
Interaction of Pelargonium sidoides Compounds with Lactoferrin and SARS-CoV-2: Insights from Molecular Simulations

 ,
,  ,
,  ,
,  ,
, 
 , ,
, , 
 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Docking of the PEL Compounds
2.2. Molecular Dynamics of the Best Docking Complexes
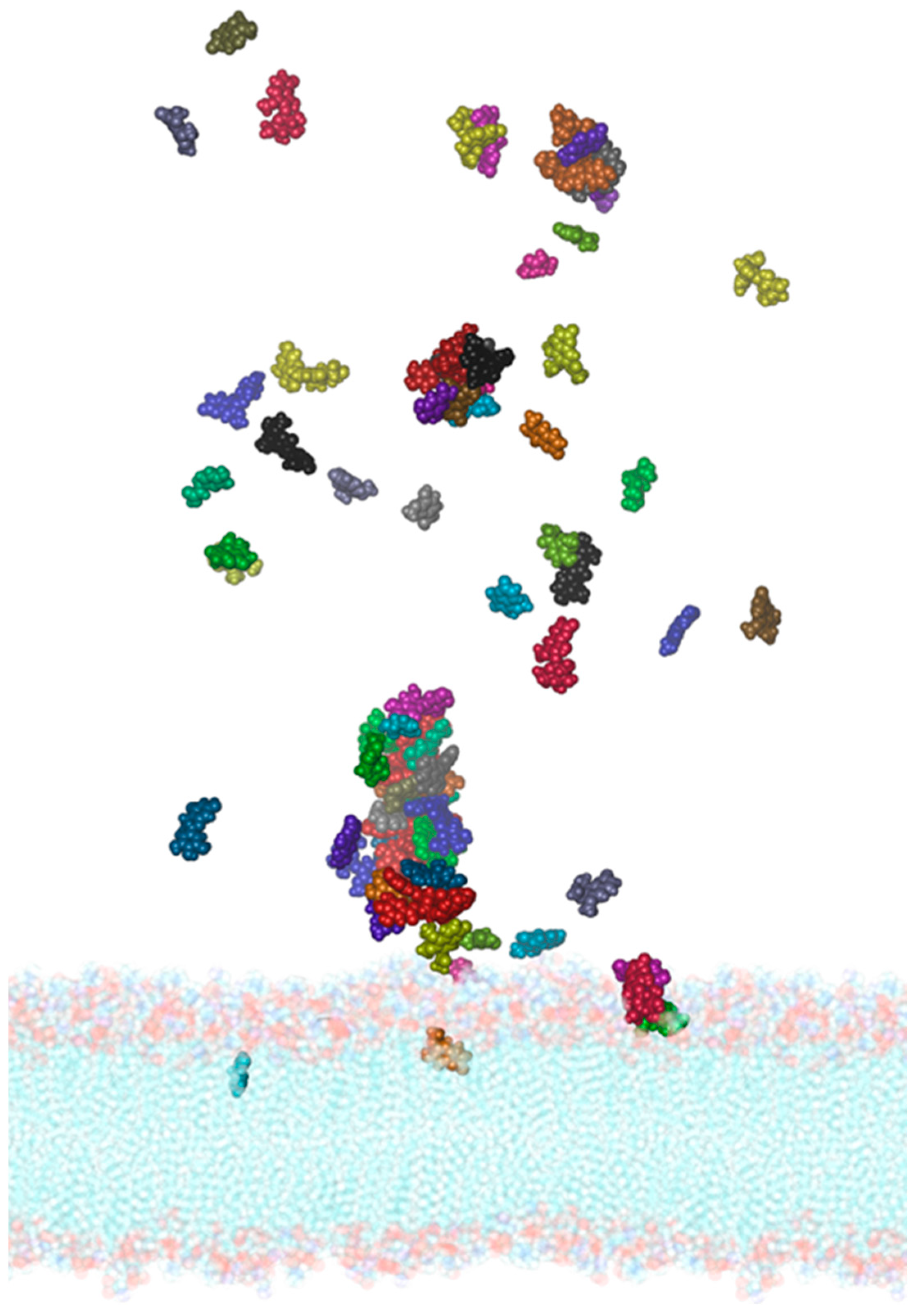
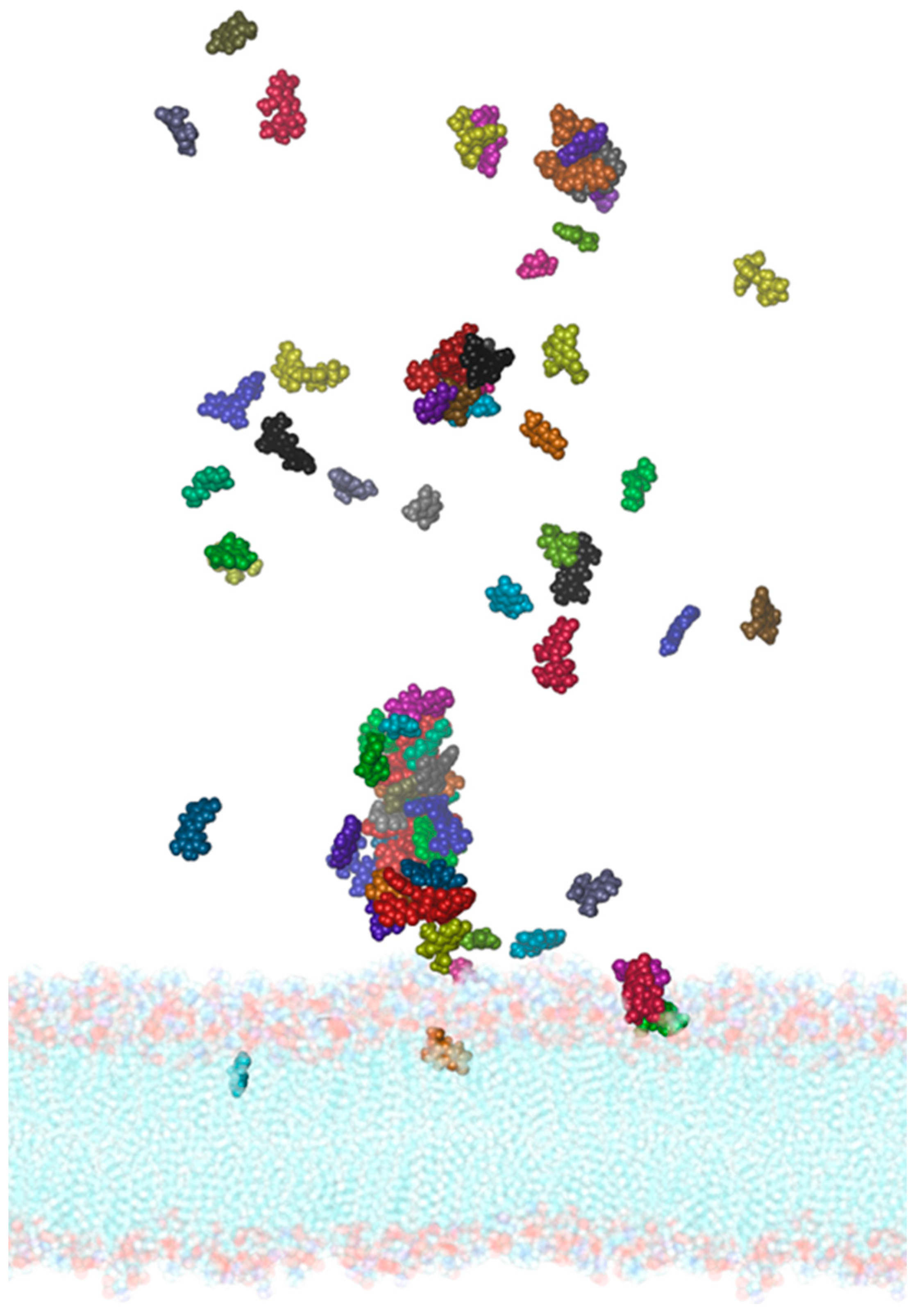
2.3. Molecular Dynamics of the SARS-CoV-2 Membrane–PEL Compounds Interaction
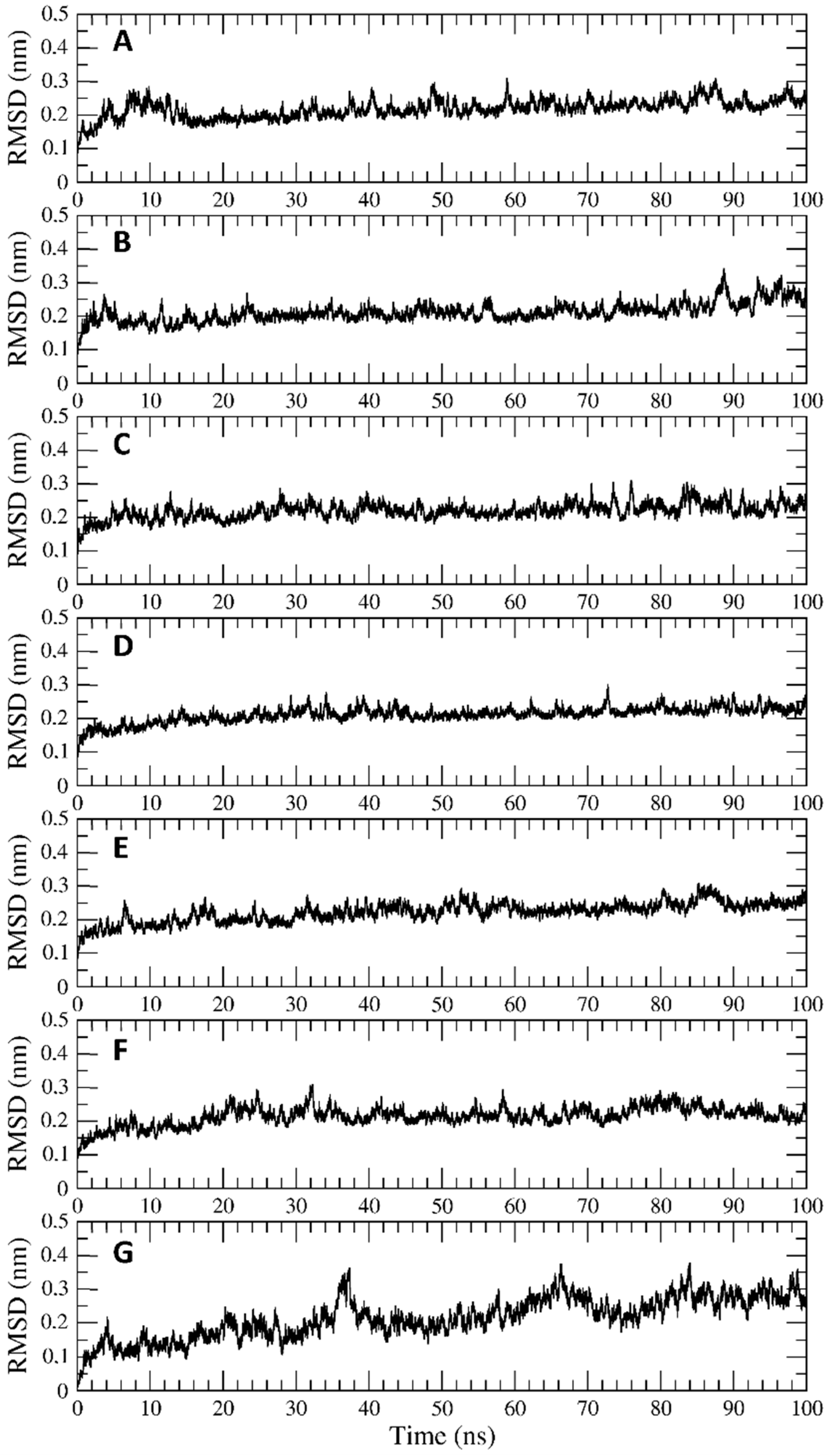
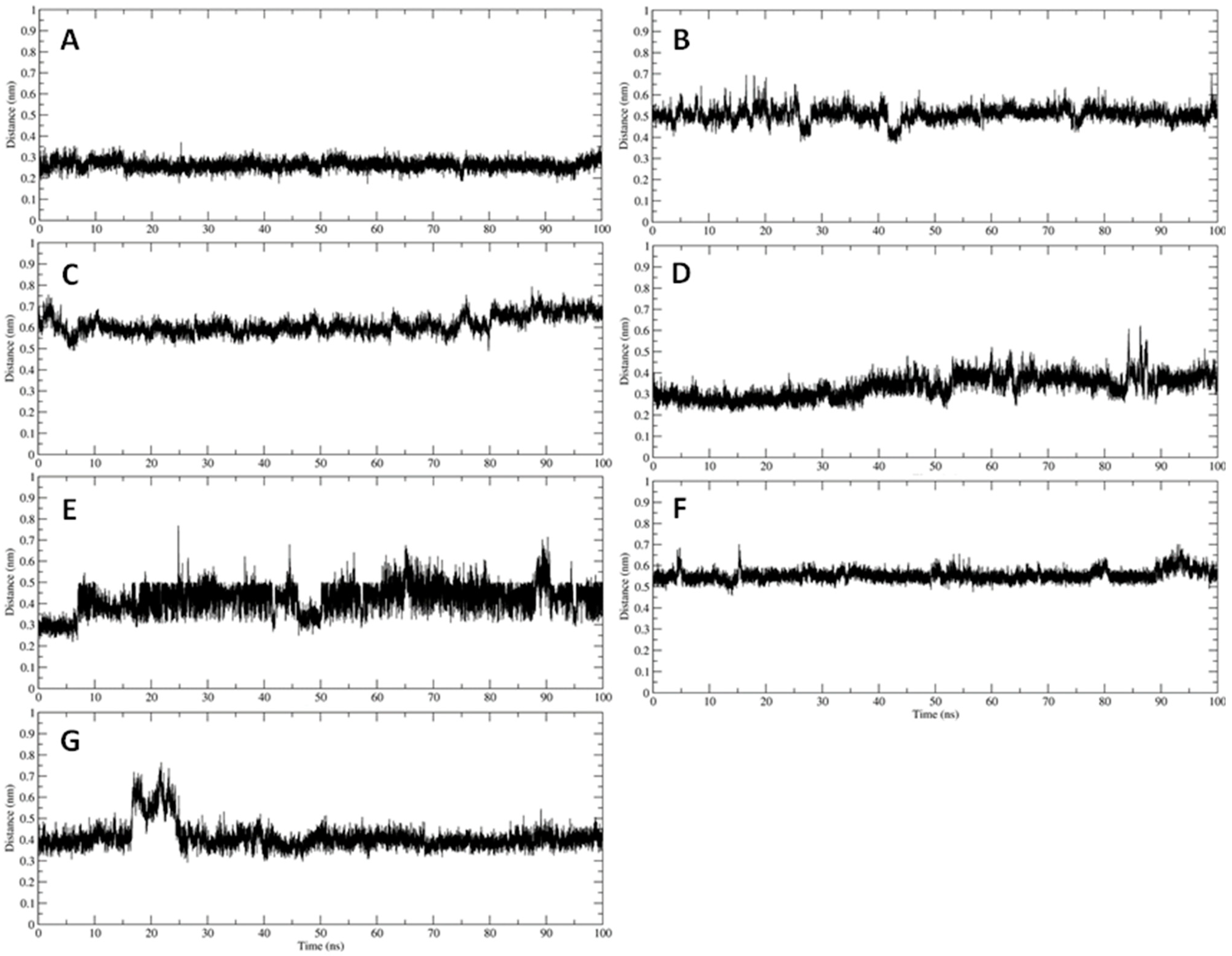
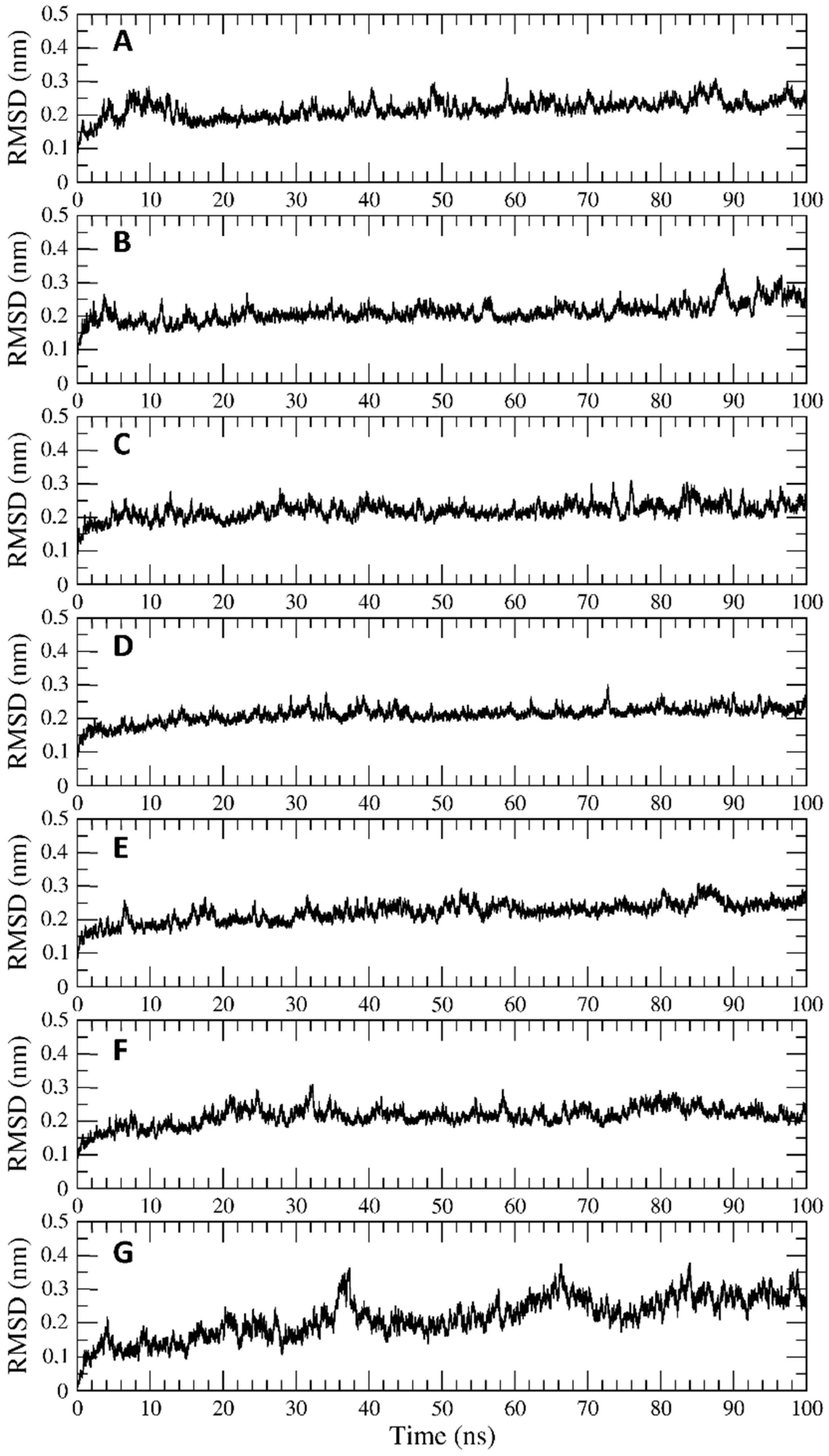
2.4. Trajectory Analysis
3. Results
3.1. Prediction of PEL Compounds Toxicity
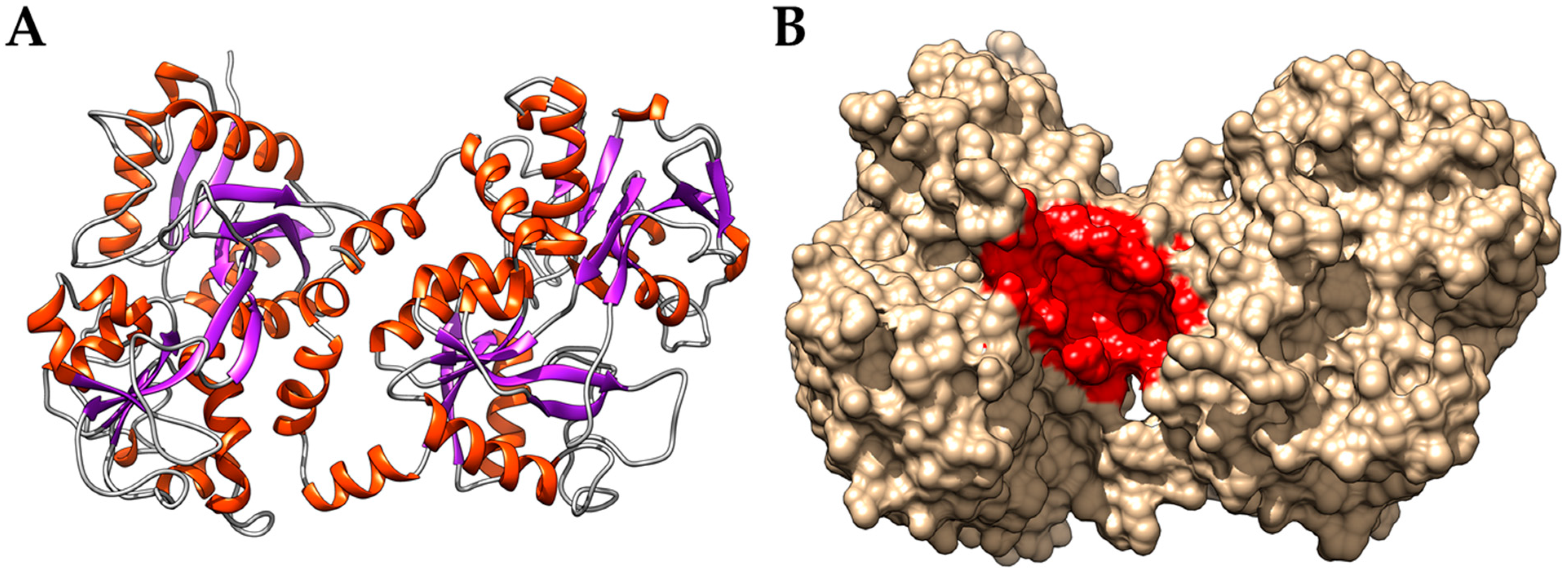
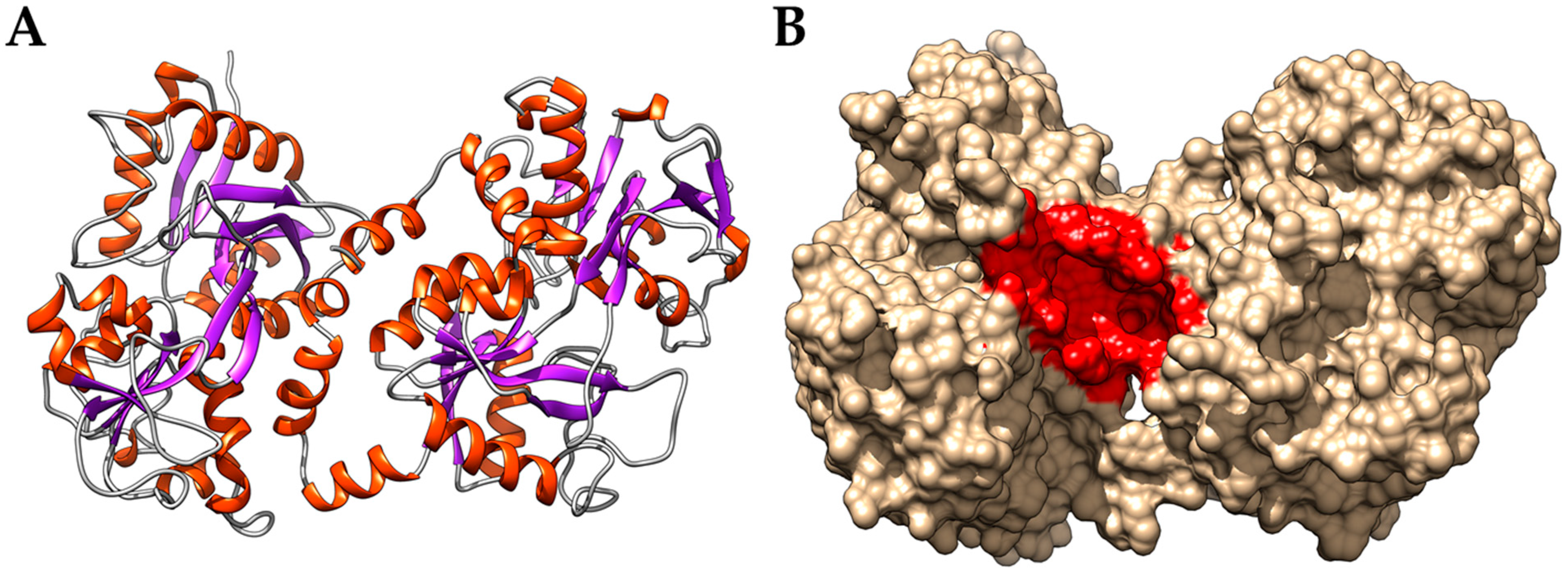
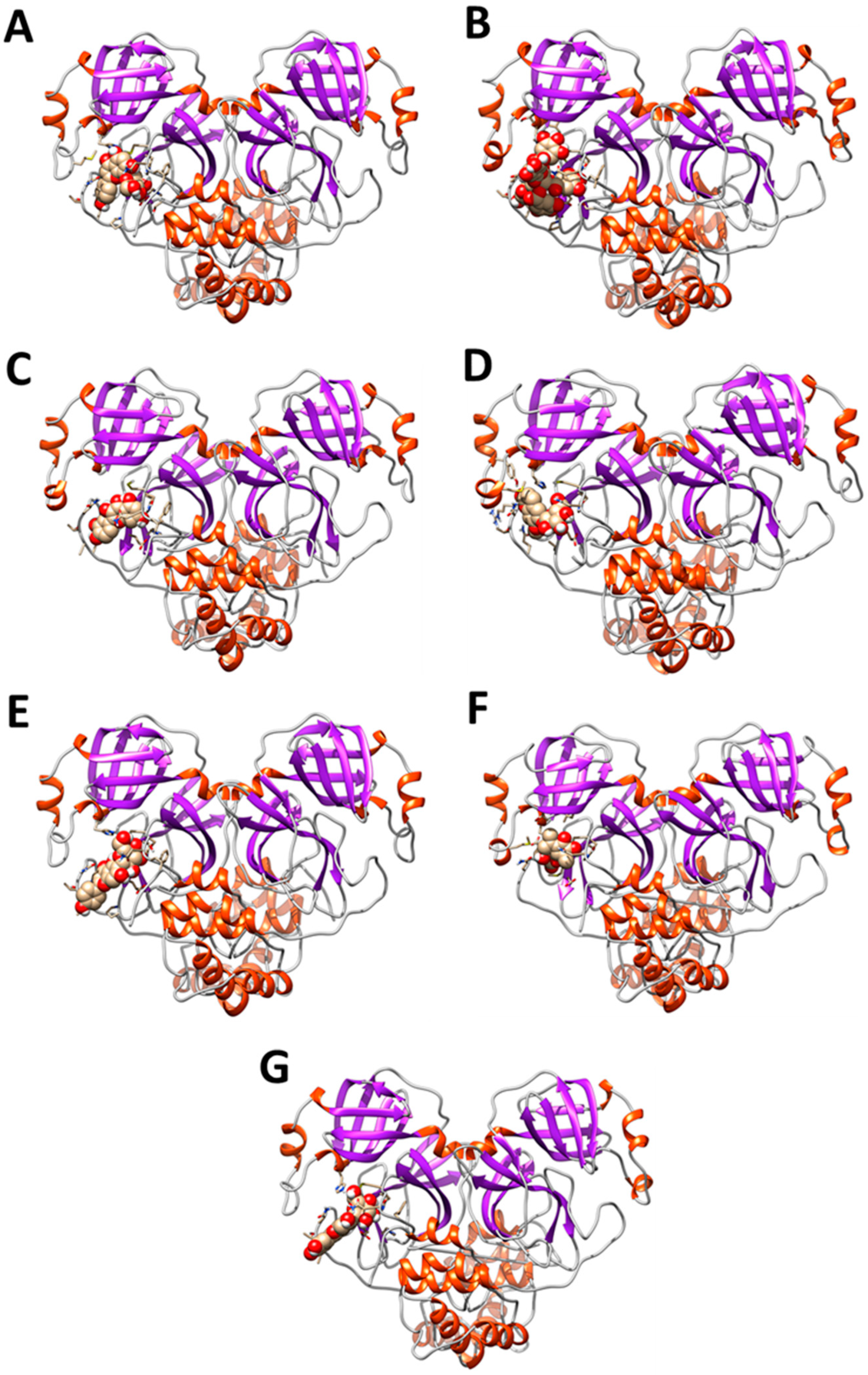
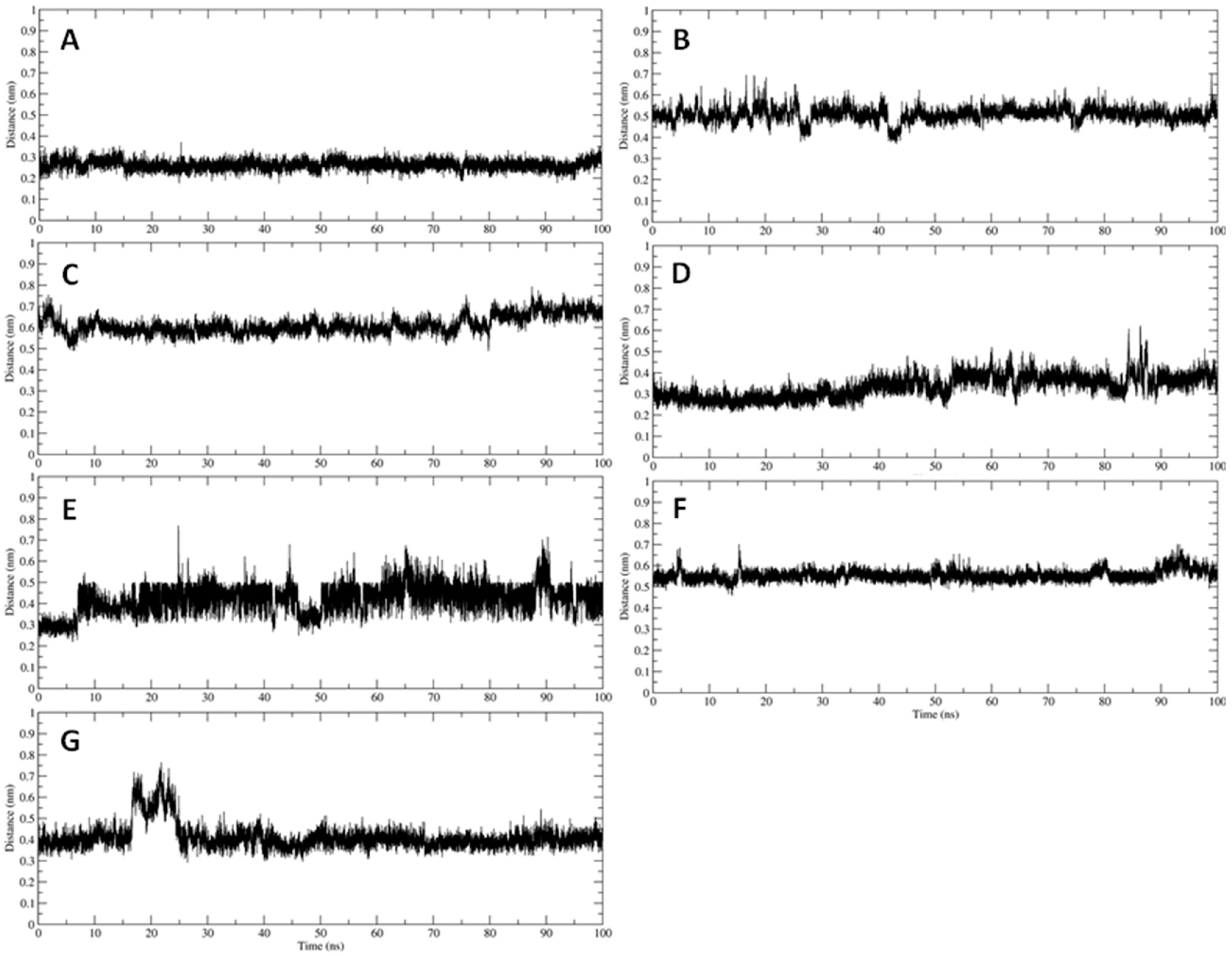
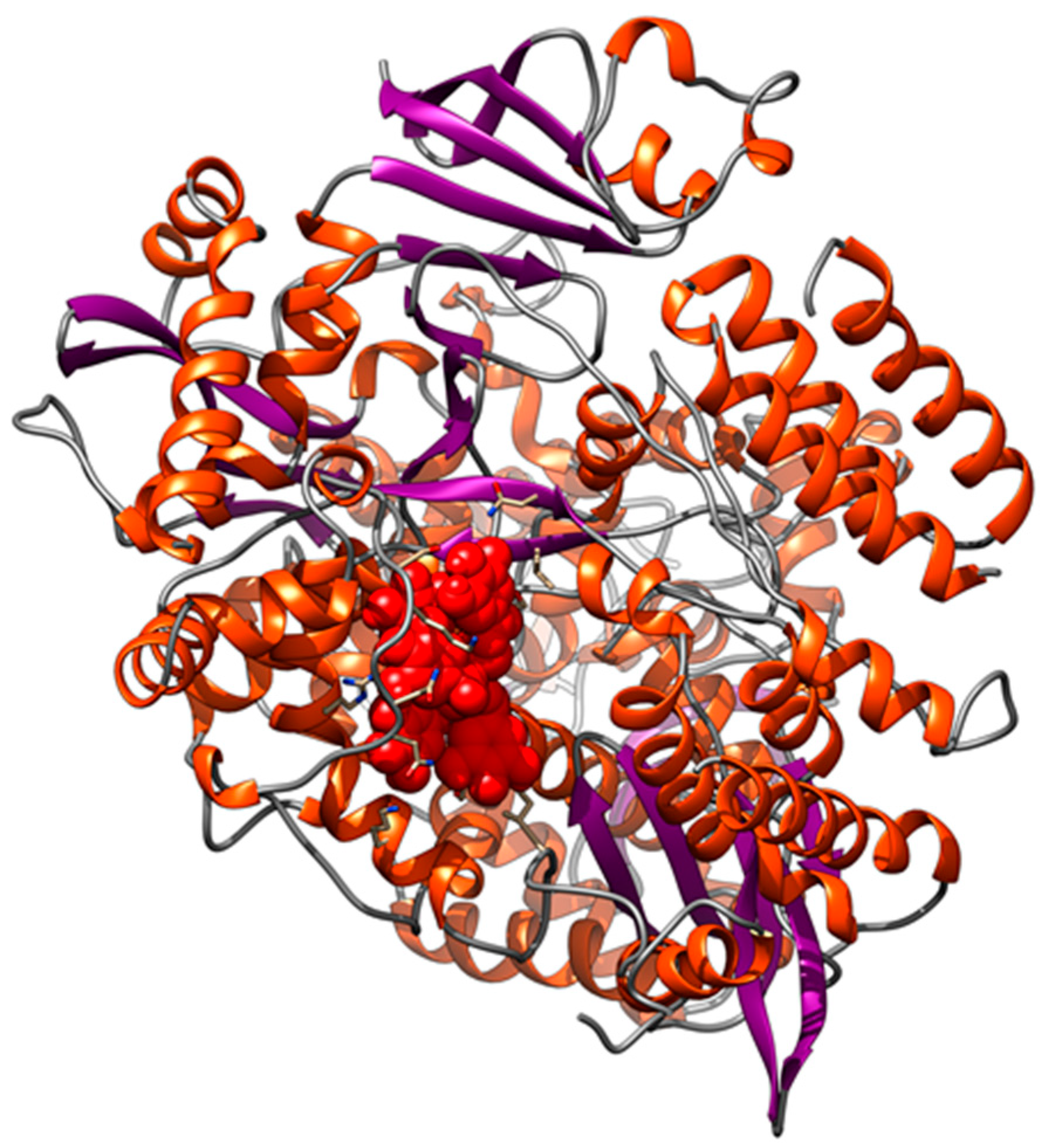
3.2. Interaction of PEL Compounds with the Bovine Lactoferrin
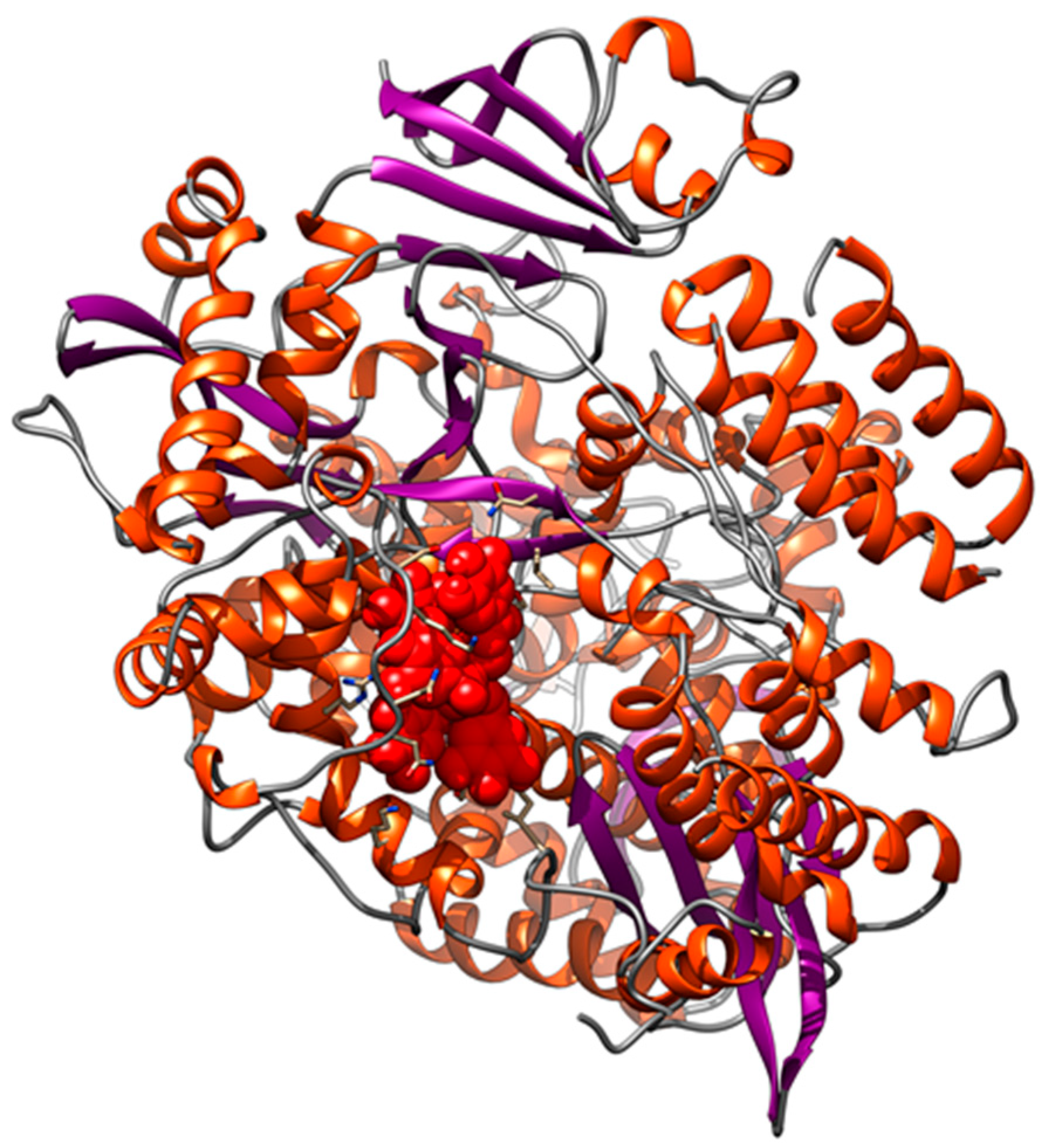
3.3. Interaction of PEL Compounds with the SARS-CoV-2 Protease (3CLpro)
3.4. Interaction of PEL Compounds with the SARS-CoV-2 Spike Glycoprotein
3.5. Interaction of PEL Compounds with the SARS-CoV-2 RdRp Polymerase
3.6. Interaction of PEL Compounds with the SARS-CoV-2 Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, D.L.; Shindo, N.; WHO Scientific and Technical Advisory Group for Infectious Hazards. COVID-19: What is next for public health? Lancet 2020, 395, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Campione, E.; Cosio, T.; Rosa, L.; Lanna, C.; Di Girolamo, S.; Gaziano, R.; Valenti, P.; Bianchi, L. Lactoferrin as Protective Natural Barrier of Respiratory and Intestinal Mucosa against Coronavirus Infection and Inflammation. Int. J. Mol. Sci. 2020, 21, 4903. [Google Scholar] [CrossRef] [PubMed]
- Nordling, L. Scientific networks are helping African countries to access coronavirus lab supplies. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Terlizzi, M.; Colarusso, C.; Di Maio, U.; Bagnulo, A.; Pinto, A.; Sorrentino, R. Antioxidant and antimicrobial properties of Pelargonium sidoides DC and lactoferrin combination. Biosci. Rep. 2020, 40, BSR20203284. [Google Scholar] [CrossRef] [PubMed]
- Campione, E.; Lanna, C.; Cosio, T.; Rosa, L.; Conte, M.P.; Iacovelli, F.; Romeo, A.; Falconi, M.; Del Vecchio, C.; Franchin, E.; et al. Lactoferrin Against SARS-CoV-2: In Vitro and In Silico Evidences. Front. Pharmacol. 2021, 12, 666600. [Google Scholar] [CrossRef]
