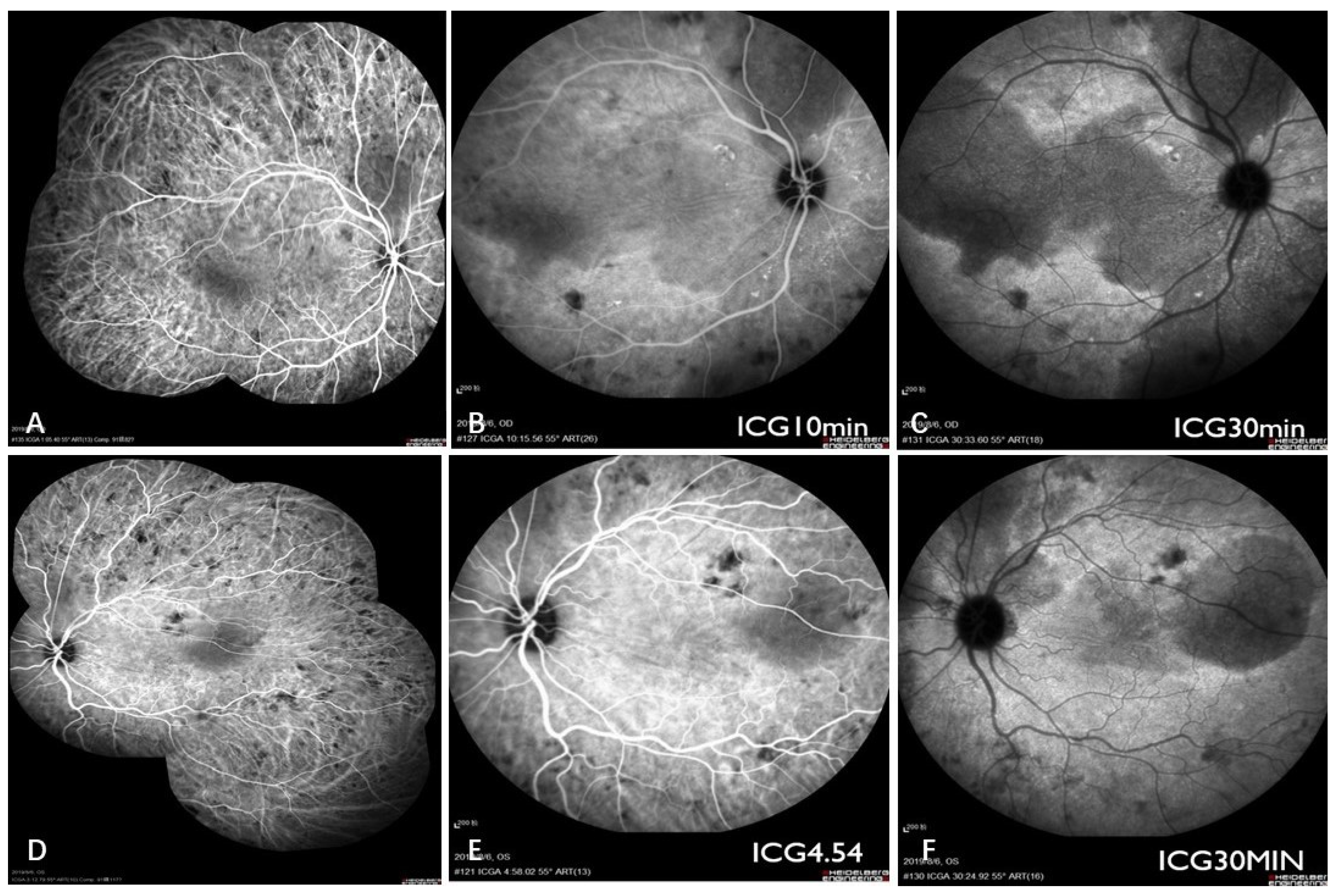
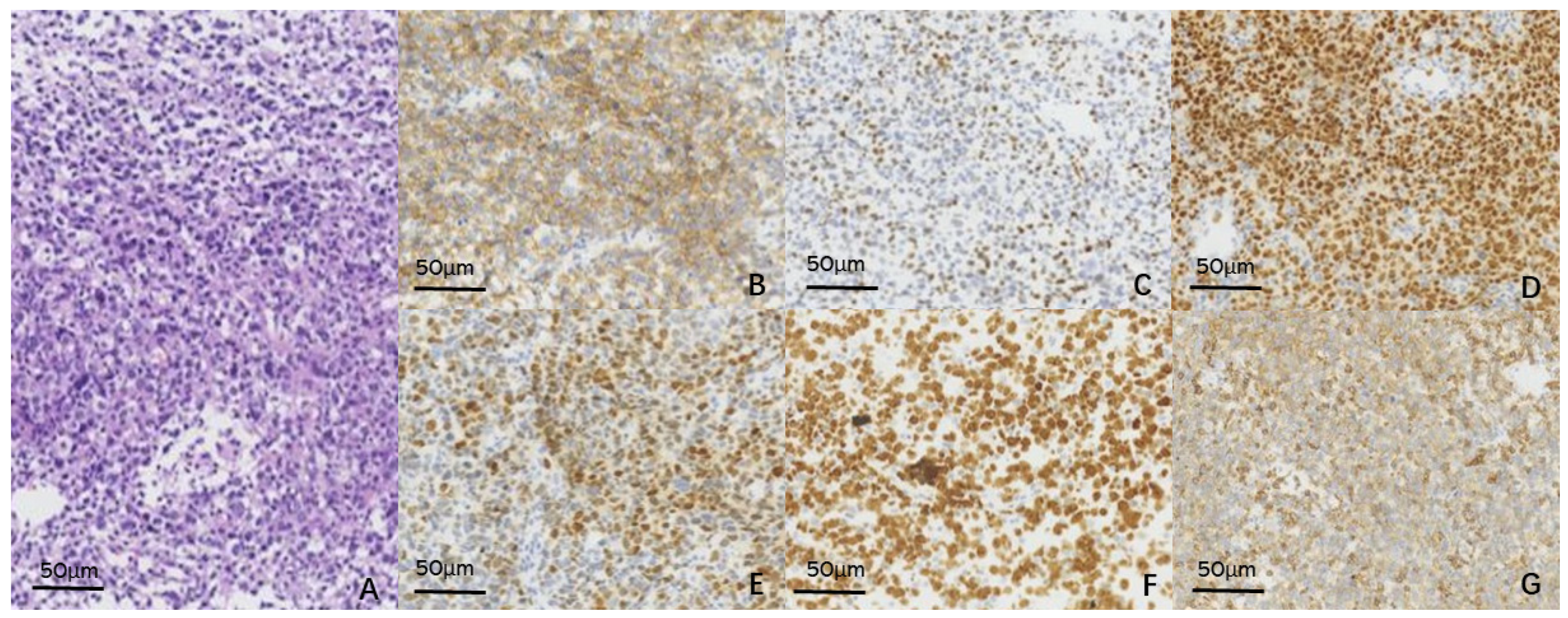
2. Case Report
3. Discussion
3.1. Brief Review of CL
3.2. Combined Diagnosis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reddy, E.K.; Bhatia, P.; Evans, R.G. Primary orbital lymphomas. Int. J. Radiat. Oncol. Biol. Phys. 1988, 15, 1239–1241. [Google Scholar] [CrossRef]
- Tang, L.J.; Gu, C.L.; Zhang, P. Intraocular lymphoma. Int. J. Ophthalmol. 2017, 10, 1301. [Google Scholar] [PubMed]
- Carole, S.; Denis, M.; Nathalie, C. Primary vitreoretinal lymphoma: A diagnostic and management challenge. Blood 2021, 138, 1519–1534. [Google Scholar] [CrossRef]
- Aronow, M.E.; Portell, C.A.; Sweetenham, J.W.; Singh, A.D. Uveal lymphoma: Clinical features, diagnostic studies, treatment selection, and outcomes. Ophthalmology 2014, 121, 334–341. [Google Scholar] [CrossRef]
- Mashayekhi, A.; Shukla, S.Y.; Shields, J.A.; Shields, C.L. Choroidal lymphoma: Clinical features and association with systemic lymphoma. Ophthalmology 2014, 121, 342–351. [Google Scholar] [CrossRef]
- Cooper, E.L.; Riker, J.L. Malignant lymphoma of the uveal tract. Am. J. Ophthalmol. 1951, 34, 1153–1158. [Google Scholar] [CrossRef]
- Vogel, M.H.; Font, R.L.; Zimmerman, L.E.; Levine, R.A. Reticulum cell sarcoma of the retina and uvea. Report of six cases and review of the literature. Am. J. Ophthalmol. 1968, 66, 205–215. [Google Scholar] [CrossRef]
- Nelson, C.C.; Hertzberg, B.S.; Klintworth, G.K. A histopathologic study of 716 unselected eyes in patients with cancer at the time of death. Am. J. Ophthalmol. 1983, 95, 788–793. [Google Scholar] [CrossRef]
- Leff, S.R.; Shields, J.A.; Augsburger, J.J.; Miller, R.V.; Liberatore, B.E. Unilateral eyelid, conjunctival, and choroidal tumours as initial presentation of diffuse large-cell lymphoma. Br. J. Ophthalmol. 1985, 69, 861–864. [Google Scholar] [CrossRef] [Green Version]
- Tavallali, A.; Shields, C.L.; Bianciotto, C.; Shields, J.A. Choroidal Lymphoma Masquerading as Anterior Ischemic Optic Neuropathy. Eur. J. Ophthalmol. 2010, 20, 959. [Google Scholar] [CrossRef]
- Coupland, S.E.; Foss, H.D.; Hidayat, A.A.; Cockerham, G.C.; Hummel, M.; Stein, H. Extranodal marginal zone B cell lymphomas of the uvea: An analysis of 13 cases. J. Pathol. 2002, 197, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Coupland, S.E.; Damato, B. Understanding intraocular lymphomas. Clin. Exp. Ophthalmol 2008, 36, 564–578. [Google Scholar] [CrossRef] [PubMed]
