Overcoming of Microenvironment Protection on Primary Chronic Lymphocytic Leukemia Cells after Treatment with BTK and MDM2 Pharmacological Inhibitors


 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Next Generation Sequencing (NGS)
2.3. Pharmacological Treatments and Evaluation of Cell Viability and Apoptosis
2.4. Mitochondrial Activity Assessment
2.5. RNA and Protein Analyses
2.6. Statistical Analysis
3. Results
3.1. Microenvironment-Activated B-CLL Cells Display Up-Regulation of c-MYC and p53
3.2. Ibrutinib and Nutlin-3 Combination Efficiently Kills Microenvironment-Activated Leukemic Cells
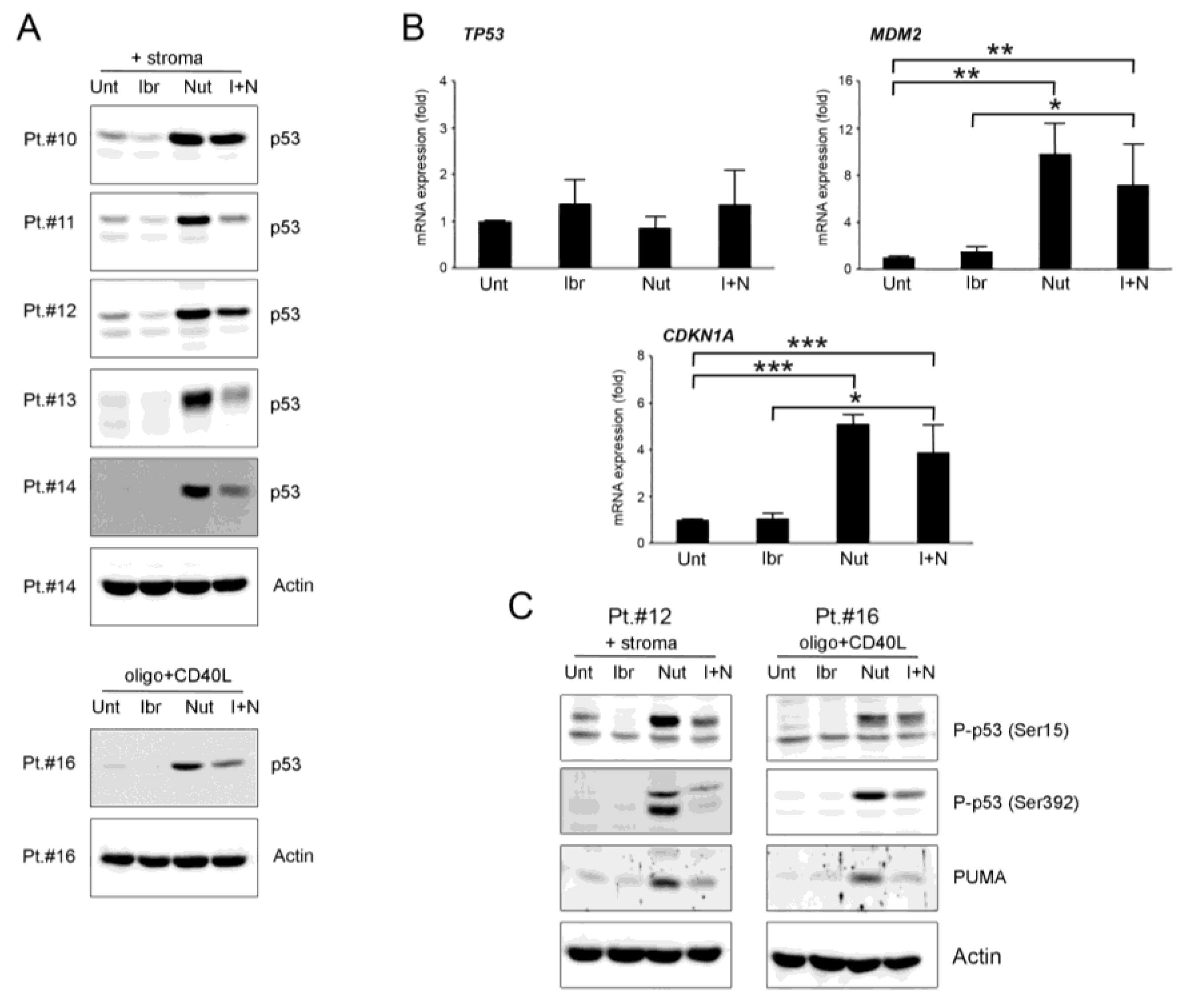
3.3. Effects of Ibrutinib and Nutlin-3 Combination on p53 Pathway
3.4. Ibrutinib Plus Nutlin-3 Combination Mediates Mitochondria-Dependent Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, M.Y.; Kashyap, M.K.; Kumar, D. The chronic lymphocytic leukemia microenvironment: Beyond the B-cell receptor. Best Pract. Res. Clin. Haematol. 2016, 29, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef]
- Vangapandu, H.V.; Ayres, M.L.; Bristow, C.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Stellrecht, C.M.; Gandhi, V. The stromal microenvironment modulates mitochondrial oxidative phosphorylation in chronic lymphocytic leukemia cells. Neoplasia 2017, 19, 762–771. [Google Scholar] [CrossRef]
- Blunt, M.D.; Koehrer, S.; Dobson, R.C.; Larrayoz, M.; Wilmore, S.; Hayman, A.; Parnell, J.; Smith, L.D.; Davies, A.; Johnson, P.W.M.; et al. The dual Syk/JAK inhibitor cerdulatinib antagonizes b-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin. Cancer. Res. 2017, 23, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Bojarczuk, K.; Sasi, B.K.; Gobessi, S.; Innocenti, I.; Pozzato, G.; Laurenti, L.; Efremov, D.G. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood 2016, 127, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef]
- Oppermann, S.; Ylanko, J.; Shi, Y.; Hariharan, S.; Oakes, C.C.; Brauer, P.M.; Zúñiga-Pflücker, J.C.; Leber, B.; Spaner, D.E.; Andrews, D.W. High-content screening identifies kinase inhibitors that overcome venetoclax resistance in activated CLL cells. Blood 2016, 128, 934–947. [Google Scholar] [CrossRef]
- Secchiero, P.; Voltan, R.; Rimondi, E.; Melloni, E.; Athanasakis, E.; Tisato, V.; Gallo, S.; Rigolin, G.M.; Zauli, G. The γ-secretase inhibitors enhance the anti-leukemic activity of ibrutinib in B-CLL cells. Oncotarget 2017, 8, 59235–59245. [Google Scholar] [CrossRef][Green Version]