- Campione, E.; Lanna, C.; Cosio, T.; Rosa, L.; Conte, M.P.; Iacovelli, F.; Romeo, A.; Falconi, M.; Del Vecchio, C.; Franchin, E.; et al. Lactoferrin as Antiviral Treatment in COVID-19 Management: Preliminary Evidence. Int. J. Environ. Res. Public Health 2021, 18, 10985. [Google Scholar] [CrossRef]
- Papies, J.; Emanuel, J.; Heinemann, N.; Kulić, Ž.; Schroeder, S.; Tenner, B.; Lehner, M.D.; Seifert, G.; Müller, M.A. Antiviral and Immunomodulatory Effects of Pelargonium sidoides DC. Root Extract EPs® 7630 in SARS-CoV-2-Infected Human Lung Cells. Front. Pharmacol. 2021, 12, 757666. [Google Scholar] [CrossRef]
- Moyo, M.; Van Staden, J. Medicinal properties and conservation of Pelargonium sidoides DC. J. Ethnopharmacol. 2014, 152, 243–255. [Google Scholar] [CrossRef]
- Heinrich, M.; Barnes, J.; Prieto-Garcia, J.; Gibbons, S.; Williamson, E.M. Fundamentals of Pharmacognosy and Phytotherapy; Churchill Livingston: London, UK, 2018. [Google Scholar]
- EMA. “Assessment Report on Pelargonium Sidoides DC and/or Pelargonium Reniforme Curt., Radix,” in EMA/HMPC/444251/2015 Committee on Herbal Medicinal Products (HMPC); European Medicines Agency: Strasbourg, France, 2018. [Google Scholar]
- COLOMBIA. Vademecum Colombiano de Plantas Medicinales; Ministerio de la Protecion Social/Imprenta Nacional Colombiana: Bogotá, Colombia, 2008.
- Michaelis, M.; Doerr, H.W.; Cinatl, J., Jr. Investigation of the influence of EPs® 7630, a herbal drug preparation from Pelargonium sidoides, on replication of a broad panel of respiratory viruses. Phytomedicine 2011, 18, 384–386. [Google Scholar] [CrossRef]
- Theisen, L.L.; Muller, C.P. EPs® 7630 (Umckaloabo®), an extract from Pelargonium sidoides roots, exerts anti-influenza virus activity in vitro and in vivo. Antiviral Res. 2012, 94, 147–156. [Google Scholar] [CrossRef]
- Roth, M.; Fang, L.; Stolz, D.; Tamm, M. Pelargonium sidoides radix extract EPs 7630 reduces rhinovirus infection through modulation of viral binding proteins on human bronchial epithelial cells. PLoS ONE 2019, 14, e0210702. [Google Scholar] [CrossRef] [Green Version]
- Kolodziej, H.; Kayser, O.; Radtke, O.A.; Kiderlen, A.F.; Koch, E. Pharmacological profile of extracts of Pelargonium sidoides and their constituents. Phytomedicine 2003, 10 (Suppl. S4), 18–24. [Google Scholar] [CrossRef]
