
Tumour Genetic Heterogeneity in Relation to Oral Squamous Cell Carcinoma and Anti-Cancer Treatment


 ,
, {kind=link}
Abstract
1. Introduction
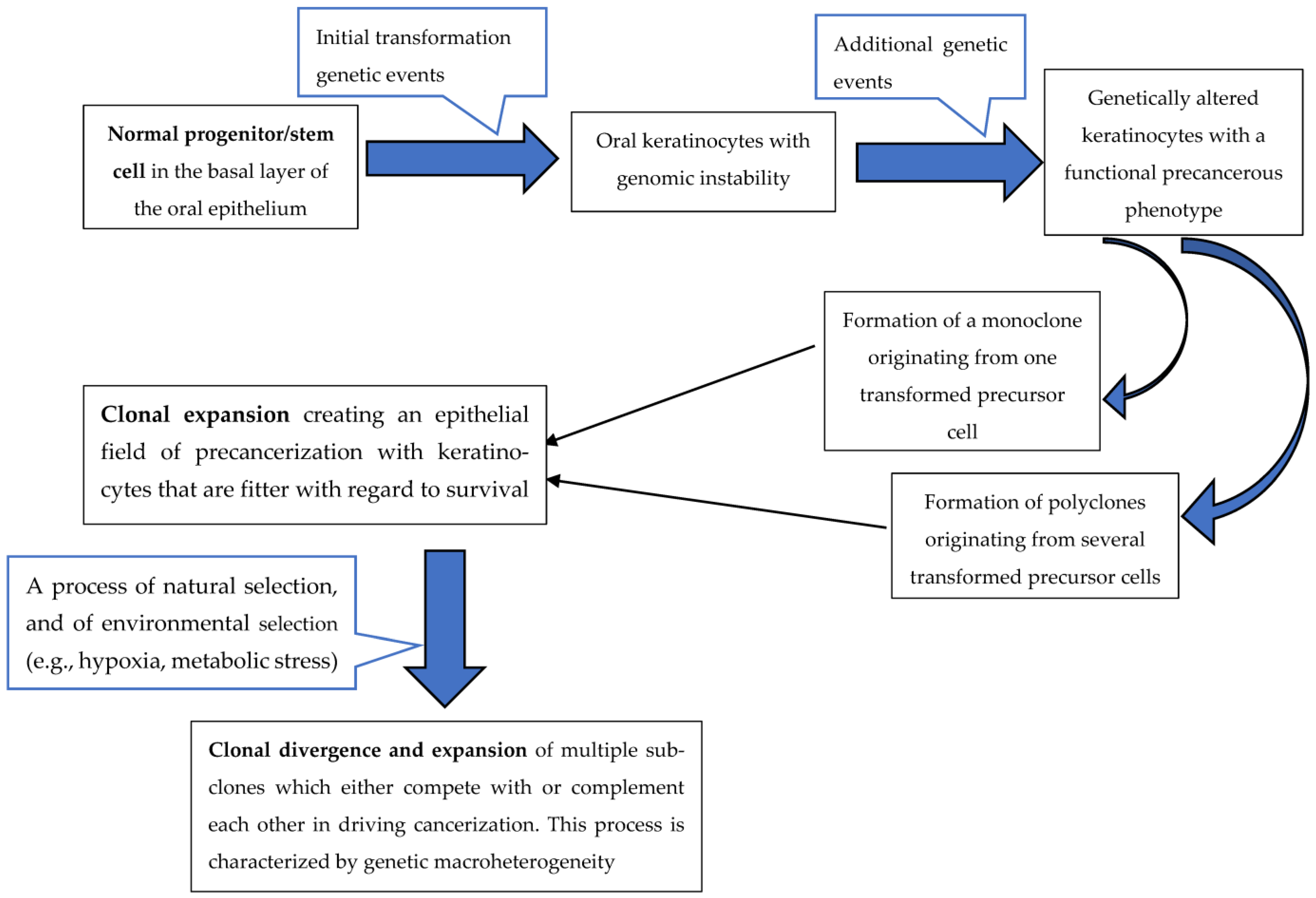
2. Oral Squamous Cell Carcinoma Genetic Heterogeneity
3. Cancer Driver Genes
4. Driver and Passenger Mutations
5. Tumour Genetic Heterogeneity and Phenotypic Plasticity in Relation to Tumour Microenvironment
6. Intratumour Heterogeneity and Resistance to Cancer Chemotherapy
7. Radiation Therapy in Relation to Tumour Heterogeneity
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Turajlic, S.; Sottoriva, A.; Graham, T.; Swanton, C. Resolving genetic heterogeneity in cancer. Nat. Rev. Genet. 2019, 20, 404–416. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Welch, D.R. Tumor heterogeneity—A ‘contemporary concept’founded on historical insights and predictions. Cancer Res. 2016, 76, 4–6. [Google Scholar] [CrossRef]
- Sottoriva, A.; Barnes, C.P.; Graham, T.A. Catch my drift? Making sense of genomic intra-tumour heterogeneity. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 95–100. [Google Scholar] [CrossRef]
- Williams, M.J.; Werner, B.; Heide, T.; Curtis, C.; Barnes, C.P.; Sottoriva, A.; Graham, T.A. Quantification of subclonal selection in cancer from bulk sequencing data. Nat. Genet. 2018, 50, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Barber, L.J.; Davies, M.N.; Gerlinger, M. Dissecting cancer evolution at the macro-heterogeneity and micro-heterogeneity scale. Curr. Opin. Genet. Dev. 2015, 30, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Van Loo, P. Timing somatic events in the evolution of cancer. Genome Biol. 2018, 19, 95. [Google Scholar] [CrossRef]
- Wodarz, D.; Newell, A.C.; Komarova, N.L. Passenger mutations can accelerate tumour suppressor gene inactivation in cancer evolution. J. R. Soc. Interface 2018, 15, 20170967. [Google Scholar] [CrossRef]
- Feller, L.; Kramer, B.; Lemmer, J. A short account of metastatic bone disease. Cancer Cell Int. 2011, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Feller, L.; Bouckaert, M.; Wood, N.; Khammissa, R.; Meyerov, R.; Lemmer, J.; Chikte, U. A short account of cancer-specifically in relation to squamous cell carcinoma: Communication. S. Afr. Dent. J. 2010, 65, 322–324. [Google Scholar]
- Feller, L.; Lemmer, J. Field cancerization and oral leukoplakia. Field Cancerization Basic Sci. Clin. Appl. 2011, 1, 95–111. [Google Scholar]
- Parsons, B.L. Multiclonal tumor origin: Evidence and implications. Mutat. Res. Rev. Mutat. Res. 2018, 777, 1–18. [Google Scholar] [CrossRef]
- Khammissa, R.A.; Meer, S.; Lemmer, J.; Feller, L. Oral squamous cell carcinoma in a South African sample: Race/ethnicity, age, gender, and degree of histopathological differentiation. J. Cancer Res. Ther. 2014, 10, 908–914. [Google Scholar] [CrossRef]
- Suwa, T.; Kobayashi, M.; Nam, J.M.; Harada, H. Tumor microenvironment and radioresistance. Exp. Mol. Med. 2021, 53, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Feller, L.; Lemmer, J. Oral squamous cell carcinoma: Epidemiology, clinical presentation and treatment. J. Cancer Ther. 2012, 3, 21591. [Google Scholar] [CrossRef]