- Brown, J.R.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.E.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Extended follow-up and impact of high-risk prognostic factors from the phase 3 RESONATE study in patients with previously treated CLL/SLL. Leukemia 2018, 32, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Chanan-Khan, A.; Cramer, P.; Demirkan, F.; Fraser, G.; Silva, R.S.; Grosicki, S.; Pristupa, A.; Janssens, A.; Mayer, J.; Bartlett, N.L.; et al. HELIOS investigators, ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): A randomised, double-blind, phase 3 study. Lancet Oncol. 2016, 17, 200–211. [Google Scholar] [PubMed]
- Ahn, I.E.; Underbayev, C.; Albitar, A.; Herman, S.E.; Tian, X.; Maric, I.; Arthur, D.C.; Wake, L.; Pittaluga, S.; Yuan, C.M.; et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017, 129, 1469–1479. [Google Scholar] [CrossRef]
- Quinquenel, A.; Fornecker, L.M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: A FILO group study. Blood 2019, 134, 641–644. [Google Scholar] [CrossRef]
- Jain, N.; Keating, M.; Thompson, P.; Ferrajoli, A.; Burger, J.; Borthakur, G.; Takahashi, K.; Estrov, Z.; Fowler, N.; Kadia, T.; et al. Ibrutinib and Venetoclax for First-Line Treatment of CLL. N. Engl. J. Med. 2019, 380, 2095–2103. [Google Scholar] [CrossRef]
- Thompson, P.A.; Burger, J.A. Bruton’s tyrosine kinase inhibitors: First and second generation agents for patients with Chronic Lymphocytic Leukemia (CLL). Expert Opin. Investig. Drugs 2018, 27, 31–42. [Google Scholar] [CrossRef]
- Voltan, R.; Rimondi, E.; Melloni, E.; Rigolin, G.M.; Casciano, F.; Arcidiacono, M.V.; Celeghini, C.; Cuneo, A.; Zauli, G.; Secchiero, P. Ibrutinib synergizes with MDM-2 inhibitors in promoting cytotoxicity in B chronic lymphocytic leukemia. Oncotarget 2016, 7, 70623–70638. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gullà, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Mol. Ther. Nucl. Acids 2014, 3, e194. [Google Scholar] [CrossRef]
- Abraham, S.A.; Hopcroft, L.E.; Carrick, E.; Drotar, M.E.; Dunn, K.; Williamson, A.J.; Korfi, K.; Baquero, P.; Park, L.E.; Scott, M.T.; et al. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature 2016, 534, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Rigolin, G.M.; Formigaro, L.; Cavallari, M.; Quaglia, F.M.; Lista, E.; Urso, A.; Guardalben, E.; Martinelli, S.; Saccenti, E.; Bassi, C.; et al. An extensive molecular cytogenetic characterization in high-risk chronic lymphocytic leukemia identifies karyotype aberrations and TP53 disruption as predictors of outcome and chemorefractoriness. Oncotarget 2017, 8, 28008–28020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Del Papa, B.; Baldoni, S.; Dorillo, E.; De Falco, F.; Rompietti, C.; Cecchini, D.; Cantelmi, M.G.; Sorcini, D.; Nogarotto, M.; Adamo, F.M.; et al. Decreased NOTCH1 Activation Correlates with Response to Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 7540–7553. [Google Scholar] [CrossRef] [PubMed]
