
Integrated SMRT and Illumina Sequencing Provide New Insights into Crocin Biosynthesis of Gardenia jasminoides

 , , and
, , and 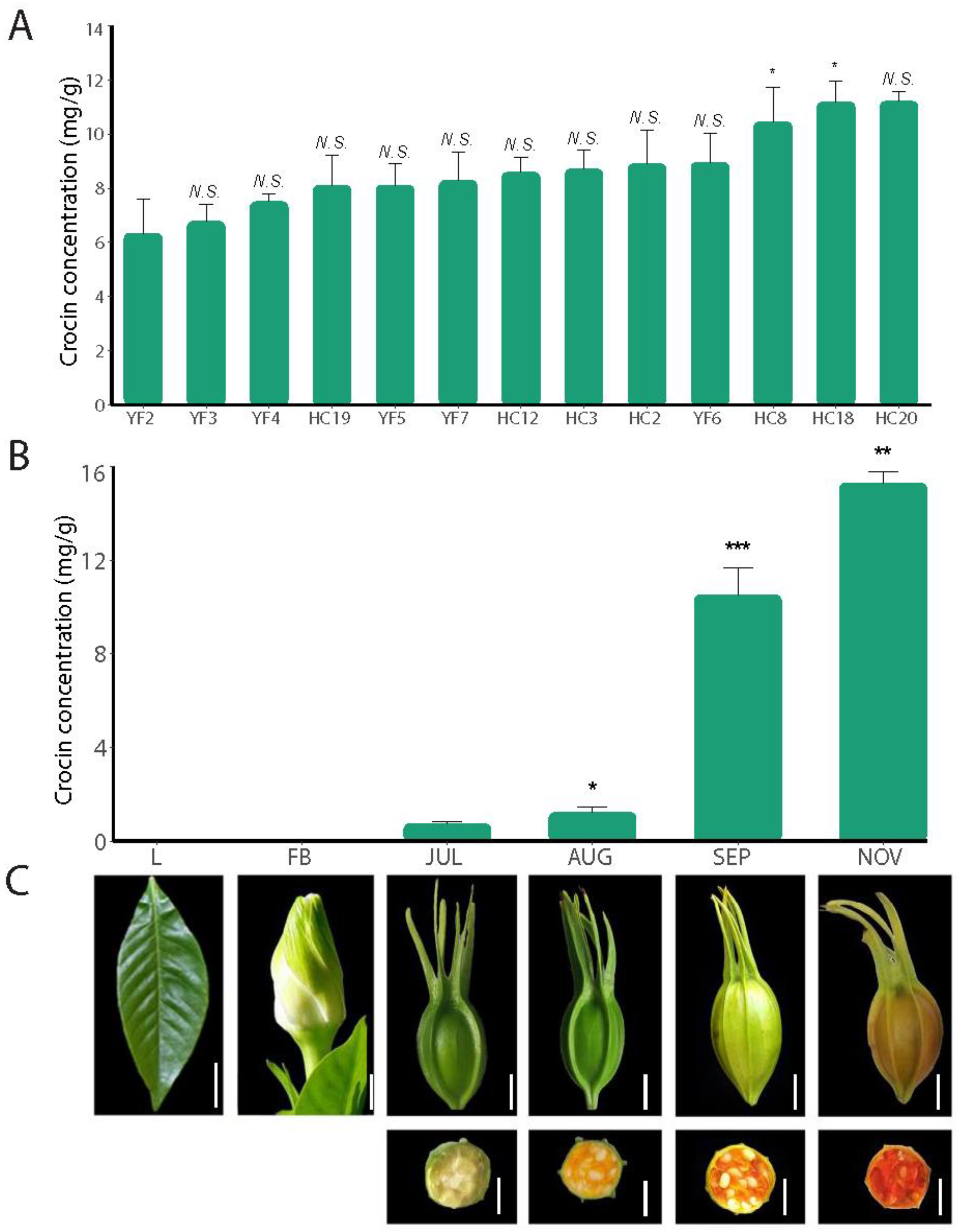{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
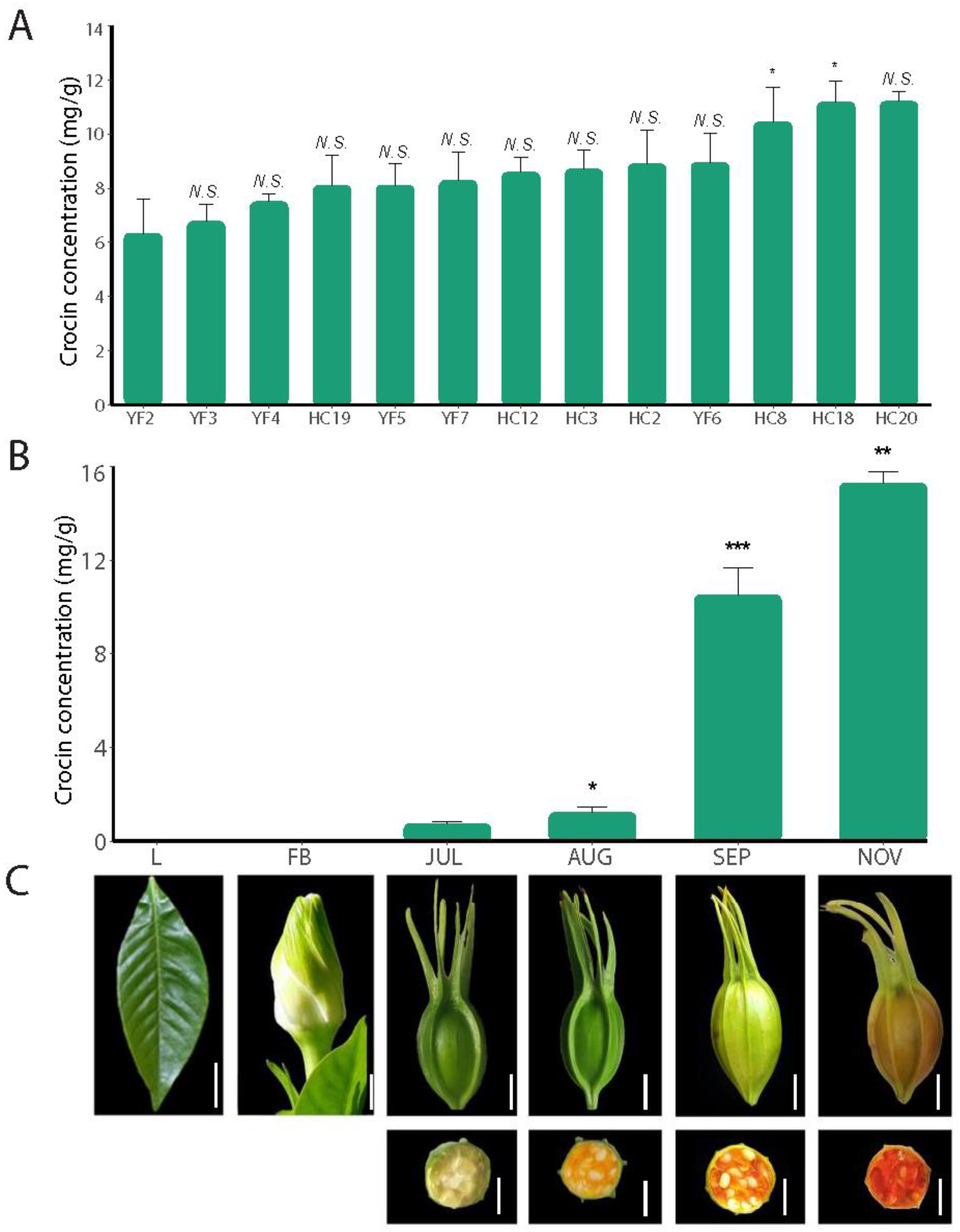
2.1. Crocin Content Changes during Fruit Development
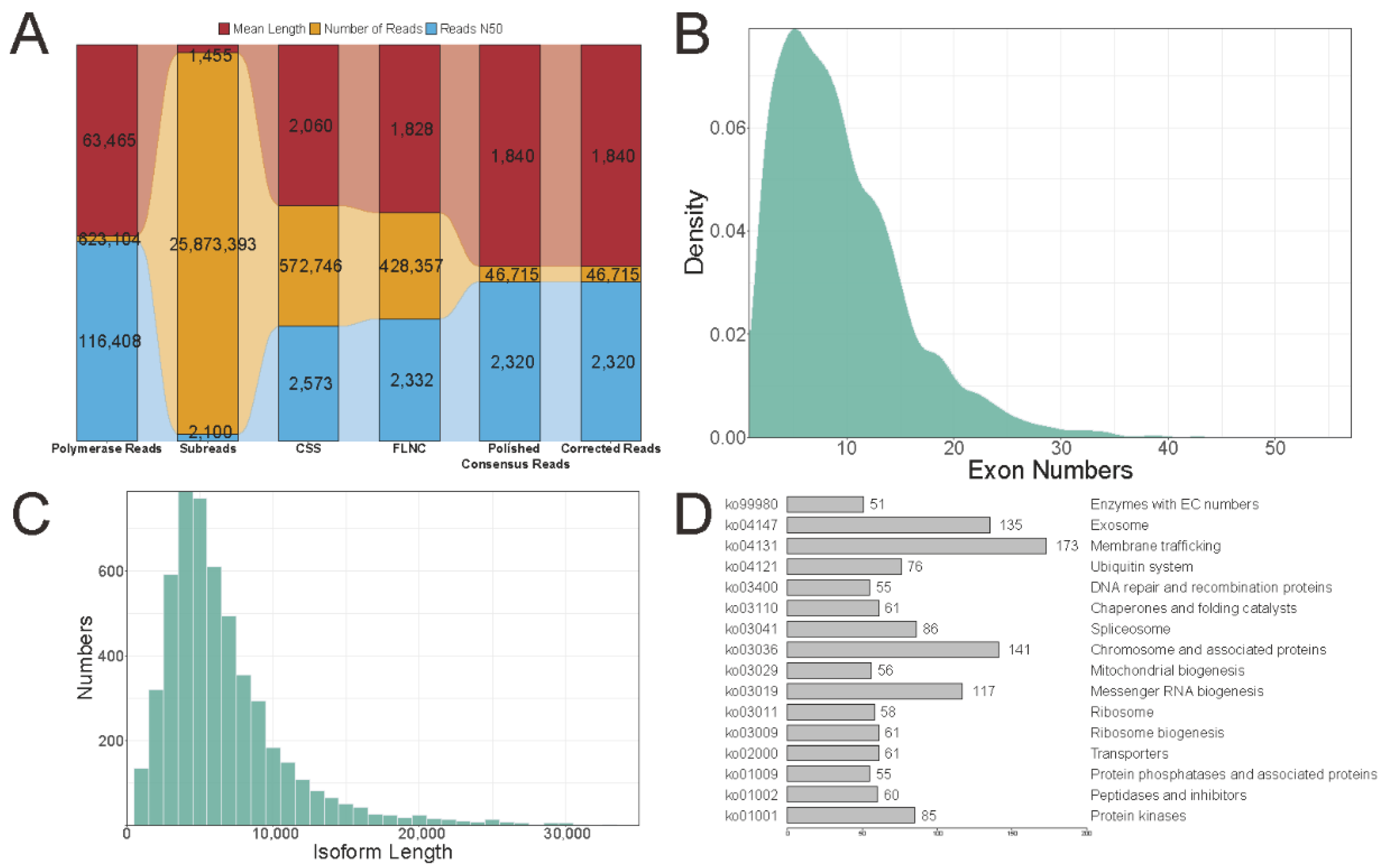
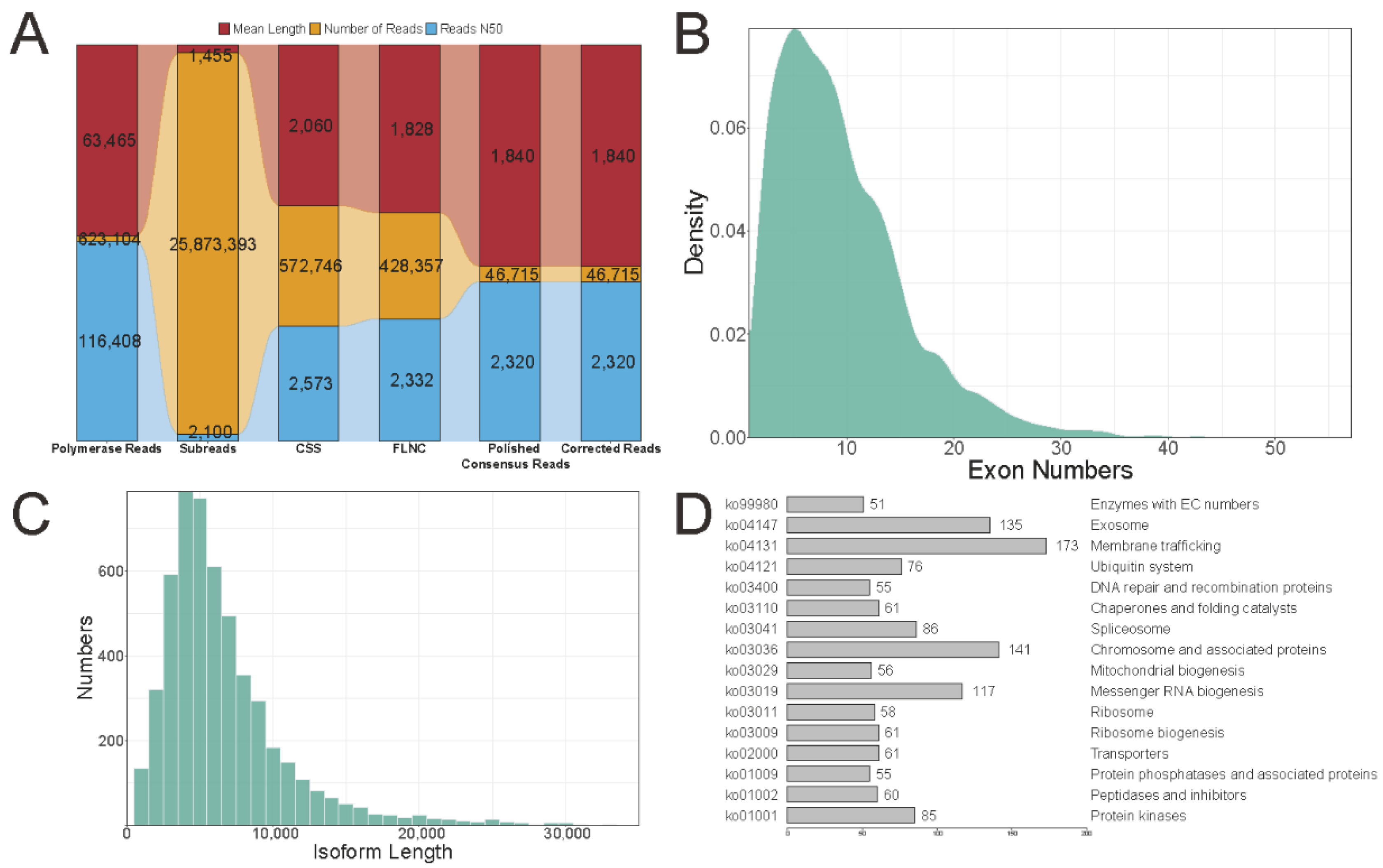
2.2. Pacbio Long-Read Sequencing Data Analysis
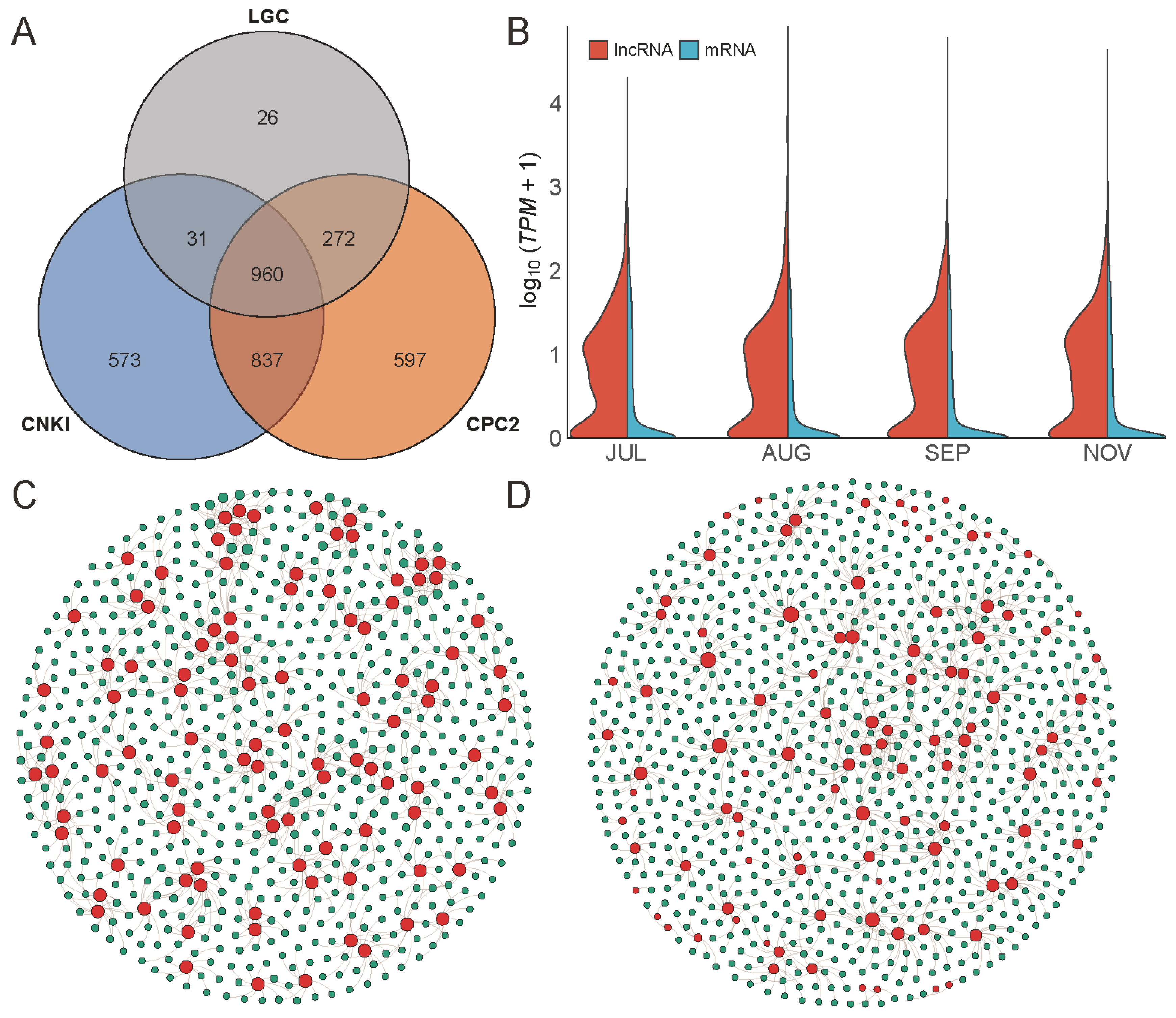
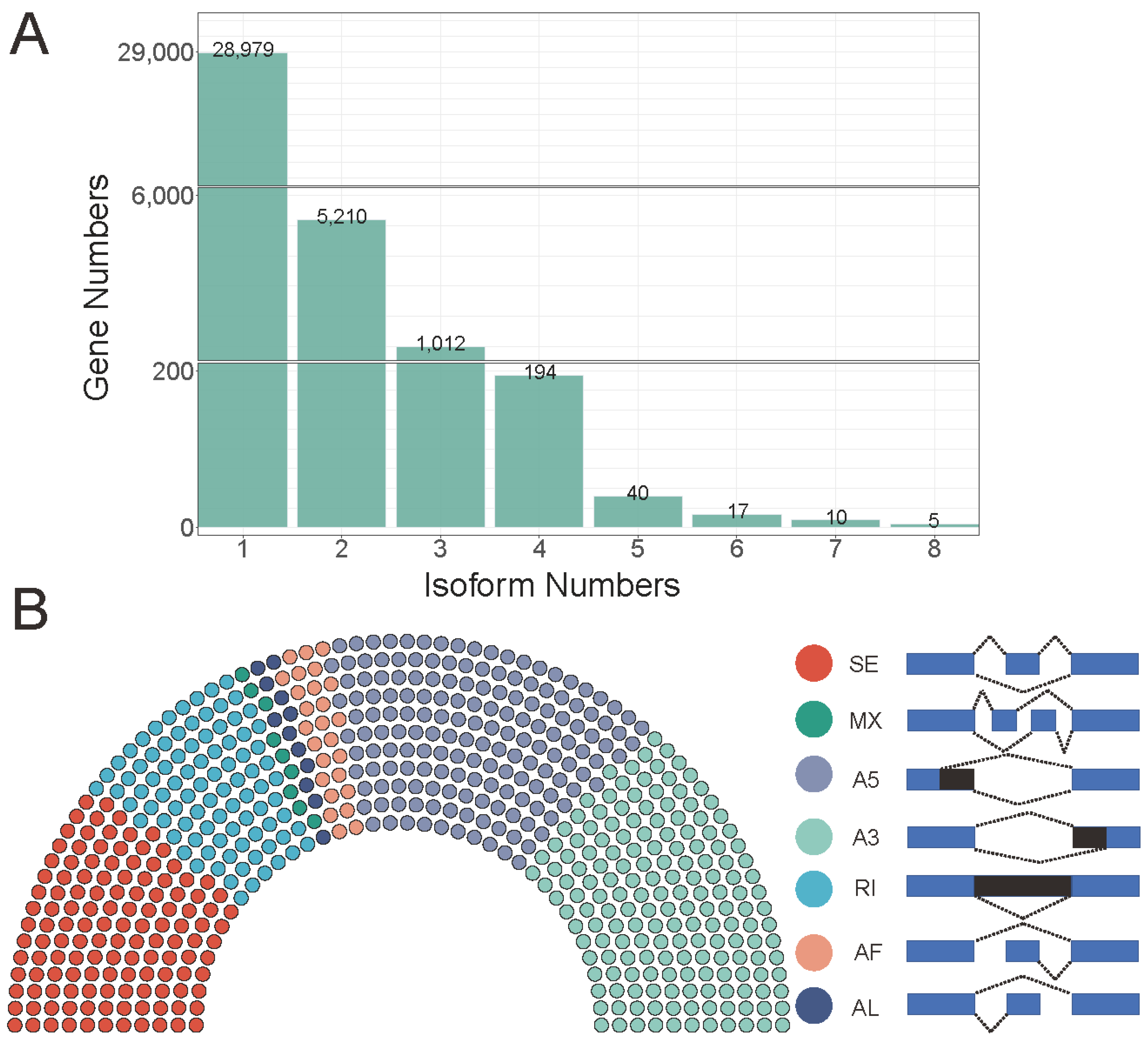
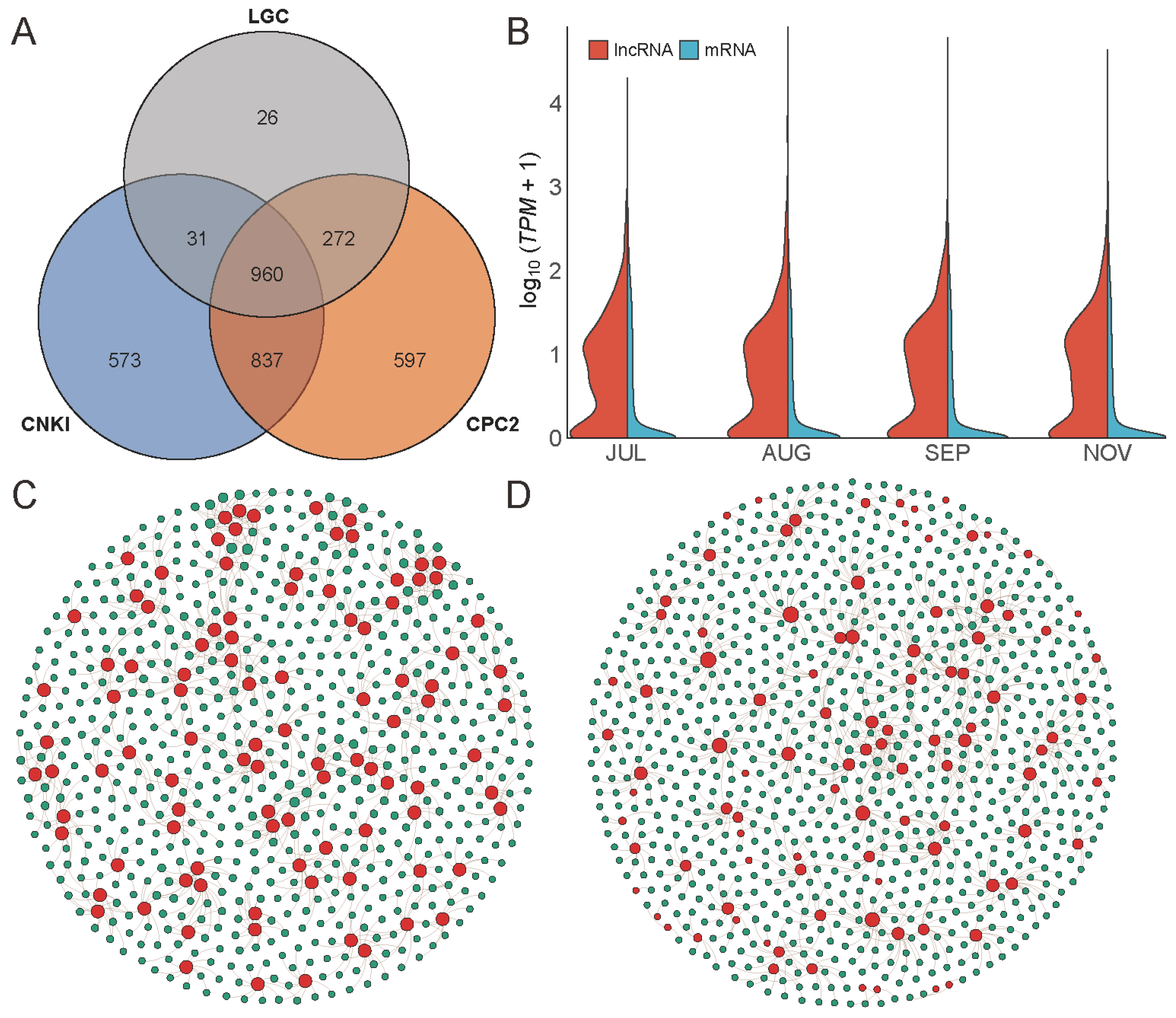
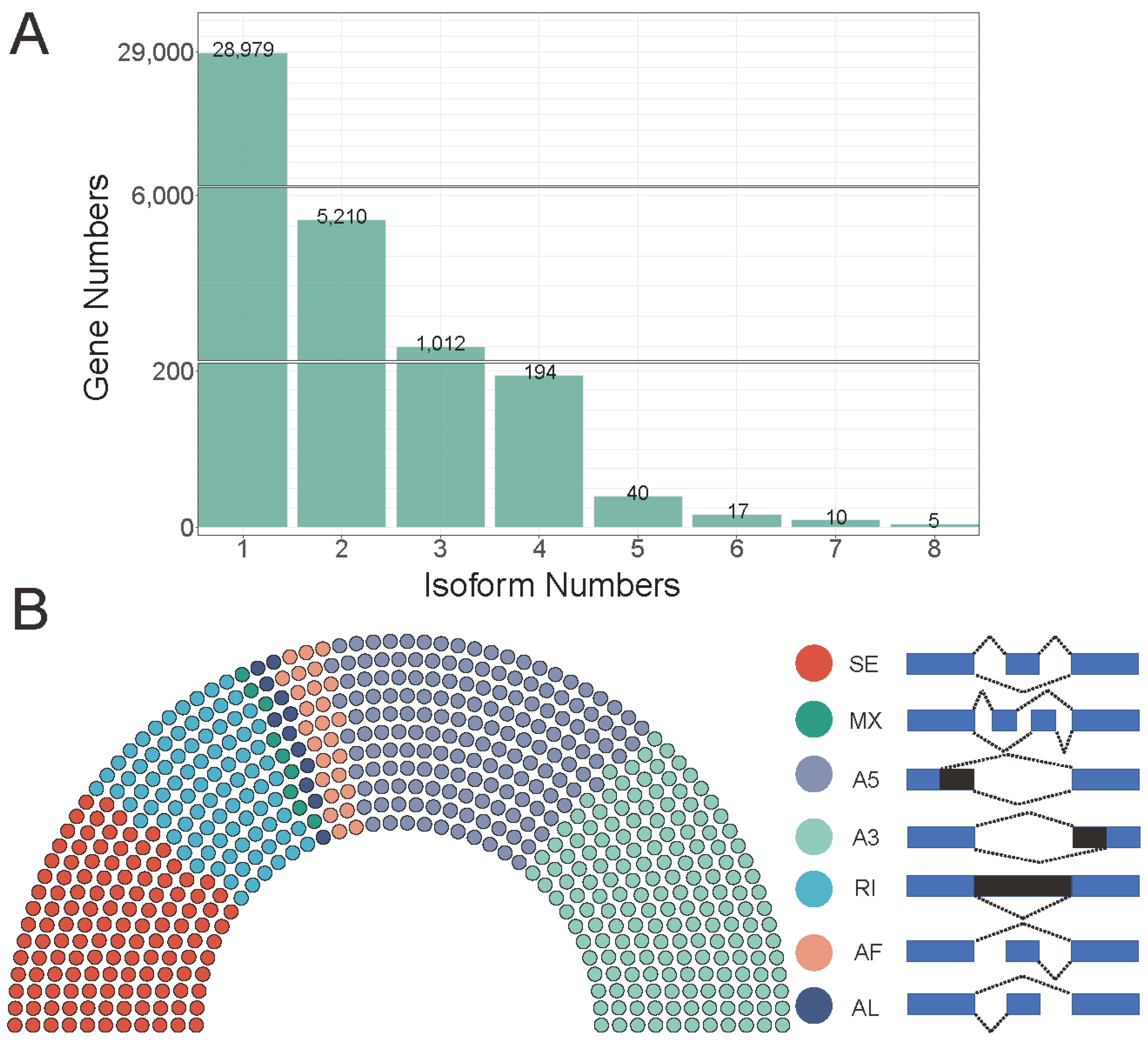
2.3. Identification and Annotation of Novel Transcripts
2.4. LncRNA and AS
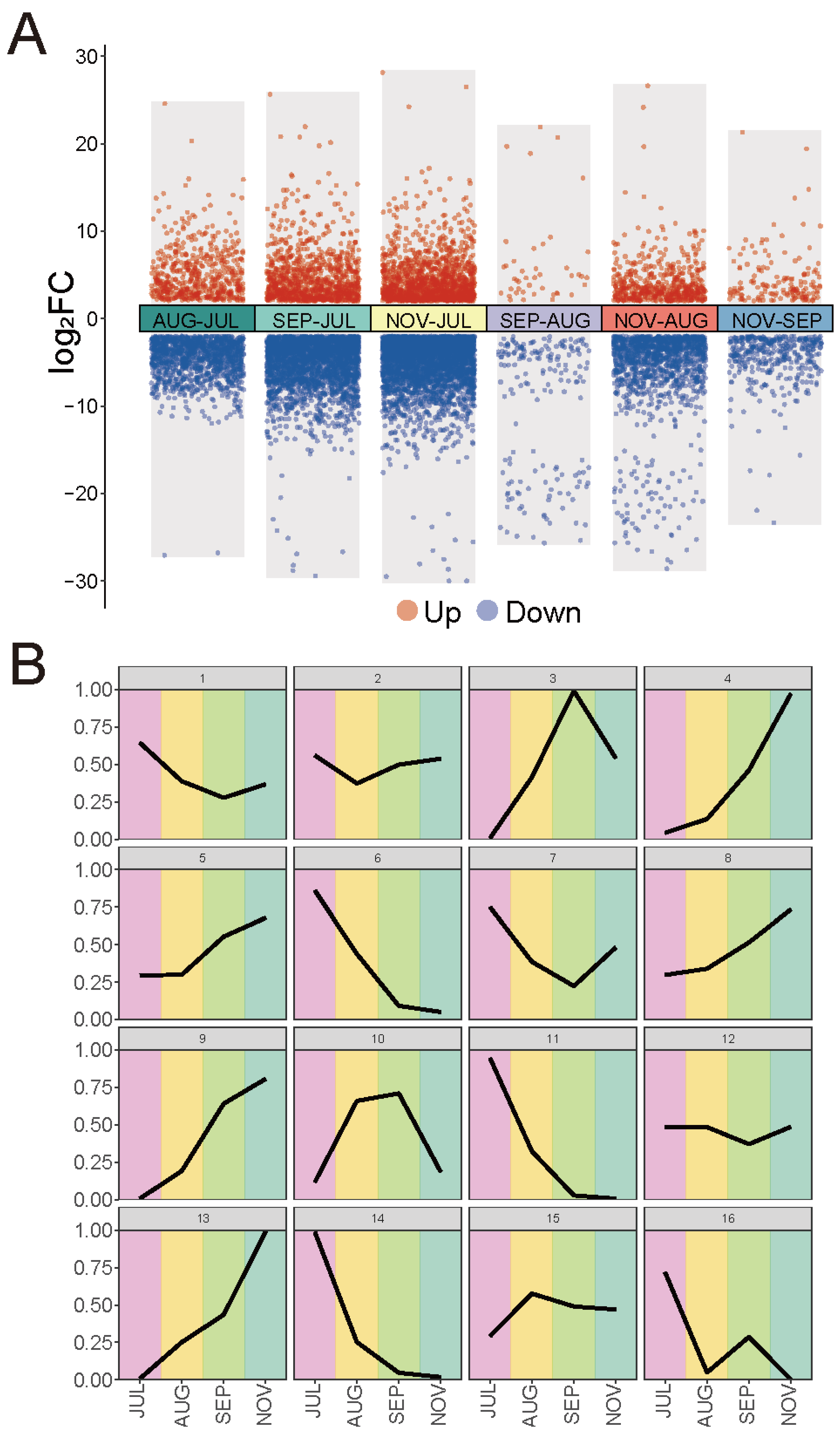
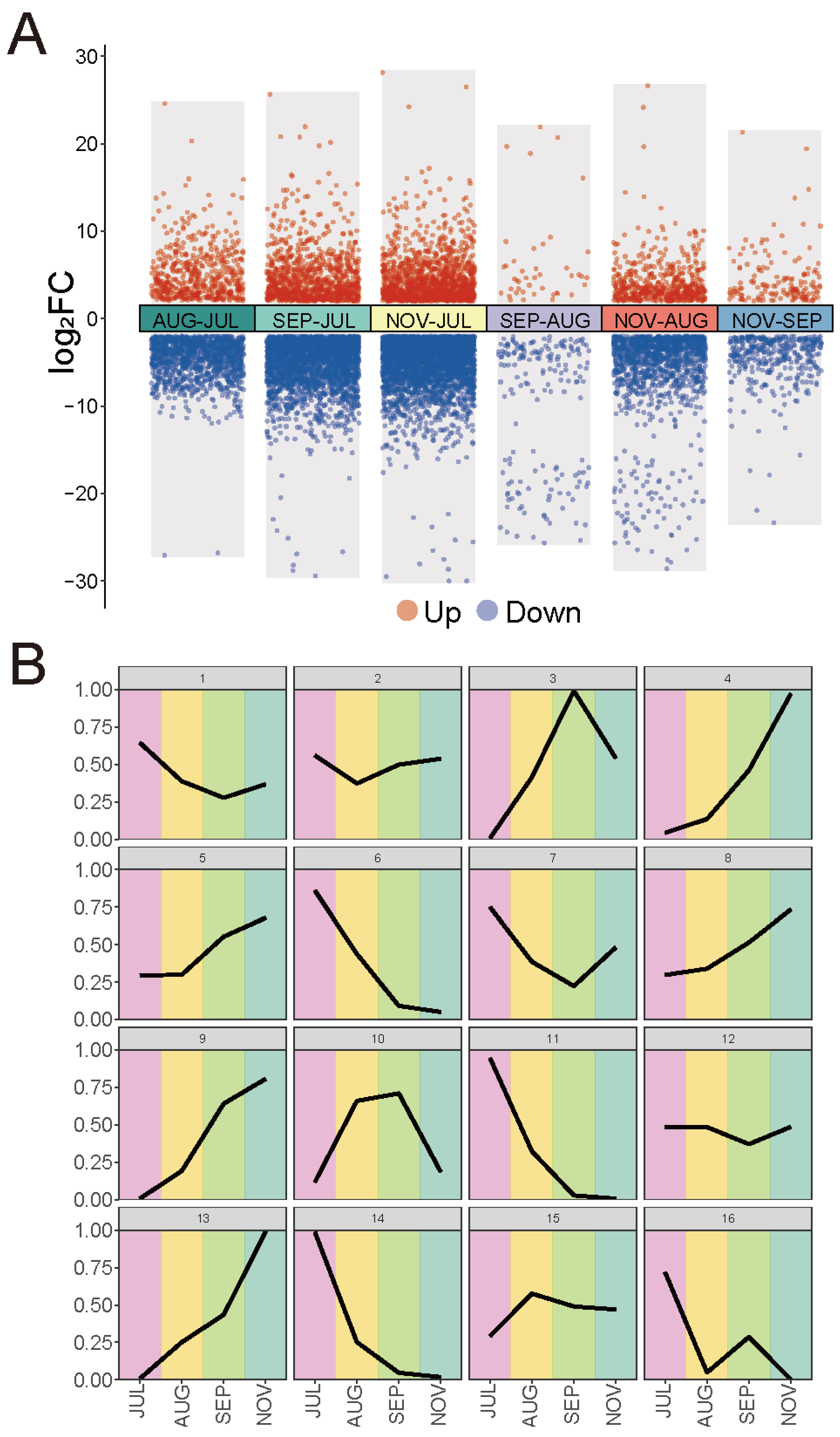
2.5. Gene Expression Profile during Fruit Development
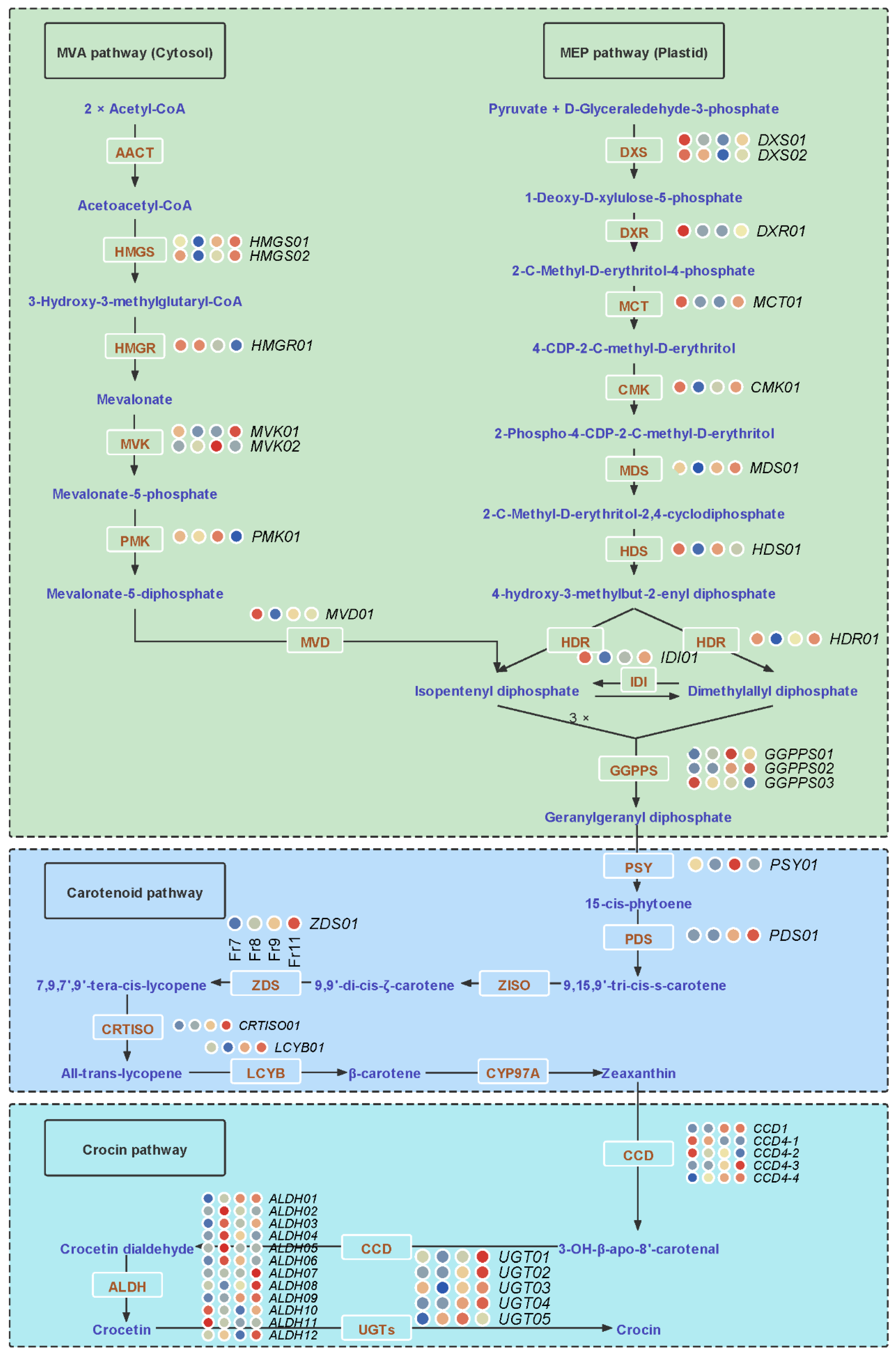
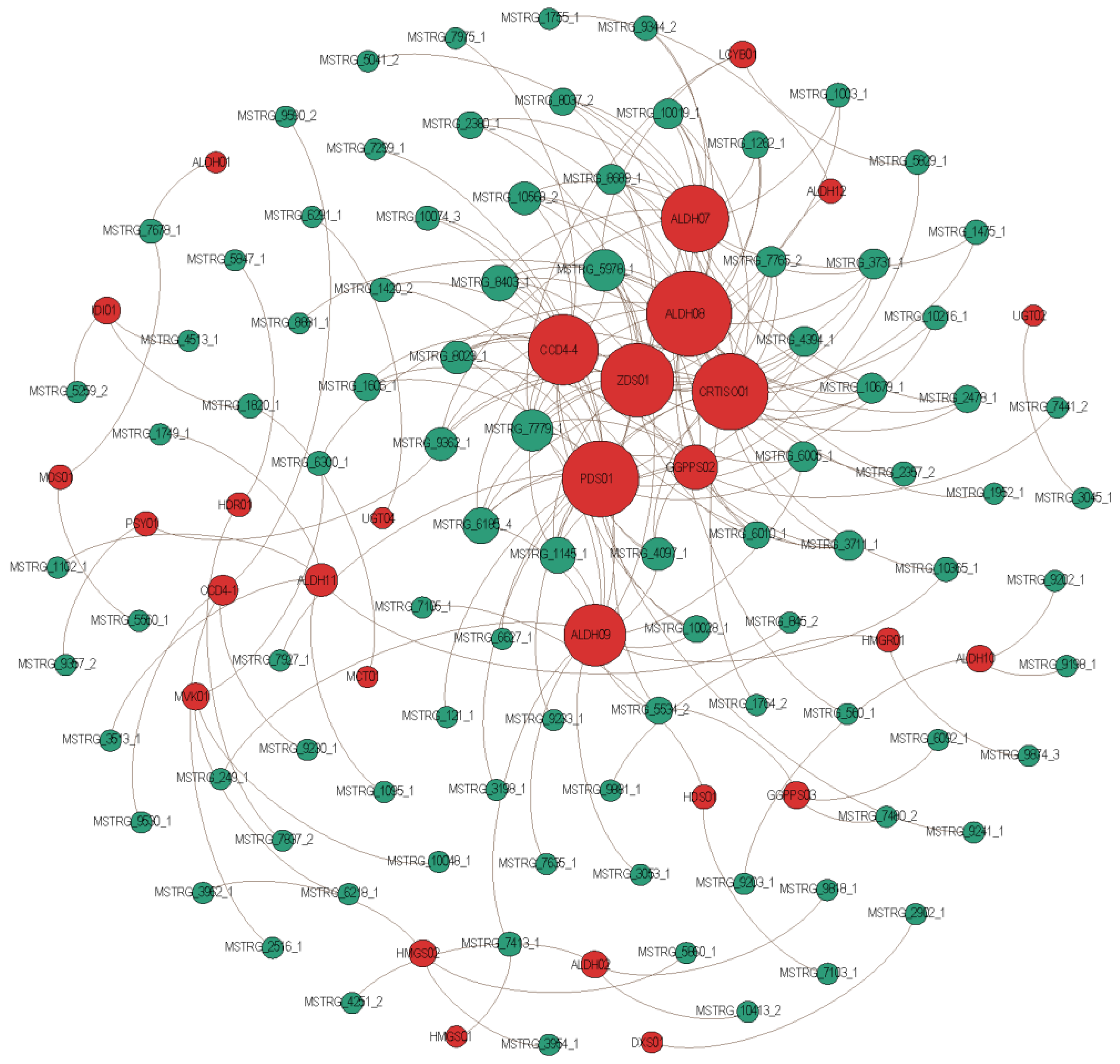
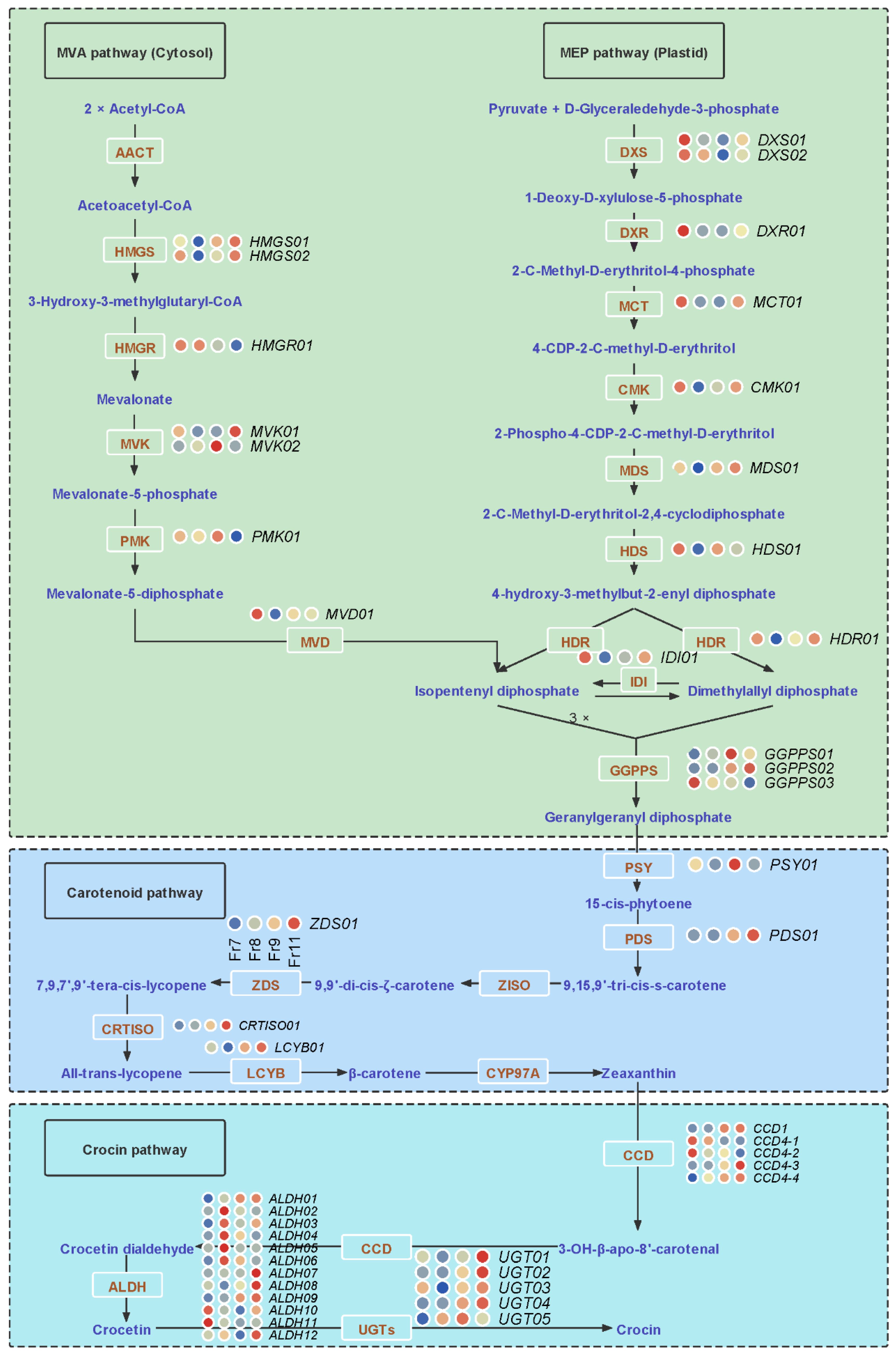
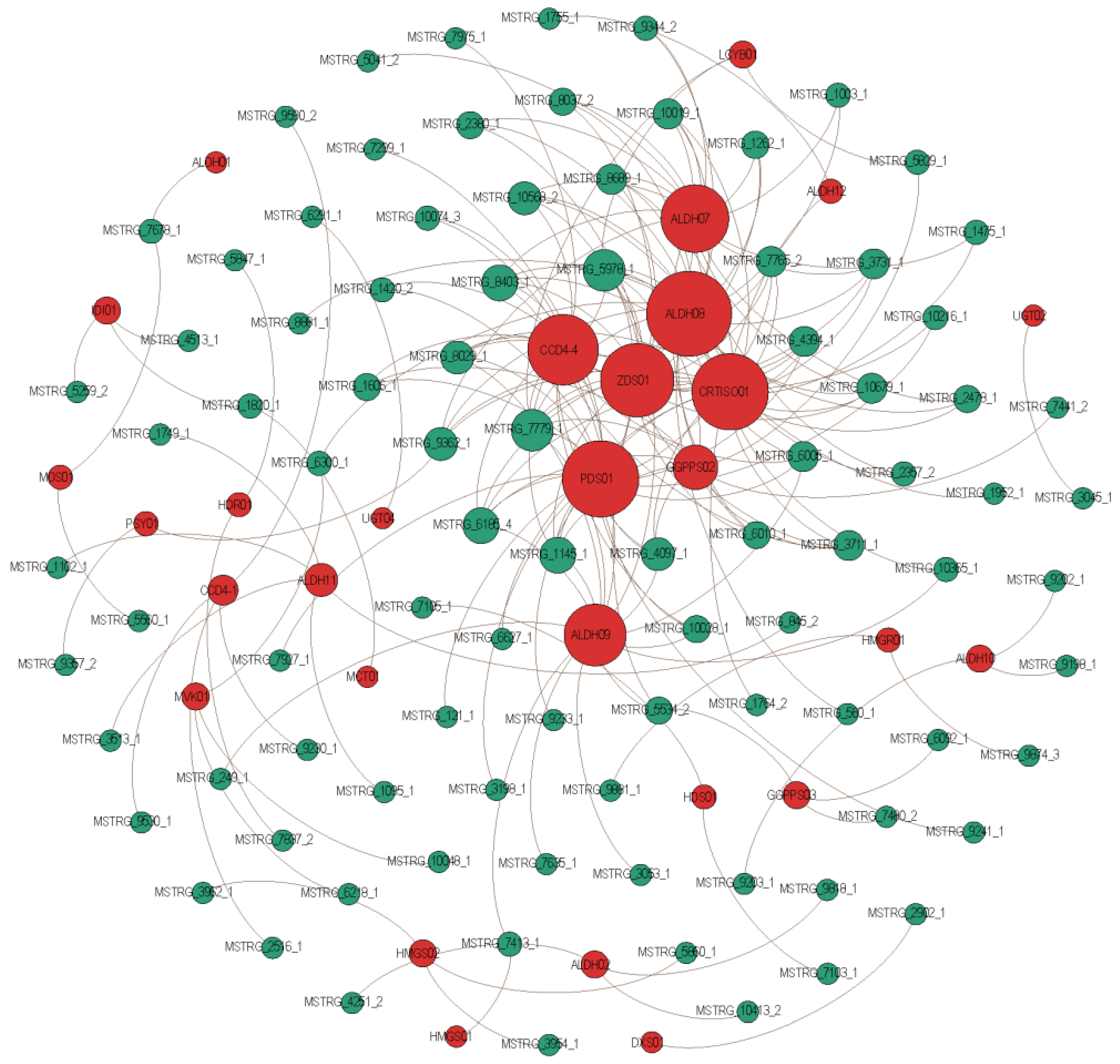
2.6. Key Genes and lncRNAs Involved in Crocin Biosynthesis
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Crocin Measurement
4.3. PacBio Library Construction and Sequencing
4.4. Transcriptome Data Analysis
4.5. Transcriptome Expression Analysis
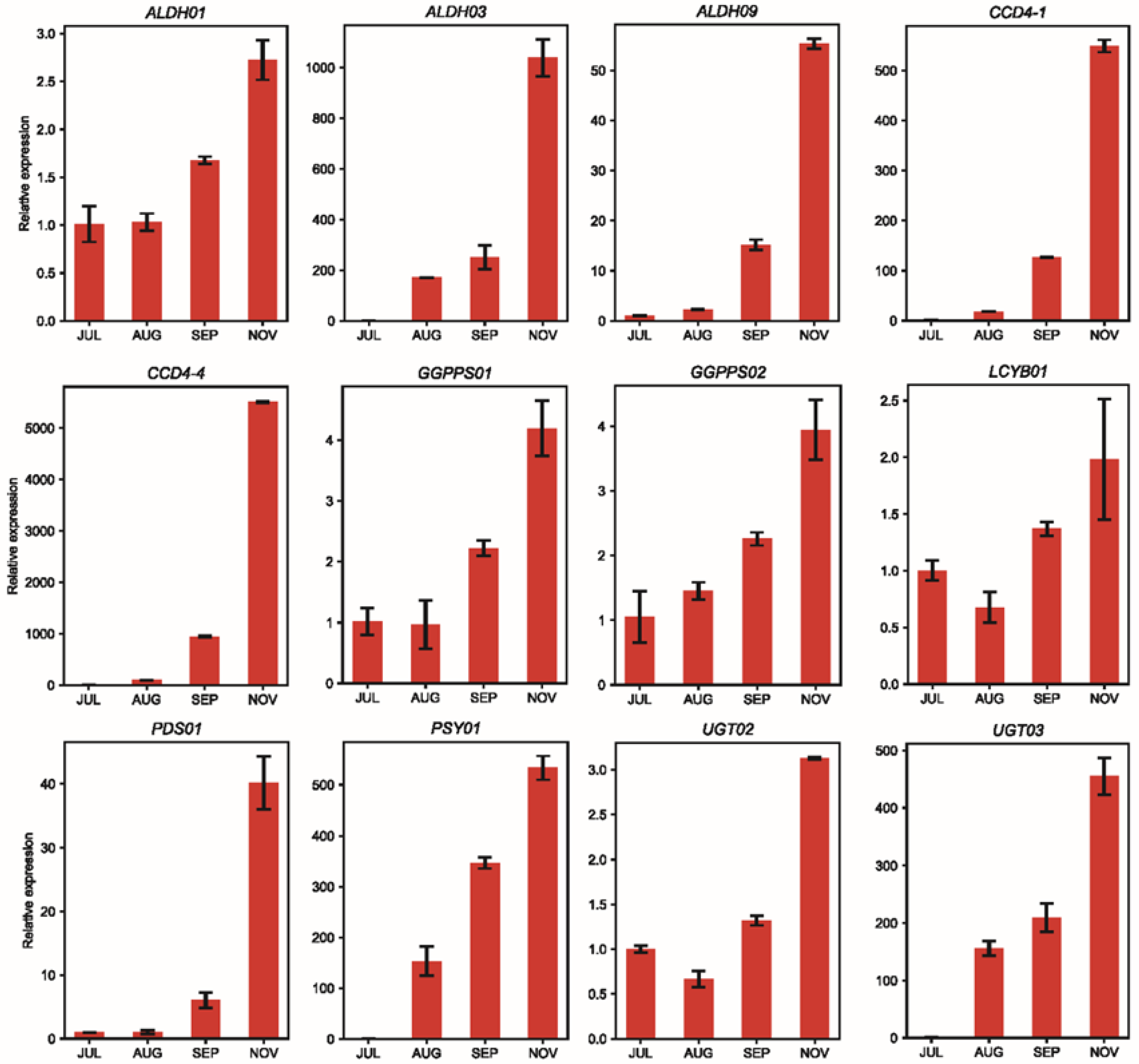
4.6. qRT-PCR
5. Conclusions
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Chang, W.-L.; Wang, H.-Y.; Shi, L.-S.; Lai, J.-H.; Lin, H.-C. Immunosuppressive Iridoids from the Fruits of Gardenia jasminoides. J. Nat. Prod. 2005, 68, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.-J.; Lim, K.-H.; Jung, H.-J.; Park, E.-H. Anti-inflammatory evaluation of gardenia extract, geniposide and genipin. J. Ethnopharmacol. 