
Psychomotor Symptoms in Chronic Cocaine Users: An Interpretative Model



Abstract
:1. Introduction
2. Case Description
3. Discussion
4. Proposal
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Baik, J.-H. Stress and the dopaminergic reward system. Exp. Mol. Med. 2020, 52, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, A.; Huber, R.; Panksepp, J. Behavioral functions of the mesolimbic dopaminergic system: An affective neuroethological perspective. Brain Res. Rev. 2007, 56, 283–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.-M.; Gainetdinov, R.R. The Physiology, Signaling, and Pharmacology of Dopamine Receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Baik, J.-H. Dopamine Signaling in reward-related behaviors. Front. Neural Circuits 2013, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, R.A.; Pryce, K.D.; Zachariou, V. The Mesolimbic Dopamine System in Chronic Pain and Associated Affective Comorbidities. Biol. Psychiatry 2020, 87, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambeitz, J.; Abi-Dargham, A.; Kapur, S.; Howes, O. Alterations in cortical and extrastriatal subcortical dopamine function in schizophrenia: Systematic review and meta-analysis of imaging studies. Br. J. Psychiatry 2014, 204, 420–429. [Google Scholar] [CrossRef]
- Toda, M.; Abi-Dargham, A. Dopamine hypothesis of schizophrenia: Making sense of it all. Curr. Psychiatry Rep. 2007, 9, 329–336. [Google Scholar] [CrossRef]
- Fiorentini, A.; Volonteri, L.S.; Dragogna, F.; Rovera, C.; Maffini, M.; Mauri, M.C.; Altamura, C.A. Substance-induced psychoses: A critical review of the literature. Curr. Drug Abus. Rev. 2011, 4, 228–240. [Google Scholar] [CrossRef]
- Molitch, M.E. Dopamine agonists and antipsychotics. Eur. J. Endocrinol. 2020, 183, C11–C13. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.J. Common cellular and molecular mechanisms in obesity and drug addiction. Nat. Rev. Neurosci. 2011, 12, 638–651. [Google Scholar] [CrossRef]
- Baik, J.-H. Dopamine signaling in food addiction: Role of dopamine D2 receptors. BMB Rep. 2013, 46, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessiglione, M.; Seymour, B.; Flandin, G.; Dolan, R.J.; Frith, C.D. Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature 2006, 442, 1042–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pessiglione, M.; Schmidt, L.; Draganski, B.; Kalisch, R.; Lau, H.; Dolan, R.J.; Frith, C.D. How the Brain Translates Money into Force: A Neuroimaging Study of Subliminal Motivation. Science 2007, 316, 904–906. [Google Scholar] [CrossRef] [Green Version]
- Schultz, W. Neuronal Reward and Decision Signals: From Theories to Data. Physiol. Rev. 2015, 95, 853–951. [Google Scholar] [CrossRef]
- Berridge, K.C.; Kringelbach, M. Pleasure Systems in the Brain. Neuron 2015, 86, 646–664. [Google Scholar] [CrossRef] [Green Version]
- Lynch, W.J.; Peterson, A.B.; Sanchez, V.; Abel, J.; Smith, M.A. Exercise as a novel treatment for drug addiction: A neurobiological and stage-dependent hypothesis. Neurosci. Biobehav. Rev. 2013, 37, 1622–1644. [Google Scholar] [CrossRef] [Green Version]
- Tritsch, N.X.; Ding, J.B.; Sabatini, B.L. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 2012, 490, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Park. Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [Green Version]
- Waddington, J.L. Psychosis in Parkinson’s disease and parkinsonism in antipsychotic-naive schizophrenia spectrum psychosis: Clinical, nosological and pathobiological challenges. Acta Pharmacol. Sin. 2020, 41, 464–470. [Google Scholar] [CrossRef]
- Khokhar, J.Y.; Dwiel, L.L.; Henricks, A.M.; Doucette, W.T.; Green, A.I. The link between schizophrenia and substance use disorder: A unifying hypothesis. Schizophr. Res. 2018, 194, 78–85. [Google Scholar] [CrossRef]