- Valenti, P.; Antonini, G. Lactoferrin: An important host defence against microbial and viral attack. Cell Mol. Life Sci. 2005, 62, 2576–2587. [Google Scholar] [CrossRef] [PubMed]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A Natural Glycoprotein Involved in Iron and Inflammatory Homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Woodman, T.; Strunk, T.; Patole, S.; Hartmann, B.; Simmer, K.; Currie, A. Effects of lactoferrin on neonatal pathogens and Bifidobacterium breve in human breast milk. PLoS ONE 2018, 13, e0201819. [Google Scholar] [CrossRef]
- Berlutti, F.; Pantanella, F.; Natalizi, T.; Frioni, A.; Paesano, R.; Polimeni, A.; Valenti, P. Antiviral properties of lactoferrin--a natural immunity molecule. Molecules 2011, 16, 6992–7018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, H.; Oda, H.; Yamauchi, K.; Abe, F. Lactoferrin for prevention of common viral infections. J. Infect. Chemother. 2014, 20, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Ashida, K.; Sasaki, H.; Suzuki, Y.A.; Lönnerdal, B. Cellular internalization of lactoferrin in intestinal epithelial cells. Biometals 2004, 17, 311–315. [Google Scholar] [CrossRef]
- Lepanto, M.S.; Rosa, L.; Paesano, R.; Valenti, P.; Cutone, A. Lactoferrin in Aseptic and Septic Inflammation. Molecules 2019, 24, 1323. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Timilsena, Y.P.; Blanch, E.; Adhikari, B. Lactoferrin: Structure, function, denaturation and digestion. Crit. Rev. Food Sci. Nutr. 2019, 59, 580–596. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Kongsted, J.; Söderhjelm, P.; Ryde, U. Nonpolar Solvation Free Energies of Protein-Ligand Complexes. J. Chem. Theory Comput. 2010, 6, 3558–3568. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.A.; Anderson, B.F.; Groom, C.R.; Haridas, M.; Baker, E.N. Three-dimensional structure of diferric bovine lactoferrin at 2.8 A resolution. J. Mol. Biol. 1997, 274, 222–236. [Google Scholar] [CrossRef]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; de Vries, A.H.; Marrink, S.J.; Berendsen, H.J. Efficient Algorithms for Langevin and DPD Dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guixà-González, R.; Rodriguez-Espigares, I.; Ramírez-Anguita, J.M.; Carrió-Gaspar, P.; Martinez-Seara, H.; Giorgino, T.; Selent, J. MEMBPLUGIN: Studying membrane complexity in VMD. Bioinformatics 2014, 30, 1478–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.P.L.S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Case, D.A.; Walker, R.C.; Cheatham, T.E.; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T. Amber 18; University California: San Francisco, CA, USA, 2018. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Lu, N.T.; Crespi, C.M.; Liu, N.M.; Vu, J.Q.; Ahmadieh, Y.; Wu, S.; Lin, S.; McClune, A.; Durazo, F.; Saab, S.; et al. A Phase I Dose Escalation Study Demonstrates Quercetin Safety and Explores Potential for Bioflavonoid Antivirals in Patients with Chronic Hepatitis C. Phytother. Res. 2016, 30, 160–168. [Google Scholar] [CrossRef]
- Küpeli Akkol, E.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Momose, Y.; Maeda-Yamamoto, M.; Nabetani, H. Systematic review of green tea epigallocatechin gallate in reducing low-density lipoprotein cholesterol levels of humans. Int. J. Food Sci. Nutr. 2016, 67, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Liu, Z.; Ma, Q.; He, Z.; Niu, Z.; Zhang, M.; Pan, L.; Qu, X.; Yu, J.; Niu, B. Studies on the Interaction between Three Small Flavonoid Molecules and Bovine Lactoferrin. Biomed. Res. Int. 2018, 2018, 7523165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Giorgino, T. Computing 1-D atomic densities in macromolecular simulations: The density profile tool for VMD. Comput. Phys. Commun. 2014, 185, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Efimova, S.S.; Zakharova, A.A.; Ostroumova, O.S. Alkaloids Modulate the Functioning of Ion Channels Produced by Antimicrobial Agents via an Influence on the Lipid Host. Front. Cell Dev. Biol. 2020, 8, 537. [Google Scholar] [CrossRef]
- Yesylevskyy, S.; Rivel, T.; Ramseyer, C. Curvature increases permeability of the plasma membrane for ions, water and the anti-cancer drugs cisplatin and gemcitabine. Sci. Rep. 2019, 9, 17214. [Google Scholar] [CrossRef] [Green Version]
- Chuchalin, A.G.; Berman, B.; Lehmacher, W. Treatment of acute bronchitis in adults with a Pelargonium sidoides preparation (EPs 7630): A randomized, double-blind, placebo-controlled trial. Explore 2005, 1, 437–445. [Google Scholar] [CrossRef]
- Timmer, A.; Günther, J.; Motschall, E.; Rücker, G.; Antes, G.; Kern, W.V. Pelargonium sidoides extract for treating acute respiratory tract infections. Cochrane Database Syst Rev. 2013, 10, CD006323. [Google Scholar] [CrossRef]