- Chanas-Larue, A.; Villalpando-Rodriguez, G.E.; Henson, E.S.; Henson, E.S.; Johnston, J.B.; Gibson, S.B. Antihistamines are synergistic with Bruton’s tyrosine kinase inhibiter ibrutinib mediated by lysosome disruption in chronic lymphocytic leukemia (CLL) cells. Leuk. Res. 2020, 96, 106423. [Google Scholar] [CrossRef] [PubMed]
- Skånland, S.S.; Cremaschi, A.; Bendiksen, H.; Hermansen, J.U.; Thimiri Govinda Raj, D.B.; Munthe, L.A.; Tjønnfjord, G.E.; Taskén, K. An in vitro assay for biomarker discovery and dose prediction applied to ibrutinib plus venetoclax treatment of CLL. Leukemia 2020, 34, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Voltan, R.; Bosco, R.; Melloni, E.; Marmiroli, S.; Rigolin, G.M.; Cuneo, A.; Secchiero, P. Dasatinib plus Nutlin-3 shows synergistic antileukemic activity in both p53 wild-type and p53 mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin. Cancer Res. 2011, 17, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Celeghini, C.; Voltan, R.; Rimondi, E.; Gattei, V.; Zauli, G. Perifosine selectively induces cell cycle block and modulates retinoblastoma and E2F1 protein levels in p53 mutated leukemic cell lines. Invest. New Drugs 2011, 29, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Rada, M.; Samuel, J.; Escorsa, J.M.; Najeeb, H.; Lee, K.G.; Lam, K.P.; Jones, G.D.; Barlev, N.A.; Macip, S. BTK modulates p53 activity to enhance apoptotic and senescent responses. Cancer Res. 2016, 76, 5405–5414. [Google Scholar] [CrossRef]
- Valente, L.J.; Aubrey, B.J.; Herold, M.J.; Kelly, G.L.; Happo, L.; Scott, C.L.; Newbold, A.; Johnstone, R.W.; Huang, D.C.; Vassilev, L.T.; et al. Therapeutic response to non-genotoxic activation of p53 by nutlin3a is driven by PUMA-mediated apoptosis in lymphoma cells. Cell Reports 2016, 14, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Xu, W.; Li, Y.; Yuan, W.W.; Yin, Y.; Song, W.W.; Wang, Y.; Huang, Q.Q.; Zhao, W.H.; Wu, J.Q. Membrane-Bound CD40L Promotes Senescence and Initiates Senescence-Associated Secretory Phenotype via NF-κB Activation in Lung Adenocarcinoma. Cell Physiol. Biochem. 2018, 48, 1793–1803. [Google Scholar] [CrossRef]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. [Google Scholar] [CrossRef]
- Moyo, T.K.; Wilson, C.S.; Moore, D.J.; Eischen, C.M. Myc enhances B-cell receptor signaling in precancerous B cells and confers resistance to Btk inhibition. Oncogene 2017, 36, 4653–4661. [Google Scholar] [CrossRef]