- Potvin, S.; Pampoulova, T.; Mancini-Marië, A.; Lipp, O.; Bouchard, R.-H.; Stip, E. Increased extrapyramidal symptoms in patients with schizophrenia and a comorbid substance use disorder. J. Neurol. Neurosurg. Psychiatry 2006, 77, 796–798. [Google Scholar] [CrossRef] [Green Version]
- Dela Peña, I.; Gevorkiana, R.; Shi, W.-X. Psychostimulants affect dopamine transmission through both dopamine transporter-dependent and independent mechanisms. Eur. J. Pharmacol. 2015, 764, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhornitsky, S.; Stip, E.; Pampoulova, T.; Rizkallah, E.; Lipp, O.; Bentaleb, L.A.; Chiasson, J.-P.; Potvin, S. Extrapyramidal symptoms in substance abusers with and without schizophrenia and in nonabusing patients with schizophrenia. Mov. Disord. 2010, 25, 2188–2194. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Jayanthi, S.; McCoy, M.T.; Beauvais, G.; Cai, N.S. Dopamine D1 receptors, regulation of gene expression in the brain, and neurodegeneration. CNS Neurol. Disord. Drug Targets 2010, 9, 526–538. [Google Scholar] [CrossRef]
- Trifilieff, P.; Martinez, D. Imaging addiction: D2 receptors and dopamine signaling in the striatum as biomarkers for impulsivity. Neuropharmacology 2014, 76, 498–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, A.H.; Mizuno, Y.; Volkow, N.D.; Howes, O.D. Association of Stimulant Use with Dopaminergic Alterations in Users of Cocaine, Amphetamine, or Methamphetamine: A Systematic Review and Meta-analysis. JAMA Psychiatry 2017, 74, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; Fowler, J.S.; Wang, G.-J.; Swanson, J.M.; Telang, F. Dopamine in Drug Abuse and Addiction: Results of imaging studies and treatment implications. Arch. Neurol. 2007, 64, 1575–1579. [Google Scholar] [CrossRef]
- Lappin, J.M.; Sara, G.E. Psychostimulant use and the brain. Addiction 2019, 114, 2065–2077. [Google Scholar] [CrossRef]
- Satel, S.L.; Swann, A.C. Extrapyramidal symptoms and cocaine abuse. Am. J. Psychiatry 1993, 150, 347. [Google Scholar] [CrossRef]
- Potvin, S.; Blanchet, P.; Stip, E. Substance abuse is associated with increased extrapyramidal symptoms in schizophrenia: A meta-analysis. Schizophr. Res. 2009, 113, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Maat, A.; Fouwels, A.; De Haan, L. Cocaine is a major risk factor for antipsychotic induced akathisia, parkinsonism and dyskinesia. Psychopharmacol. Bull. 2008, 41, 5–10. [Google Scholar]
- Green, A.I. Schizophrenia and comorbid substance use disorder: Effects of antipsychotics. J. Clin. Psychiatry 2005, 66, 21–26. [Google Scholar] [PubMed]
- United Nations Office on Drugs and Crime (UNODC). World Drug Report 2021; United Nations Office on Drugs and Crime (UNODC): Vienna, Austria, 2021. [Google Scholar]
- Drake, L.R.; Scott, P.J.H. DARK Classics in Chemical Neuroscience: Cocaine. ACS Chem. Neurosci. 2018, 9, 2358–2372. [Google Scholar] [CrossRef] [PubMed]
- Illés, A.; Balicza, P.; Molnár, V.; Bencsik, R.; Szilvási, I.; Molnar, M.J. Dynamic interaction of genetic risk factors and cocaine abuse in the background of Parkinsonism—A case report. BMC Neurol. 2019, 19, 21–60. [Google Scholar] [CrossRef] [Green Version]
- Vaillancourt, D.E.; Bschonfeld, D.; Kwak, Y.; Bohnen, N.I.; Seidler, R. Dopamine overdose hypothesis: Evidence and clinical implications. Mov. Disord. 2013, 28, 1920–1929. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.L.; Brannock, C. Free radicals and the pathobiology of brain dopamine systems. Neurochem. Int. 1998, 32, 117–131. [Google Scholar] [CrossRef]
- Granado, N.; Ares-Santos, S.; Moratalla, R. Methamphetamine and Parkinson’s Disease. Park. Dis. 2013, 2013, 38052. [Google Scholar] [CrossRef]