- Schapowal, A.; Dobos, G.; Cramer, H.; Ong, K.C.; Adler, M.; Zimmermann, A.; Brandes-Schramm, J.; Lehmacher, W. Treatment of signs and symptoms of the common cold using EPs 7630—results of a meta-analysis. Heliyon 2019, 5, e02904. [Google Scholar] [CrossRef]
- Valenti, P.; Frioni, A.; Rossi, A.; Ranucci, S.; De Fino, I.; Cutone, A.; Rosa, L.; Bragonzi, A.; Berlutti, F. Aerosolized bovine lactoferrin reduces neutrophils and pro-inflammatory cytokines in mouse models of Pseudomonas aeruginosa lung infections. Biochem. Cell Biol. 2017, 95, 41–47. [Google Scholar] [CrossRef]
- Cutone, A.; Lepanto, M.S.; Rosa, L.; Scotti, M.J.; Rossi, A.; Ranucci, S.; De Fino, I.; Bragonzi, A.; Valenti, P.; Musci, G.; et al. Aerosolized Bovine Lactoferrin Counteracts Infection, Inflammation and Iron Dysbalance in A Cystic Fibrosis Mouse Model of Pseudomonas aeruginosa Chronic Lung Infection. Int. J. Mol. Sci. 2019, 20, 2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, M.; Pisani, S.; Antonini, G.; Valenti, P.; Seganti, L.; Orsi, N. Metal complexes of bovine lactoferrin inhibit in vitro replication of herpes simplex virus type 1 and 2. Biometals 1998, 11, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.; Superti, F.; Ammendolia, M.G.; Rossi, P.; Valenti, P.; Seganti, L. Inhibition of poliovirus type 1 infection by iron-, manganese- and zinc-saturated lactoferrin. Med. Microbiol. Immunol. 1999, 187, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, R.; Rega, B.; Marchetti, M.; Seganti, L.; Antonini, G.; Valenti, P. Bovine lactoferrin peptidic fragments involved in inhibition of herpes simplex virus type 1 infection. Biochem. Biophys. Res. Commun. 1999, 264, 19–23. [Google Scholar] [CrossRef]
- Superti, F.; Siciliano, R.; Rega, B.; Giansanti, F.; Valenti, P.; Antonini, G. Involvement of bovine lactoferrin metal saturation, sialic acid and protein fragments in the inhibition of rotavirus infection. Biochim. Biophys. Acta 2001, 1528, 107–115. [Google Scholar] [CrossRef]
- Marchetti, M.; Longhi, C.; Conte, M.P.; Pisani, S.; Valenti, P.; Seganti, L. Lactoferrin inhibits herpes simplex virus type 1 adsorption to Vero cells. Antiviral. Res. 1996, 29, 221–231. [Google Scholar] [CrossRef]
- El Yazidi-Belkoura, I.; Legrand, D.; Nuijens, J.; Slomianny, M.C.; van Berkel, P.; Spik, G. The binding of lactoferrin to glycosaminoglycans on enterocyte-like HT29-18-C1 cells is mediated through basic residues located in the N-terminus. Biochim. Biophys. Acta 2001, 1568, 197–204. [Google Scholar] [CrossRef]