- Zanoni, D.K.; Montero, P.H.; Migliacci, J.C.; Shah, J.P.; Wong, R.J.; Ganly, I.; Patel, S.G. Survival outcomes after treatment of cancer of the oral cavity (1985–2015). Oral Oncol. 2019, 90, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Almangush, A.; Makitie, A.A.; Triantafyllou, A.; de Bree, R.; Strojan, P.; Rinaldo, A.; Hernandez-Prera, J.C.; Suarez, C.; Kowalski, L.P.; Ferlito, A.; et al. Staging and grading of oral squamous cell carcinoma: An update. Oral. Oncol. 2020, 107, 104799. [Google Scholar] [CrossRef] [PubMed]
- Quinlan-Davidson, S.R.; Mohamed, A.S.; Myers, J.N.; Gunn, G.B.; Johnson, F.M.; Skinner, H.; Beadle, B.M.; Gillenwater, A.M.; Phan, J.; Frank, S.J. Outcomes of oral cavity cancer patients treated with surgery followed by postoperative intensity modulated radiation therapy. Oral Oncol. 2017, 72, 90–97. [Google Scholar] [CrossRef]
- Garzino-Demo, P.; Zavattero, E.; Franco, P.; Fasolis, M.; Tanteri, G.; Mettus, A.; Tosco, P.; Chiusa, L.; Airoldi, M.; Ostellino, O.; et al. Parameters and outcomes in 525 patients operated on for oral squamous cell carcinoma. J. Craniomaxillofac. Surg. 2016, 44, 1414–1421. [Google Scholar] [CrossRef]
- Hayes, T.F.; Benaich, N.; Goldie, S.J.; Sipila, K.; Ames-Draycott, A.; Cai, W.; Yin, G.; Watt, F.M. Integrative genomic and functional analysis of human oral squamous cell carcinoma cell lines reveals synergistic effects of FAT1 and CASP8 inactivation. Cancer Lett. 2016, 383, 106–114. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef] [PubMed]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed]
- Morjaria, S. Driver mutations in oncogenesis. Int. J. Mol. Immuno. Oncol. 2021, 6, 100–102. [Google Scholar] [CrossRef]
- Waks, Z.; Weissbrod, O.; Carmeli, B.; Norel, R.; Utro, F.; Goldschmidt, Y. Driver gene classification reveals a substantial overrepresentation of tumor suppressors among very large chromatin-regulating proteins. Sci. Rep. 2016, 6, 38988. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Deininger, P. Genetic instability in cancer: Caretaker and gatekeeper genes. Ochsner J. 1999, 1, 206–209. [Google Scholar]
- Broaddus, V.C.; Ernst, J.D.; King Jr, T.E.; Lazarus, S.C.; Sarmiento, K.F.; Schnapp, L.M.; Stapleton, R.D.; Gotway, M.B. Murray & Nadel’s Textbook of Respiratory Medicine e-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Emmert-Streib, F.; de Matos Simoes, R.; Mullan, P.; Haibe-Kains, B.; Dehmer, M. The gene regulatory network for breast cancer: Integrated regulatory landscape of cancer hallmarks. Front. Genet. 2014, 5, 15. [Google Scholar] [CrossRef]
- India Project Team of the International Cancer Genome Consortium; Maitra, A.; Biswas, N.K.; Amin, K.; Kowtal, P.; Kumar, S.; Das, S.; Sarin, R.; Majumder, P.P.; Bagchi, I. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat. Commun. 2013, 4, 2873. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Koyama, R.; Fukushima, H.; Ohashi, T.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T. Profiling cancer-related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget 2017, 8, 59113. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.-C.; Hsien, S.-I.; Hsu, J.-T.; Chen, M.Y. Impact on patients with oral squamous cell carcinoma in different anatomical subsites: A single-center study in Taiwan. Sci. Rep. 2021, 11, 15446. [Google Scholar] [CrossRef]
