Abstract
We report a case of a patient with a long history of pyoderma gangrenosum (PG), in whom large vessel vasculitis, most likely Takayasu arteritis (TA), manifested many years after the diagnosis of PG and showed a rapid progression within four years, resulting in two invasive interventions. We describe the challenges of the management of the disease in our patient, the application of new pharmacological agents and the natural history of this rare disorder.
A 52-year-old male was admitted to our hospital with a fever of unknown origin and raised inflammatory parameters (CRP 251 mg/l, ESR 50 mm/h) in March 2006. He had a history of recurrent pyoderma gangrenosum (PG) of the lower extremities since the age of 14 years old. After numerous attempts with various immunosuppressive agents, stable remission was finally achieved using local tacrolimus applications (Figure 1). Physical examination except for scarring after PG lesions in the lower limbs was otherwise unremarkable. To search for the possible causes of inflammatory syndrome thoraco-abdominal computed tomography (CT) was performed. It revealed thoracic and abdominal aortic wall thickening (Figure 2, panel A) with involvement of epiaortic branches (Figure 2, panel C), the coeliac trunk, the superior mesenteric artery and iliac arteries (Figure 2, panel B). 18FDG positron emission tomography (PET) revealed the presence of active inflammatory aortic wall processes at the level of the thoracic and abdominal aorta as well as the aortic arch branches, the left coronary artery and the iliac arteries. The duplex scan of the temporal arteries was negative for the signs of giant cell arteritis (GCA), and the patient did not present any typical clinical signs of this vasculitis (e.g., localised headache, visual disturbance, tenderness of the temporal artery or jaw muscle pain). Therefore, radiological findings of large vessel vasculitis without evidence of other inflammatory disorders (ANCA, ANA, HLA B5, HLA B25 negative) in association with PG made the diagnosis of Takayasu arteritis (TA) the most likely one.
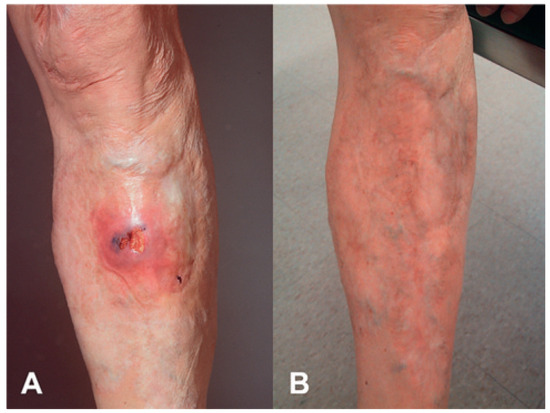
Figure 1.
Pyoderma gangrenosum of the lower extremities (A) before and (B) after local application of tacrolimus.
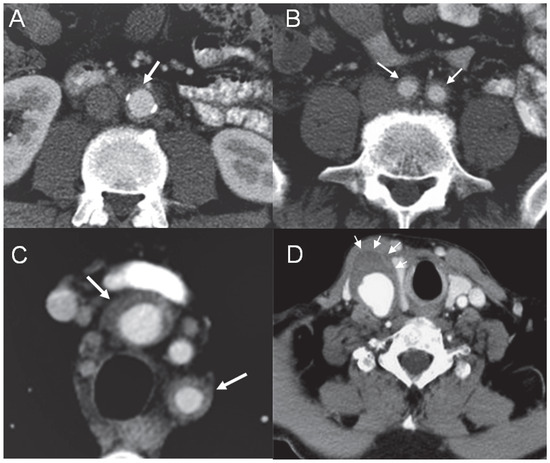
Figure 2.
Abdominal aortic wall thickening (A) with involvement of the iliac arteries (B) and epiaortic branches (C). CT angiography showing the aneurysm of the right common carotid artery with thickening of the wall and suspected thrombus (D).
Therapy with steroids, mycophenolate and cyclosporin did not result in a desirable improvement. On the basis of the fact that PG was responsive to local tacrolimus, the probative treatment with oral tacrolimus at a dosage of 3 mg/b.i.d., on top of steroids and mycophenolate, was attempted. It resulted in a rapid clinical improvement and regression of inflammation documented with 18FDG PET after four months. Six months later, the patient presented with pain in the right supraclavicular region and presence of a pulsating tumour. CT angiography revealed an aneurysm of the innominate artery (maximal diameter of 24 mm) expanding to the right carotid commune artery (maximal dimensions of 24 × 33 mm) with wall thickening and suspected thrombus adherent to the wall of the aneurysm (Figure 2, panel D). Mild dilatation of the ascending aorta (44 mm), not observed on the previous CT scan, was also documented. The patient continued immunosuppressive treatment and clopidogrel was introduced. The possibility of endovascular retrograde covered stenting was discussed with the patient. Due to further expansion of the right carotid commune artery aneurysm (increased to maximal dimensions of 35 x 46 mm), the intervention was successfully performed two months later (January 2007) with good positioning of the stent and without signs of leaking (Figure 3). The patient continued immunosuppressive therapy with subsequent periodical radiological and duplex controls. He had two relapses of PG, which required hospitalisation, in the next two years. In 2009, routine echocardiography revealed expansion of the ascending aorta from 44 to 50 mm leading to severe aortic regurgitation, and dilatation of the left ventricle. Coronary angiography was performed and showed a co-lateralised occlusion of the right coronary artery and non-significant plaque in the left anterior descending artery. In November 2009, aortic valve and ascending aorta replacement as well as single aorto-coronary by-pass graft surgery was performed. Histological analysis of an aortic specimen showed degeneration of media, adventitial fibrosis with lymphocyte infiltration. Although these findings were not specific for TA and could be found in other forms of vasculitis, it is known that a negative biopsy does not definitely rule out the diagnosis of TA.