- Superti, F.; Ammendolia, M.G.; Valenti, P.; Seganti, L. Antirotaviral activity of milk proteins: Lactoferrin prevents rotavirus infection in the enterocyte-like cell line HT-29. Med. Microbiol. Immunol. 1997, 186, 83–91. [Google Scholar] [CrossRef]
- Puddu, P.; Borghi, P.; Gessani, S.; Valenti, P.; Belardelli, F.; Seganti, L. Antiviral effect of bovine lactoferrin saturated with metal ions on early steps of human immunodeficiency virus type 1 infection. Int. J. Biochem. Cell Biol. 1998, 30, 1055–1062. [Google Scholar] [CrossRef]
- Loschwitz, J.; Jäckering, A.; Keutmann, M.; Olagunju, M.; Eberle, R.J.; Coronado, M.A.; Olabisi, O.O.; Strodel, B. Novel inhibitors of the main protease enzyme of SARS-CoV-2 identified via molecular dynamics simulation-guided in vitro assay. Bioorg. Chem. 2021, 111, 104862. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Epigallocatechin-3-gallate | −8.1 (0.3) |
| Taxifolin-3-glucoside | −7.9 (0.2) |
| Gallocatechin | −7.7 (0.2) |
| Artelin | −7.7 (0.2) |
| Pentagalloylglucose | −7.6 (0.4) |
| Vitexin | −7.6 (0.3) |
| Isoorientin | −7.5 (0.2) |
| Isovitexin | −7.3 (0.2) |
| Orientin | −7.2 (0.3) |
| Magnolioside | −7.2 (0.3) |
| Quercetin | −7.1 (0.2) |
| 6-8-dihydroxy-7-methoxycoumarin | −7.0 (0.3) |
| 7-acetoxy-5-6-dimethoxycoumarin | −6.8 (0.2) |
| Dimethoxycoumarin | −6.4 (0.3) |
| Fraxetin | −6.4 (0.2) |
| 6-7-8-trihydroxycoumarin | −6.2 (0.4) |
| Isofraxoside | −6.2 (0.2) |
| Umckalin | −6.2 (0.2) |
| Scopoletin | −6.0 (0.2) |
| 5-6-7-trimethoxycoumarin | −6.0 (0.2) |
| Caffeic acid | −5.7 (0.3) |
| Ferulic acid | −5.7 (0.2) |
| Vanillic acid | −5.4 (0.3) |
| Gallic acid | −5.3 (0.2) |
| Apocynin | −5.3 (0.2) |
| Homovanillic acid | −5.3 (0.1) |
| Dihydroxybenzoic acid | −4.7 (0.2) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Taxifolin-3-glucoside | −39.1 (4.5) |
| Isoorientin | −33.7 (3.6) |
| Gallocatechin | −30.8 (2.8) |
| Artelin | −30.0 (3.4) |
| Vitexin | −24.6 (4.8) |
| Epigallocatechin-3-gallate | −21.3 (2.5) |
| Pentagalloyl glucose | −16.6 (4.2) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Vitexin | −8.7 (0.3) |
| Pentagalloylglucose | −8.3 (0.4) |
| Quercetin | −8.1 (0.2) |
| Magnolioside | −8.0 (0.3) |
| Isovitexin | −8.0 (0.2) |
| Artelin | −8.0 (0.2) |
| Isoorientin | −8.0 (0.2) |
| Orientin | −7.4 (0.3) |
| Taxifolin-3-glucoside | −7.4 (0.1) |
| Epigallocatechin-3-gallate | −7.3 (0.3) |