2006, 103, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wang, Q.; Luo, C.; Tan, C.; Yuan, X. Iridoid glycosides extracted from Zhizi (Fructus Gardeniae) decrease collagen-induced platelet aggregation and reduce carotid artery thrombosis in an in vivo rat model. J. Tradit. Chin. Med. Chung I Tsa Chih Ying Wen Pan/Spons. All-China Assoc. Tradit. Chin. Med. Acad. Tradit. Chin. Med. 2013, 33, 531–534. [Google Scholar] [CrossRef] [Green Version]
- Gardner, E. CEO IT Achievement Award. Pulling it together. Pelham’s leadership put Trinity at cutting edge. Mod. Healthc. 2004, (Suppl. S8–S9). [Google Scholar]
- Singer, A.C.; Thompson, I.P.; Bailey, M.J. The tritrophic trinity: A source of pollutant-degrading enzymes and its implications for phytoremediation. Curr. Opin. Microbiol. 2004, 7, 239–244. [Google Scholar] [CrossRef]
- Amerizadeh, F.; Rezaei, N.; Rahmani, F.; Hassanian, S.M.; Moradi-Marjaneh, R.; Fiuji, H.; Boroumand, N.; Nosrati-Tirkani, A.; Ghayour-Mobarhan, M.; Ferns, G.A.; et al. Crocin synergistically enhances the antiproliferative activity of 5-flurouracil through Wnt/PI3K pathway in a mouse model of colitis-associated colorectal cancer. J. Cell. Biochem. 2018, 119, 10250–10261. [Google Scholar] [CrossRef]
- Milani, A.; Basirnejad, M.; Shahbazi, S.; Bolhassani, A. Carotenoids: Biochemistry, pharmacology and treatment. Br. J. Pharmacol. 2016, 174, 1290–1324. [Google Scholar] [CrossRef] [Green Version]
- Demurtas, O.; Frusciante, S.; Ferrante, P.; Diretto, G.; Hosseinpor Azad, N.; Pietrella, M.; Aprea, G.; Taddei, A.; Romano, E.; Mi, J.; et al. Candidate Enzymes for Saffron Crocin Biosynthesis Are Localized in Multiple Cellular Compartments. Plant Physiol. 2018, 177, 990–1006. [Google Scholar] [CrossRef] [Green Version]