- Castrogiovanni, C.; Waterschoot, B.; De Backer, O.; Dumont, P. Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 2018, 25, 190–203. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, A. Survival signaling goes BAD. Dev. Cell 2002, 3, 607–608. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. p53 and Bad: Remote strangers become close friends. Cell Res. 2007, 17, 283–285. [Google Scholar] [CrossRef]
- Cao, X.; Deng, X.; May, W.S. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood 2003, 102, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2017, 92, 946–965. [Google Scholar] [CrossRef]
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J. Clin. Oncol. 2017, 35, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Patel, A.; Wang, Y.J.; Zhang, Y.K.; Kathawala, R.J.; Qiu, L.H.; Patel, B.A.; Huang, L.H.; Shukla, S.; Yang, D.H.; et al. The BTK inhibitor ibrutinib (PCI-32765) overcomes paclitaxel resistance in ABCB1- and ABCC10-overexpressing cells and tumors. Mol. Cancer Ther. 2017, 16, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Grassilli, E.; Pisano, F.; Cialdella, A.; Bonomo, S.; Missaglia, C.; Cerrito, M.G.; Masiero, L.; Ianzano, L.; Giordano, F.; Cicirelli, V.; et al. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene 2016, 35, 4368–4378. [Google Scholar] [CrossRef]
- Zhou, Y.; Perez, R.E.; Duan, L.; Maki, C.G. DZNep represses Bcl-2 expression and modulates apoptosis sensitivity in response to Nutlin-3a. Cancer Biol. Ther. 2018, 19, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, X.; Li, Y.; Xiao, Q.; Cui, X.H.; Xiao, G.D.; Wang, J.C.; Xu, C.W.; Ren, H.; Liu, D. Nutlin-3-induced Sensitization of Non-Small Cell Lung Cancer Stem Cells to Axitinib-Induced Apoptosis through Repression of Akt1/Wnt Signaling. Oncol. Res. 2019, 27, 987. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Demographics | B-CLL Characterization | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pt. # | Age | Sex | WBC × 103/Lymphocytes (%) | CD38+ † | IgHV status | Cytogenetic Abnormalities * | TP53 Status (%) | Therapy | |
| 1 | 76 | M | 67.1/84.7 | pos | mut | del13q | c.380C>T (39.6%), c.920-2A>G (splicing) (26.2%) | R-ibr | |
| 2 | 58 | M | 84.3/84.2 | neg | unmut | neg | unmut | FCR | |
| 3 | 85 | F | 54.8/90.2 | pos | mut | del13q, trisomy 12 | unmut | steroids | |
| 4 | 68 | M | 122/89.5 | neg | unmut | del11q | unmut | Ibr | |
| 5 | 57 | F | 181/97.8 | neg | mut | del13q | unmut | R-ibr | |
| 6 | 84 | F | 99/94.4 | neg | mut | del13q | unmut | Chl + Pdn | |
| 7 | 84 | M | 69.6/88.2 | neg | na | neg | unmut | untreated | |
| 8 | 36 | F | 197/80.3 | pos | unmut | neg | unmut | FCR | |
| 9 | 66 | M | 23.9/83.7 | neg | unmut | del13q | unmut | Ibr | |
| 10 | 76 | F | na | pos | unmut | del13q | unmut | R-benda | |
| 11 | 59 | M | 194.9/90.6 | neg | unmut | trisomy 12 | unmut | FCR | |