- White, V.A. Understanding and classification of ocular lymphomas. Ocul. Oncol. Pathol. 2019, 5, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Preziosa, C.; Yaghy, A.; Ruben, M.; Invernizzi, A.; Fung, A.T.; Staurenghi, G.; Shields, C.L. Choroidal Lymphoma: Diagnostic Value of Combined Indocyanine Green Angiography and Optical Coherence Tomography. Ocul. Immunol. Inflamm. 2022, 1–8. [Google Scholar] [CrossRef]
- Shields, J.A.; Shields, C.L. Intraocular Tumors: An Atlas and Textbook, 2nd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; pp. 504–533. [Google Scholar]
- Heiferman, M.J.; Yu, M.D.; Mruthyunjaya, P. Update in Molecular Testing for Intraocular Lymphoma. Cancers 2022, 14, 4546. [Google Scholar] [CrossRef]
- Rodriguez, E.F.; Sepah, Y.J.; Jang, H.S.; Ibrahim, M.; Nguyen, Q.D.; Rodriguez, F.J. Cytologic features in vitreous preparations of patients with suspicion of intraocular lymphoma. Diagn. Cytopathol. 2014, 42, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Schrijver, B.; Kolijn, P.M.; Ten Berge, J.C.; Nagtzaam, N.M.; van Rijswijk, A.L.; Swagemakers, S.M.; van der Spek, P.J.; Missotten, T.O.; van Velthoven, M.E.; de Hoog, J.; et al. Vitreous proteomics, a gateway to improved understanding and stratification of diverse uveitis aetiologies. Acta Ophthalmol. 2022, 100, 403–413. [Google Scholar] [CrossRef]
- Hernández-Pons, A.; Gómez-Beltrán, E.; Fernández-Zarzoso, M.; Albert-Fort, M.; Martínez-Costa, L. Choroidal lymphoma diagnosed by polymerase chain reaction–based clonality assessment. Eur. J. Ophthalmol. 2020, 31, 112067212091133. [Google Scholar] [CrossRef]
- Fung, C.Y.; Tarbell, N.J.; Lucarelli, M.J.; Goldberg, S.I.; Linggood, R.M.; Harris, N.L.; Ferry, J.A. Ocular adnexal lymphoma: Clinical behavior of distinct World Health Organization classification subtypes. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 1382–1391. [Google Scholar] [CrossRef]
- Mashayekhi, A.; Hasanreisoglu, M.; Shields, C.L.; Shields, J.A. External beam radiation for choroidal lymphoma: Efficacy and complications. Retina 2016, 36, 2006–2012. [Google Scholar] [CrossRef]