- Frusciante, S.; Diretto, G.; Bruno, M.; Ferrante, P.; Pietrella, M.; Prado-Cabrero, A.; Rubio-Moraga, A.; Beyer, P.; Gomez-Gomez, L.; Al-Babili, S.; et al. Novel carotenoid cleavage dioxygenase catalyzes the first dedicated step in saffron crocin biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 12246. [Google Scholar] [CrossRef] [Green Version]
- Ahrazem, O.; Diretto, G.; Argandoña, J.; Rubio-Moraga, Á.; Julve, J.M.; Orzáez, D.; Granell, A.; Gómez-Gómez, L. Evolutionarily distinct carotenoid cleavage dioxygenases are responsible for crocetin production in Buddleja davidii. J. Exp. Bot. 2017, 68, 4663–4677. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Pu, X.; Gao, R.; Demurtas, O.C.; Fleck, S.J.; Richter, M.; He, C.; Ji, A.; Sun, W.; Kong, J.; et al. Tandem gene duplications drive divergent evolution of caffeine and crocin biosynthetic pathways in plants. BMC Biol. 2020, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 2009, 583, 3303–3309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demurtas, O.C.; de Brito Francisco, R.; Diretto, G.; Ferrante, P.; Frusciante, S.; Pietrella, M.; Aprea, G.; Borghi, L.; Feeney, M.; Frigerio, L.; et al. ABCC Transporters Mediate the Vacuolar Accumulation of Crocins in Saffron Stigmas. Plant Cell 2019, 31, 2789–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagatoshi, M.; Terasaka, K.; Owaki, M.; Sota, M.; Inukai, T.; Nagatsu, A.; Mizukami, H. UGT75L6 and UGT94E5 mediate sequential glucosylation of crocetin to crocin in Gardenia jasminoides. FEBS Lett. 2012, 586, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, P. Organization and regulation of gene transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef]