| 12 | 58 | M | 24.6/81.3 | neg | mut | del13q | unmut | untreated | |
| 13 | 71 | M | 73.6/91.5 | neg | mut | del13q | unmut | untreated | |
| 14 | 59 | F | 52.2/90.2 | pos | na | del11q | unmut | FCR | |
| 15 | 56 | M | 82.6/94.3 | pos | unmut | trisomy 12 | unmut | FCR | |
| 16 | 61 | M | 49.5/66.5 | neg | unmut | trisomy 12 | c.G743>A (1%), c.G527>T (11.3%) | ibr | |
| 17 | 60 | M | 34/80.1 | pos | na | del13q, del11q, trisomy 12 | c.G733>A (17.4%) | ibr + ofatum | |
| 18 | 75 | M | 10.4/88.3 | neg | mut | del13q | unmut | untreaed | |
| 19 | 69 | F | 156/92.8 | pos | unmut | neg | unmut | R-Benda | |
| 20 | 61 | M | 17.2/63.2 | neg | na | del13q | unmut | untreated | |
| 21 | 76 | M | 168/87.2 | neg | mut | del13q, del11q | unmut | untreated | |
| 22 | 81 | M | 12.7/33 | neg | na | neg | unmut | untreated | |
| 23 | 50 | F | 35.1/88.4 | neg | na | del13q | unmut | untreated | |
| 24 | 72 | M | 33.2/87.3 | pos | na | del11q, trisomy 12 | na | untreated | |
| 25 | 73 | M | 78.4/92.2 | neg | mut | neg | unmut | untreated | |
| 26 | 63 | F | 40.7/84.1 | pos | unmut | del13q | c.G475C (3.6%) | FCR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimondi, E.; Melloni, E.; Romani, A.; Tisato, V.; Casciano, F.; Rigolin, G.M.; Milani, D.; Celeghini, C.; Zauli, G.; Secchiero, P.; et al. Overcoming of Microenvironment Protection on Primary Chronic Lymphocytic Leukemia Cells after Treatment with BTK and MDM2 Pharmacological Inhibitors. Curr. Oncol. 2021, 28, 2439-2451. https://doi.org/10.3390/curroncol28040223
Rimondi E, Melloni E, Romani A, Tisato V, Casciano F, Rigolin GM, Milani D, Celeghini C, Zauli G, Secchiero P, et al. Overcoming of Microenvironment Protection on Primary Chronic Lymphocytic Leukemia Cells after Treatment with BTK and MDM2 Pharmacological Inhibitors. Current Oncology. 2021; 28(4):2439-2451. https://doi.org/10.3390/curroncol28040223
Chicago/Turabian StyleRimondi, Erika, Elisabetta Melloni, Arianna Romani, Veronica Tisato, Fabio Casciano, Gian Matteo Rigolin, Daniela Milani, Claudio Celeghini, Giorgio Zauli, Paola Secchiero, and et al. 2021. "Overcoming of Microenvironment Protection on Primary Chronic Lymphocytic Leukemia Cells after Treatment with BTK and MDM2 Pharmacological Inhibitors" Current Oncology 28, no. 4: 2439-2451. https://doi.org/10.3390/curroncol28040223
APA StyleRimondi, E., Melloni, E., Romani, A., Tisato, V., Casciano, F., Rigolin, G. M., Milani, D., Celeghini, C., Zauli, G., Secchiero, P., & Voltan, R. (2021). Overcoming of Microenvironment Protection on Primary Chronic Lymphocytic Leukemia Cells after Treatment with BTK and MDM2 Pharmacological Inhibitors. Current Oncology, 28(4), 2439-2451. https://doi.org/10.3390/curroncol28040223