- Doycheva, D.; Zierhut, M.; Süsskind, D.; Bartz-Schmidt, K.U.; Deuter, C. Diagnostics and treatment of choroidal lymphoma. Ophthalmologe 2015, 112, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sewit, T.; Joachim, Y. Primary intraocular lymphoma: Treatment outcomes with ocular radiation therapy alone. Leuk Lymphoma 2014, 55, 795–801. [Google Scholar]
- Yang, X.; Dalvin, L.A.; Lim, L.A.; Mashayekhi, A.; Shields, J.A.; Shields, C.L. Ultra-low-dose (boom-boom) radiotherapy for choroidal lymphoma in three consecutive cases. Eur. J. Ophthalmol. 2021, 31, NP91–NP96. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Usui, Y.; Goto, H. Clinical features and diagnostic significance of the intraocular fluid of 217 patients with intraocular lymphoma. Jpn. J. Ophthalmol. 2012, 56, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Fukutsu, K.; Namba, K.; Iwata, D.; Mizuuchi, K.; Kase, S.; Suzuki, K.; Shimizu, H.; Shibata, Y.; Yamawaki, F.; Onozawa, M.; et al. Pseudo-inflammatory manifestations of choroidal lymphoma resembling Vogt-Koyanagi-Harada disease: Case report based on multimodal imaging. BMC Ophthalmol. 2020, 20, 94. [Google Scholar] [CrossRef]
- Marko, L.; Mark, W. Unilateral Idiopathic Choroidal Effusion in a Patient Who Takes Sulfonamides. Ocul. Immunol. Inflamm. 2021, 29, 1032–1034. [Google Scholar]
- Shields, C.L.; Arepalli, S.; Pellegrini, M.; Mashayekhi, A.; Shields, J.A. Choroidal lymphoma shows calm, rippled, or undulating topography on enhanced depth imaging optical coherence tomography in 14 eyes. Retina 2014, 34, 1347–1353. [Google Scholar] [CrossRef]
- Portell, C.A.; Aronow, M.E.; Rybicki, L.A.; Macklis, R.; Singh, A.D.; Sweetenham, J.W. Clinical Characteristics of 95 Patients With Ocular Adnexal and Uveal Lymphoma: Treatment Outcomes in Extranodal Marginal Zone Subtype. Clin. Lymphoma Myeloma Leuk. 2014, 14, 203–210. [Google Scholar] [CrossRef]
- Kam, A.W.; Galvin, J.; Cherepanoff, S.; Miller, A.A.; Fung, A.T. Primary Choroidal Lymphoma Diagnosed with 27-Gauge Pars Plana Vitrectomy Choroidal Biopsy. Case Rep. Ophthalmol. 2019, 10, 10–213. [Google Scholar] [CrossRef]
- Whitcup, S.M.; Stark-Vancs, V.; Wittes, R.E.; Solomon, D.; Podgor, M.J.; Nussenblatt, R.B.; Chan, C.C. Association of interleukin 10 in the vitreous and cerebrospinal fluid and primary central nervous system lymphoma. Arch. Ophthalmol. 1997, 115, 1157–1160. [Google Scholar] [CrossRef]