- Diamantopoulos, M.A.; Tsiakanikas, P.; Scorilas, A. Non-coding RNAs: The riddle of the transcriptome and their perspectives in cancer. Ann. Transl. Med. 2018, 6, 241. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, Z.; Wang, Y.; Li, L.; Yang, X. MicroRNA annotation in plants: Current status and challenges. Brief. Bioinform. 2021, 22, bbab075. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of S alvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Li, J.; Harata-Lee, Y.; Denton, M.D.; Feng, Q.; Rathjen, J.R.; Qu, Z.; Adelson, D.L. Long read reference genome-free reconstruction of a full-length transcriptome from Astragalus membranaceus reveals transcript variants involved in bioactive compound biosynthesis. Cell Discov. 2017, 3, 17031. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Wang, X.; Zhong, L.; Tang, Y.; Zhou, X.; Liu, Y.; Zhan, R.; Zheng, H.; Chen, W.; et al. Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol. 2019, 19, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Liu, K.; Yu, R.; Zhou, B.; Huang, P.; Cao, Z.; Zhou, Y.; Wang, J. From “Dark Matter” to “Star”: Insight Into the Regulation Mechanisms of Plant Functional Long Non-Coding RNAs. Front. Plant Sci. 2021, 12, 650926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, E.; Tang, Y.; Cai, T.; Zhang, L.; Wang, J.; Hao, Y.; Zhang, B.; Zhou, Y.; Guo, X.; et al. Deeply Mining a Universe of Peptides Encoded by Long Noncoding RNAs. Mol. Cell. Proteom. 2021, 20, 100109. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Blevins, T.; Swiezewski, S. Long Noncoding RNAs in Plants. Annu. Rev. Plant Biol. 2021, 72, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Liu, Y.; Xia, R. Long Non-Coding RNAs, the Dark Matter: An Emerging Regulatory Component in Plants. Int. J. Mol. Sci. 2020, 22, 86. [Google Scholar] [CrossRef] [PubMed]