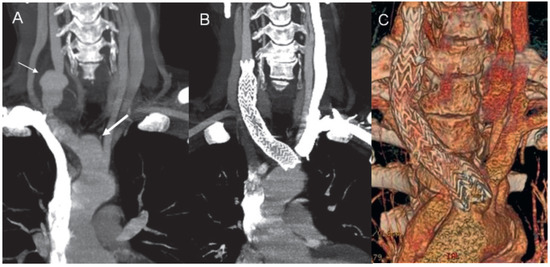
Figure 3.
A) CT angiography in a minimal intensity projection modality showing the aneurysm of the innominate artery (thick arrow) and the right commune carotid artery (thin arrow). CT angiography in a minimal intensity projection modality (B) and a 3D volume rendering reconstruction (C) demonstrate the correct positioning of the stent.
Currently, one year after intervention the patient is clinically stable, without the signs of inflammation, continuing immunosuppressive therapy, and with a normally functioning aortic prosthesis.
Discussion
TA is a rare inflammatory vasculitis predominantly involving the aorta and its main branches. It is a rare disorder more frequently seen in the Asian population than in other races [1,2]. However, as the recent study by Arnaud et al. [3] has shown, white patients tend to have late-onset disease compared with other races. This could be the case for our patient.
TA typically presents with non-specific systemic signs and symptoms such as arthralgia, fever, fatigue, headaches, rashes and weight loss [2]. Chronic inflammation of the aorta and its major branches, including subclavian, common carotid, coronary, pulmonary and renal arteries, may result in localised stenoses, and vascular occlusion. However, it is established that in 20% of lesions more acute inflammation combined with inadequate fibrotic response may destroy the arterial wall leading to aneurysm formation. In addition to substantial morbidity, mortality has been reported to be as high as 35% at 5 years.
Diagnosis of TA in patients with early phase nonocclusive disease may be challenging as there is no diagnostic serologic test and symptoms are generally constitutional. The American College of Rheumatology classification remains, however, the most widely applied (table) with 3 or more criteria associated with a sensitivity of 90.5% and a specificity of 97.8%. However, it is reported that the principal limitation of the current criteria for the diagnosis of TA is that patients with early phase non-occlusive disease or with rapid course disease leading to aneurysm formation may fail to fulfil them. We suppose that this could be the case in our patient, whose physical examination on admission was unremarkable. The diagnosis of TA is made on the basis of clinical presentation and imaging results, and histopathologic confirmation can be obtained in patients who undergo vascular surgery.

Table.
American College of Rheumatology 1991 criteria for the classification of Takayasu arteritis.
Table.
American College of Rheumatology 1991 criteria for the classification of Takayasu arteritis.

Considerable advances have been made in recent years in vascular imaging. Thus, MRI and MR angiography, CT and CT angiography, positron emission tomography (PET), CT-PET and high resolution ultrasound are becoming more widely available for the investigation of patients presenting with symptoms suggestive of TA and for disease monitoring. The advantage of PET is its ability to detect pre-stenotic disease in patients presenting with non-specific features commonly associated with early TA, as could be true for our case.
Treatment regimens depend on the disease activity. It has been suggested that 50% of patients respond to corticosteroid therapy alone, but 50.5% of patients relapse during corticosteroid treatment. In those who relapse on corticosteroids monotherapy, it is common practice to add a further immunosuppressive drug. The most commonly used immunosuppressive drugs are methotrexate, azathioprine and cyclophosphamide. However, there are no data that steroid or indeed other immunosuppressive therapies alter the outcome in TA.
Data are emerging in support of tumour necrosis factor α (TNF-α) antagonists [4] as well as biological therapies designed to inhibit growth factors [2] in the treatment of TA.