- Nair, S.; Singh, B.; Pawar, P.V.; Datta, S.; Nair, D.; Kane, S.; Chaturvedi, P. Squamous cell carcinoma of tongue and buccal mucosa: Clinico-pathologically different entities. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 3921–3928. [Google Scholar] [CrossRef]
- Su, W.W.Y.; Su, C.W.; Chang, D.C.; Chuang, S.L.; Chen, S.L.S.; Hsu, C.Y.; Yen, A.M.F.; Chiu, S.Y.H.; Fann, J.C.Y.; Lee, Y.H. Impact of varying anatomic sites on advanced stage and survival of oral cancer: 9-year prospective cohort of 27 717 cases. Head Neck 2019, 41, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Kim, J.H. Increasing incidence and improving survival of oral tongue squamous cell carcinoma. Sci. Rep. 2020, 10, 7877. [Google Scholar] [CrossRef]
- Mohsen, H.; Gunasekharan, V.; Qing, T.; Seay, M.; Surovtseva, Y.; Negahban, S.; Szallasi, Z.; Pusztai, L.; Gerstein, M.B. Network propagation-based prioritization of long tail genes in 17 cancer types. Genome Biol. 2021, 22, 287. [Google Scholar] [CrossRef]
- Iranzo, J.; Martincorena, I.; Koonin, E.V. Cancer-mutation network and the number and specificity of driver mutations. Proc. Natl. Acad. Sci. USA 2018, 115, E6010–E6019. [Google Scholar] [CrossRef]
- Aparisi, F.; Amado-Labrador, H.; Calabuig-Fariñas, S.; Torres, S.; Herreros-Pomares, A. Passenger mutations in cancer evolution. Cancer Rep. 2019, 3. [Google Scholar] [CrossRef]
- McFarland, C.D.; Korolev, K.S.; Kryukov, G.V.; Sunyaev, S.R.; Mirny, L.A. Impact of deleterious passenger mutations on cancer progression. Proc. Natl. Acad. Sci. USA 2013, 110, 2910–2915. [Google Scholar] [CrossRef]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef]
- McFarland, C.D.; Mirny, L.A.; Korolev, K.S. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc. Natl. Acad. Sci. USA 2014, 111, 15138–15143. [Google Scholar] [CrossRef]
- Li, X.; Thirumalai, D. Interplay of Driver, Mini-Driver, and Deleterious Passenger Mutations on Cancer Progression. bioRxiv 2016, 084392. [Google Scholar] [CrossRef]
- Feller, L.; Khammissa, R.A.G.; Lemmer, J. Biomechanical cell regulatory networks as complex adaptive systems in relation to cancer. Cancer Cell Int. 2017, 17, 16. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ramon y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Zellmer, V.R.; Zhang, S. Evolving concepts of tumor heterogeneity. Cell Biosci. 2014, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Black, J.R.; McGranahan, N. Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer 2021, 21, 379–392. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; De, S. Drivers of dynamic intratumor heterogeneity and phenotypic plasticity. Am. J. Physiol. Cell Physiol. 2021, 320, C750–C760. [Google Scholar] [CrossRef]
- Feller, L.; Altini, M.; Lemmer, J. Inflammation in the context of oral cancer. Oral Oncol. 2013, 49, 887–892. [Google Scholar] [CrossRef]
- Feller, G.; Khammissa, R.A.G.; Nemutandani, M.S.; Feller, L. Biological consequences of cancer radiotherapy in the context of oral squamous cell carcinoma. Head Face Med. 2021, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Gluzman, M.; Scott, J.G.; Vladimirsky, A. Optimizing adaptive cancer therapy: Dynamic programming and evolutionary game theory. Proc. Biol. Sci. 2020, 287, 20192454. [Google Scholar] [CrossRef]