| Gallocatechin | −7.3 (0.2) |
| Isofraxoside | −7.2 (0.2) |
| Fraxetin | −6.9 (0.2) |
| 7-acetoxy-5-6-dimethoxycoumarin | −6.7 (0.2) |
| Dimethoxycoumarin | −6.4 (0.3) |
| 6-8-dihydroxy-7-methoxycoumarin | −6.2 (0.3) |
| Dihydroxybenzoic acid | −6.2 (0.2) |
| Scopoletin | −5.9 (0.2) |
| 6-7-8-trihydroxycoumarin | −5.7 (0.4) |
| Caffeic acid | −5.7 (0.3) |
| 5-6-7-trimethoxycoumarin | −5.7 (0.2) |
| Umckalin | −5.7 (0.2) |
| Ferulic acid | −5.6 (0.2) |
| Gallic acid | −5.6 (0.2) |
| Vanillic acid | −5.3 (0.3) |
| Apocynin | −5.3 (0.2) |
| Homovanillic acid | −4.6 (0.1) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Vitexin | −29.87 (3.40) |
| Pentagalloyl-glucose | −25.74 (5.99) |
| Magnolioside | −25.44 (4.05) |
| Isoorientin | −23.14 (3.95) |
| Artelin | −21.76 (5.59) |
| Quercetin | −15.21 (2.69) |
| Isovitexin | −15.08 (3.59) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Pentagalloylglucose | −10.4 (0.3) |
| Vitexin | −9.5 (0.2) |
| 6-8-Dihydroxy-7-methoxycoumarin | −9.3 (0.3) |
| Quercetin | −9.3 (0.2) |
| Taxifolin-3-glucoside | −9.1 (0.1) |
| Isofraxoside | −8.7 (0.2) |
| Isovitexin | −8.6 (0.2) |
| Gallocatechin | −8.6 (0.2) |
| Isoorientin | −8.5 (0.2) |
| Epigallocatechin-3-gallate | −8.3 (0.3) |
| Artelin | −8.2 (0.2) |
| Magnolioside | −8.0 (0.1) |
| Orientin | −7.4 (0.3) |
| Fraxetin | −7.4 (0.2) |
| Scopoletin | −7.0 (0.2) |
| 7-acetoxy-5-6-dimethoxycoumarin | −6.8 (0.2) |
| Umckalin | −6.8 (0.2) |
| Caffeic acid | −6.6 (0.3) |
| 5-6-7-trimethoxycoumarin | −6.6 (0.2) |
| Ferulic acid | −6.4(0.2) |
| Dimethoxycoumarin | −6.4 (0.3) |
| Homovanillic acid | −6.1 (0.1) |
| Gallic acid | −5.7(0.2) |
| 6-7-8-trihydroxycoumarin | −5.7 (0.4) |
| Vanillic acid | −5.6 (0.3) |
| Apocynin | −5.3 (0.2) |
| Dihydroxybenzoic acid | −5.2 (0.2) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Taxifolin-3-glucoside | −43.2 (5.8) |
| Pentagalloylglucose | −38.7 (5.5) |
| Magnolioside | −32.2 (4.2) |
| Isovitexin | −26.3 (4.7) |
| Epigallocatechin-3-gallate | −25.8 (3.4) |
| Gallocatechin | −24.7 (3.9) |
| Isofraxoside | −23.3 (4.7) |
| Isoorientin | −20.9 (6.8) |
| 6-8-dihydroxy-7-methoxycoumarin | −19.4 (4.5) |
| Quercetin | −19.3 (4.5) |
| Vitexin | −18.9 (6.1) |
| Compound | Interaction Energy (kcal/mol ± SD) |
|---|---|
| Pentagalloyl glucose | −9.0 (0.5) |
| Vitexin | −7.4 (0.1) |
| Taxifolin-3-glucoside | −7.4 (0.1) |
| Magnolioside | −7.3 (0.2) |