- Shao-yong, D.; Quan, C.A.O.; Lin, Y.U.; Pei-lin, Z.H.U.; Xian-rong, W. Phenotypic Diversity of Leaf and Fruit Traits of Natural Gardenia jasminoides Population. For. Res. 2015, 28, 289–296. [Google Scholar]
- Liu, T.; Yu, S.; Xu, Z.; Tan, J.; Wang, B.; Liu, Y.-G.; Zhu, Q. Prospects and progress on crocin biosynthetic pathway and metabolic engineering. Comput. Struct. Biotechnol. J. 2020, 18, 3278–3286. [Google Scholar] [CrossRef]
- De Filippis, L.F. Plant secondary metabolites. In Plant-Environment Interaction; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 263–299. [Google Scholar]
- Henneberg, R.; Otuki, M.F.; Furman, A.E.F.; Hermann, P.; do Nascimento, A.J.; Leonart, M.S.S. Protective effect of flavonoids against reactive oxygen species production in sickle cell anemia patients treated with hydroxyurea. Rev. Bras. Hematol. Hemoter. 2013, 35, 52–55. [Google Scholar] [CrossRef] [Green Version]
- Shitan, N.; Hayashida, M.; Yazaki, K. Translocation and accumulation of nicotine via distinct spatio-temporal regulation of nicotine transporters in Nicotiana tabacum. Plant Signal Behav. 2015, 10, e1035852. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhong, Y.; Yu, F.; Xu, M. Deep sequencing identifies miRNAs and their target genes involved in the biosynthesis of terpenoids in Cinnamomum camphora. Ind. Crops Prod. 2020, 145, 111853. [Google Scholar] [CrossRef]