Revascularisation should only be considered if stenotic or occlusive lesions lead to significant haemodynamic effects, or if aneurysmal enlargement results in a risk of rupture or dissection, as in our patient. Other indications include severe stenoses of the cervicocranial circulation considered to increase the risk of cerebrovascular accident, significant coronary artery disease, coarctation of the aorta, aortic regurgitation, severe limb claudication and significant renal artery stenosis. If possible, surgery should be postponed until the acute phase of the disease has passed.
TA is a chronic, progressive disease. Its degree of activity varies over the time: the intensity of its inflammatory processes typically fluctuates between exacerbation and remission. Vascular complications of the cardiac, renal, and central nervous system are the chief causes of morbidity and mortality [1].
As with the other inflammatory diseases, TA is associated with increased cardiovascular risk and premature atherosclerosis. Therefore, it is imperative that cardiovascular risk factors such as hypertension, hyperlipidemia and life style factors such as smoking status are screened for and aggressively managed.
Various skin lesions can be associated with TA, including erymatosous nodules with necrotising or granulomatous vasculitis, and, rarely, with pyoderma gangrenosum [5]. PG is one of the neutrophilic dermatoses frequently related to ulcerative colitis, rheumatoid arthritis, and, rarely, to TA. It mainly affects the lower extremities of middle-aged patients, forming recalcitrant ulcers. Several cases of TA associated with PG have been reported without any relationship to clinical severity [6]. However, this association is described to be very rare in the European population. Topical tacrolimus represents a first-line monotherapy in the treatment of selected cases of local PG [7].
Tacrolimus is a macrolide antibiotic with immunosuppressive properties which acts as calcineurin inhibitor. The drug inhibits calcium-dependent events, such as interleukin-2 gene transcription, nitric oxide synthase activation, cell degranulation, T-cell proliferation and apoptosis [8]. The use of systemic tacrolimus in TA is not well established. In our patient, the probative treatment with oral tacrolimus was attempted mostly on the basis of the responsiveness of PG to this agent.
Only one case report of successful treatment of TA associated with rheumatoid arthritis has been described to date [9].
Conclusions
As illustrated by our case, the management of patients who suffer from large vessel vasculitis can be very complex and requires a multidisciplinary approach as well as close follow-up. Moreover, we show that administration of systemic tacrolimus which had earlier shown to be effective for PG, also helped to achieve remission for TA, thus, underlying its role as a possible treatment option for selected patients. To our best knowledge, the successful treatment of both diseases with tacrolimus is described for the first time.
Funding/potential competing interests
No financial support and no other potential conflict of interest relevant to this article were reported.
References
- Saab, F.; Giugliano, R.P.; Giugliano, G.R. Takayasu arteritis in a young woman: a 4-year case history. Tex Heart Inst J. 2009, 36, 470–474. [Google Scholar] [PubMed]
- Andrews, J.; Mason, J.C. Takayasu’s arteritis-recent advances in imaging offer promise. Rheumatology (Oxford) 2007, 46, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Haroche, J.; Limal, N.; Toledano, D.; Gambotti, L.; Costedoat Chalumeau, N.; et al. Takayasu arteritis in France: a single-center retrospective study of 82 cases comparing white, North African, and black patients. Medicine (Baltimore) 2010, 89, 1–17. [Google Scholar] [PubMed]
- Tatò, F.; Rieger, J.; Hoffmann, U. Refractory Takayasu’s arteritis successfully treated with the human, monoclonal anti-tumor necrosis factor antibody adalimumab. Int Angiol. 2005, 24, 304–307. [Google Scholar] [PubMed]
- Futaki, K.; Komine, M.; Hosoda, S.; Hirashima, M.; Yokokura, H.; Yamada, T.; et al. Pyoderma gangrenosum associated with Takayasu’s arteritis without typical symptoms. Eur J Dermatol. 2009, 19, 266–267. [Google Scholar]
- Ujiie, H.; Sawamura, D.; Yokota, K.; Nishie, W.; Shichinohe, R.; Shimizu, H. Pyoderma gangrenosum associated with Takayasu’s arteritis. Clin Exp Dermatol. 2004, 29, 357–359. [Google Scholar] [PubMed]
- Marzano, A.V.; Trevisan, V.; Lazzari, R.; Crosti, C. Topical tacrolimus for the treatment of localized, idiopathic, newly diagnosed pyoderma gangrenosum. J Dermatolog Treat. 2010, 21, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Bonham, C.A.; Zeevi, A. Mode of action of tacrolimus (FK506): molecular and cellular mechanisms. Ther Drug Monit. 1995, 17, 584–919. [Google Scholar] [CrossRef] [PubMed]
- Yokoe, I.; Haraoka, H.; Harashima, H. A patient with Takayasu’s arteritis and rheumatoid arthritis who responded to tacrolimus hydrate. Intern Med. 2007, 46, 1873–1877. [Google Scholar] [CrossRef][Green Version]
© 2011 by the author. Attribution - Non-Commercial - NoDerivatives 4.0.