- Pepper, J.W.; Scott Findlay, C.; Kassen, R.; Spencer, S.L.; Maley, C.C. Cancer research meets evolutionary biology. Evol. Appl. 2009, 2, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-Induced Mutagenesis and Selective Pressures Sculpt Cancer Evolution. Cold Spring Harb Perspect. Med. 2017, 7, a026617. [Google Scholar] [CrossRef]
- Somarelli, J.A. The Hallmarks of Cancer as Ecologically Driven Phenotypes. Front. Ecol. Evol. 2021, 9, 661583. [Google Scholar] [CrossRef]
- Ma, Y.; West, J.; Newton, P.K. Competitive release in tumors. bioRxiv 2018, 263335. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Chemotherapy: Preventing competitive release. Nat. Rev. Cancer 2016, 16, 199. [Google Scholar] [CrossRef]
- McDermott, N.; Meunier, A.; Mooney, B.; Nortey, G.; Hernandez, C.; Hurley, S.; Lynam-Lennon, N.; Barsoom, S.H.; Bowman, K.J.; Marples, B.; et al. Fractionated radiation exposure amplifies the radioresistant nature of prostate cancer cells. Sci. Rep. 2016, 6, 34796. [Google Scholar] [CrossRef] [PubMed]
- Galeaz, C.; Totis, C.; Bisio, A. Radiation Resistance: A Matter of Transcription Factors. Front. Oncol. 2021, 11, 662840. [Google Scholar] [CrossRef]
- Usman, S.; Jamal, A.; Teh, M.T.; Waseem, A. Major Molecular Signaling Pathways in Oral Cancer Associated with Therapeutic Resistance. Front. Oral Health 2020, 1, 603160. [Google Scholar] [CrossRef]
- Alfonso, J.; Berk, L. Modeling the effect of intratumoral heterogeneity of radiosensitivity on tumor response over the course of fractionated radiation therapy. Radiat. Oncol. 2019, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Lewin, T.D.; Maini, P.K.; Moros, E.G.; Enderling, H.; Byrne, H.M. The Evolution of Tumour Composition During Fractionated Radiotherapy: Implications for Outcome. Bull. Math. Biol. 2018, 80, 1207–1235. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feller, G.; Khammissa, R.A.G.; Ballyram, R.; Beetge, M.-M.; Lemmer, J.; Feller, L. Tumour Genetic Heterogeneity in Relation to Oral Squamous Cell Carcinoma and Anti-Cancer Treatment. Int. J. Environ. Res. Public Health 2023, 20, 2392. https://doi.org/10.3390/ijerph20032392
Feller G, Khammissa RAG, Ballyram R, Beetge M-M, Lemmer J, Feller L. Tumour Genetic Heterogeneity in Relation to Oral Squamous Cell Carcinoma and Anti-Cancer Treatment. International Journal of Environmental Research and Public Health. 2023; 20(3):2392. https://doi.org/10.3390/ijerph20032392
Chicago/Turabian StyleFeller, Gal, Razia Abdool Gafaar Khammissa, Raoul Ballyram, Mia-Michaela Beetge, Johan Lemmer, and Liviu Feller. 2023. "Tumour Genetic Heterogeneity in Relation to Oral Squamous Cell Carcinoma and Anti-Cancer Treatment" International Journal of Environmental Research and Public Health 20, no. 3: 2392. https://doi.org/10.3390/ijerph20032392
APA StyleFeller, G., Khammissa, R. A. G., Ballyram, R., Beetge, M.-M., Lemmer, J., & Feller, L. (2023). Tumour Genetic Heterogeneity in Relation to Oral Squamous Cell Carcinoma and Anti-Cancer Treatment. International Journal of Environmental Research and Public Health, 20(3), 2392. https://doi.org/10.3390/ijerph20032392