- Nakano, K.; Shiroma, A.; Shimoji, M.; Tamotsu, H.; Ashimine, N.; Ohki, S.; Shinzato, M.; Minami, M.; Nakanishi, T.; Teruya, K.; et al. Advantages of genome sequencing by long-read sequencer using SMRT technology in medical area. Hum. Cell 2017, 30, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.; Sun, S.; Wei, J.; Li, Y.; Sun, C. Iso-Seq analysis of the Taxus cuspidata transcriptome reveals the complexity of Taxol biosynthesis. BMC Plant Biol. 2019, 19, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Xu, M.; Liu, F.; Cui, C.; Zhou, B. Reconstruction of the full-length transcriptome atlas using PacBio Iso-Seq provides insight into the alternative splicing in Gossypium australe. BMC Plant Biol. 2019, 19, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Jiang, X.; Wang, L.; Wang, W.; Fu, C.; Yan, X.; Geng, X. A survey of transcriptome complexity using PacBio single-molecule real-time analysis combined with Illumina RNA sequencing for a better understanding of ricinoleic acid biosynthesis in Ricinus communis. BMC Genom. 2019, 20, 456. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Liu, H.; Ren, J.; Ye, X.; Feng, H.; Liu, Z. Single-molecule real-time sequencing facilitates the analysis of transcripts and splice isoforms of anthers in Chinese cabbage (Brassica rapa L. ssp. pekinensis). BMC Plant Biol. 2019, 19, 517. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, X.; Wang, Y.; Tan, J.; Chen, D.-X. Metabolomics integrated with transcriptomics reveals the distribution of iridoid and crocin metabolic flux in Gardenia jasminoides Ellis. PLoS ONE 2021, 16, e0256802. [Google Scholar] [CrossRef]
- Ji, A.; Jia, J.; Xu, Z.; Li, Y.; Bi, W.; Ren, F.; He, C.; Liu, J.; Hu, K.; Song, J. Transcriptome-Guided Mining of Genes Involved in Crocin Biosynthesis. Front. Plant Sci. 2017, 8, 518. [Google Scholar] [CrossRef] [Green Version]