| Isovitexin | −7.3 (0.2) |
| Isoorientin | −7.3 (0.3) |
| Quercetin | −7.2 (0.2) |
| Isofraxoside | −7.2 (0.2) |
| Epigallocatechin-3-gallate | −6.9 (0.3) |
| Gallocatechin | −6.9 (0.2) |
| Orientin | −6.8 (0.1) |
| Artelin | −6.4 (0.2) |
| 6-8-dihydroxy-7-methoxycoumarin | −6.2 (0.3) |
| Fraxetin | −6.0 (0.1) |
| 7-acetoxy-5-6-dimethoxycoumarin | −5.9 (0.2) |
| Gallic acid | −5.9 (0.2) |
| Caffeic acid | −5.8 (0.4) |
| Scopoletin | −5.8 (0.1) |
| 6-7-8-trihydroxycoumarin | −5.7 (0.3) |
| Vanillic acid | −5.7 (0.3) |
| Umckalin | −5.7 (0.2) |
| Ferulic acid | −5.6 (0.2) |
| Dimethoxycoumarin | −5.5 (0.3) |
| Apocynin | −5.5 (0.2) |
| Dihydroxybenzoic acid | −5.4 (0.3) |
| 5-6-7-trimethoxycoumarin | −5.4 (0.1) |
| Homovanillic acid | −5.4 (0.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacovelli, F.; Costanza, G.; Romeo, A.; Cosio, T.; Lanna, C.; Bagnulo, A.; Di Maio, U.; Sbardella, A.; Gaziano, R.; Grelli, S.; et al. Interaction of Pelargonium sidoides Compounds with Lactoferrin and SARS-CoV-2: Insights from Molecular Simulations. Int. J. Environ. Res. Public Health 2022, 19, 5254. https://doi.org/10.3390/ijerph19095254
Iacovelli F, Costanza G, Romeo A, Cosio T, Lanna C, Bagnulo A, Di Maio U, Sbardella A, Gaziano R, Grelli S, et al. Interaction of Pelargonium sidoides Compounds with Lactoferrin and SARS-CoV-2: Insights from Molecular Simulations. International Journal of Environmental Research and Public Health. 2022; 19(9):5254. https://doi.org/10.3390/ijerph19095254
Chicago/Turabian StyleIacovelli, Federico, Gaetana Costanza, Alice Romeo, Terenzio Cosio, Caterina Lanna, Antonino Bagnulo, Umberto Di Maio, Alice Sbardella, Roberta Gaziano, Sandro Grelli, and et al. 2022. "Interaction of Pelargonium sidoides Compounds with Lactoferrin and SARS-CoV-2: Insights from Molecular Simulations" International Journal of Environmental Research and Public Health 19, no. 9: 5254. https://doi.org/10.3390/ijerph19095254
APA StyleIacovelli, F., Costanza, G., Romeo, A., Cosio, T., Lanna, C., Bagnulo, A., Di Maio, U., Sbardella, A., Gaziano, R., Grelli, S., Squillaci, E., Miani, A., Piscitelli, P., Bianchi, L., Falconi, M., & Campione, E. (2022). Interaction of Pelargonium sidoides Compounds with Lactoferrin and SARS-CoV-2: Insights from Molecular Simulations. International Journal of Environmental Research and Public Health, 19(9), 5254. https://doi.org/10.3390/ijerph19095254