- Wolf, L.A.; Reed, G.F.; Buggage, R.R.; Nussenblatt, R.B.; Chan, C.C. Vitreous cytokine levels. Ophthalmology 2003, 110, 1671–1672. [Google Scholar] [CrossRef]
- Mathai, A.; Lall, A.; Jain, R.; Pathengay, A. Systemic non-Hodgkin’s lymphoma masquerading as Vogt–Koyanagi–Harada disease in an HIV-positive patient. Clin. Exp. Ophthalmol. 2006, 34, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Gyoten, D.; Ueno, S.; Okado, S.; Chaya, T.; Yasuda, S.; Morimoto, T.; Kondo, M.; Kimura, K.; Hayashi, T.; Leroy, B.P.; et al. Broad locations of antigenic regions for anti-TRPM1 autoantibodies in paraneoplastic retinopathy with retinal ON bipolar cell dysfunction. Exp. Eye Res. 2021, 212, 108770. [Google Scholar] [CrossRef] [PubMed]
- Pefkianaki, M.; Agrawal, R.; Desai, P.; Pavesio, C.; Sagoo, M.S. Bilateral Diffuse Uveal Melanocytic Proliferation (BDUMP) associated with B-cell lymphoma: Report of a rare case. BMC Cancer 2015, 15, 23. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Items | Result | Unit | Reference Range | Test Method |
|---|---|---|---|---|
| IL-2 | 0 | Pg/mL | Cytometric Bead Array | |
| IL-4 | 0 | Pg/mL | Cytometric Bead Array | |
| IL-6 | 23.1 | Pg/mL | 1.0~50.0 | Cytometric Bead Array |
| IL-10 | 0 | Pg/mL | 0~5.0 | Cytometric Bead Array |
| TNF-α | 0 | Pg/mL | 0~5.0 | Cytometric Bead Array |
| IFN-γ | 0.8 | Pg/mL | - | Cytometric Bead Array |
| IL-10/IL-6 | 0 | - | <1 | Cytometric Bead Array |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Zhang, T.; Yan, B.; Huang, Y. Value of Combined Diagnosis for Choroidal Lymphoma: A Case Report. Curr. Oncol. 2022, 29, 8835-8845. https://doi.org/10.3390/curroncol29110695
Yang M, Zhang T, Yan B, Huang Y. Value of Combined Diagnosis for Choroidal Lymphoma: A Case Report. Current Oncology. 2022; 29(11):8835-8845. https://doi.org/10.3390/curroncol29110695
Chicago/Turabian StyleYang, Ming, Taoran Zhang, Bojing Yan, and Yingxiang Huang. 2022. "Value of Combined Diagnosis for Choroidal Lymphoma: A Case Report" Current Oncology 29, no. 11: 8835-8845. https://doi.org/10.3390/curroncol29110695
APA StyleYang, M., Zhang, T., Yan, B., & Huang, Y. (2022). Value of Combined Diagnosis for Choroidal Lymphoma: A Case Report. Current Oncology, 29(11), 8835-8845. https://doi.org/10.3390/curroncol29110695