- Bhogireddy, S.; Mangrauthia, S.K.; Kumar, R.; Pandey, A.K.; Singh, S.; Jain, A.; Budak, H.; Varshney, R.K.; Kudapa, H. Regulatory non-coding RNAs: A new frontier in regulation of plant biology. Funct. Integr. Genom. 2021, 21, 313–330. [Google Scholar] [CrossRef]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Shen, T.; Ran, N.; Zhang, H.; Pan, H.; Su, X.; Xu, M. Integrated Degradome and Srna Sequencing Revealed miRNA-mRNA Regulatory Networks between the Phloem and Developing Xylem of Poplar. Int. J. Mol. Sci. 2022, 23, 4537. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Xu, M.; Qi, H.; Feng, Y.; Yang, Z.; Xu, M. Uncovering miRNA-mRNA Regulatory Modules in Developing Xylem of Pinus massoniana via Small RNA and Degradome Sequencing. Int. J. Mol. Sci. 2021, 22, 10154. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Du, Q.; Xiao, L.; Lu, W.; Wang, L.; Xie, J.; Song, Y.; Xu, B.; Zhang, D. Genetic architecture underlying the lignin biosynthesis pathway involves noncoding RNAs and transcription factors for growth and wood properties in Populus. Plant Biotechnol. J. 2019, 17, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Bordoloi, K.S.; Baruah, P.M.; Das, M.; Agarwala, N. Unravelling lncRNA mediated gene expression as potential mechanism for regulating secondary metabolism in Citrus limon. Food Biosci. 2022, 46, 101448. [Google Scholar] [CrossRef]
- Salmela, L.; Rivals, E. LoRDEC: Accurate and efficient long read error correction. Bioinformatics 2014, 30, 3506–3514. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yin, H.; Li, B.; Yu, C.; Wang, F.; Xu, X.; Cao, J.; Bao, Y.; Wang, L.; Abbasi, A.A.; et al. Characterization and identification of long non-coding RNAs based on feature relationship. Bioinformatics 2019, 35, 2949–2956. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trincado, J.L.; Entizne, J.C.; Hysenaj, G.; Singh, B.; Skalic, M.; Elliott, D.J.; Eyras, E. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 2018, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2004, 21, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. Mclust 5: Clustering, classification and density estimation using Gaussian finite mixture models. R J. 2016, 8, 205–233. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, T.; Zheng, Y.; Liu, Q.; Chen, C.; Huang, L.; Deng, S.; Xu, M.; Yang, C. Integrated SMRT and Illumina Sequencing Provide New Insights into Crocin Biosynthesis of Gardenia jasminoides. Int. J. Mol. Sci. 2022, 23, 6321. https://doi.org/10.3390/ijms23116321
Shen T, Zheng Y, Liu Q, Chen C, Huang L, Deng S, Xu M, Yang C. Integrated SMRT and Illumina Sequencing Provide New Insights into Crocin Biosynthesis of Gardenia jasminoides. International Journal of Molecular Sciences. 2022; 23(11):6321. https://doi.org/10.3390/ijms23116321
Chicago/Turabian StyleShen, Tengfei, Yongjie Zheng, Qian Liu, Caihui Chen, Lili Huang, Shaoyong Deng, Meng Xu, and Chunxia Yang. 2022. "Integrated SMRT and Illumina Sequencing Provide New Insights into Crocin Biosynthesis of Gardenia jasminoides" International Journal of Molecular Sciences 23, no. 11: 6321. https://doi.org/10.3390/ijms23116321
APA StyleShen, T., Zheng, Y., Liu, Q., Chen, C., Huang, L., Deng, S., Xu, M., & Yang, C. (2022). Integrated SMRT and Illumina Sequencing Provide New Insights into Crocin Biosynthesis of Gardenia jasminoides. International Journal of Molecular Sciences, 23(11), 6321. https://doi.org/10.3390/ijms23116321