- Granado, N.; Ares-Santos, S.; O’Shea, E.; Vicario-Abejón, C.; Colado, M.I.; Moratalla, R. Selective Vulnerability in Striosomes and in the Nigrostriatal Dopaminergic Pathway After Methamphetamine Administration: Early loss of TH in striosomes after methamphetamine. Neurotox. Res. 2010, 18, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Granado, N.; O’Shea, E.; Bove, J.; Vila, M.; Colado, M.I.; Moratalla, R. Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J. Neurochem. 2008, 107, 1102–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, B.K.; Zhu, W. The effects of methamphetamine on the production of free radicals and oxidative stress. J. Pharmacol. Exp. Ther. 1998, 287, 107–114. [Google Scholar] [PubMed]
- Büttner, A. Review: The neuropathology of drug abuse. Neuropathol. Appl. Neurobiol. 2011, 37, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.I.; Stout, K.A.; Miller, G.W. The vesicular monoamine transporter 2: An underexplored pharmacological target. Neurochem. Int. 2014, 73, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Little, K.Y.; Ramssen, E.; Welchko, R.; Volberg, V.; Roland, C.J.; Cassin, B. Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res. 2009, 168, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Villalba, R.M.; Smith, Y. Differential striatal spine pathology in Parkinson’s disease and cocaine addiction: A key role of dopamine? Neuroscience 2013, 251, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitriu, D.; LaPlant, Q.; Grossman, Y.S.; Dias, C.; Janssen, W.G.; Russo, S.J.; Morrison, J.H.; Nestler, E.J. Subregional, Dendritic Compartment, and Spine Subtype Specificity in Cocaine Regulation of Dendritic Spines in the Nucleus Accumbens. J. Neurosci. 2012, 32, 6957–6966. [Google Scholar] [CrossRef]
- Dobi, A.; Seabold, G.K.; Christensen, C.H.; Bock, R.; Alvarez, V.A. Cocaine-Induced Plasticity in the Nucleus Accumbens Is Cell Specific and Develops without Prolonged Withdrawal. J. Neurosci. 2011, 31, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, B.; Gorny, G.; Li, Y.; Samaha, A.-N.; Robinson, T.E. Amphetamine or cocaine limits the ability of later experience to promote structural plasticity in the neocortex and nucleus accumbens. Proc. Natl. Acad. Sci. USA 2003, 100, 10523–10528. [Google Scholar] [CrossRef] [Green Version]
- Pulipparacharuvil, S.; Renthal, W.; Hale, C.F.; Taniguchi, M.; Xiao, G.; Kumar, A.; Russo, S.J.; Sikder, D.; Dewey, C.M.; Davis, M.M.; et al. Cocaine Regulates MEF2 to Control Synaptic and Behavioral Plasticity. Neuron 2008, 59, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, N.; Lu, K.; Zhang, L.; Gu, J.; Guo, F.; An, S.; Zhang, L.; Zhang, L. Cocaine-induced dendritic remodeling occurs in both D1 and D2 dopamine receptor-expressing neurons in the nucleus accumbens. Neurosci. Lett. 2012, 517, 118–122. [Google Scholar] [CrossRef]
- Lee, K.-W.; Kim, Y.; Kim, A.M.; Helmin, K.; Nairn, A.C.; Greengard, P. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc. Natl. Acad. Sci. USA 2006, 103, 3399–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalba, R.M.; Smith, Y. Striatal Spine Plasticity in Parkinson’s Disease. Front. Neuroanat. 2010, 4, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Kai, L.; Hockberger, P.E.; Wokosin, D.L.; Surmeier, D.J. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell. Neurosci. 2010, 44, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clare, K.; Pan, C.; Kim, G.; Park, K.; Zhao, J.; Volkow, N.D.; Lin, Z.; Du, C. Cocaine Reduces the Neuronal Population While Upregulating Dopamine D2-Receptor-Expressing Neurons in Brain Reward Regions: Sex-Effects. Front. Pharmacol. 2021, 12, 347. [Google Scholar] [CrossRef]
- Friedman, J.H.; Chang, V. Crack cocaine use due to dopamine agonist therapy in Parkinson disease. Neurology 2013, 80, 2269–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mash, D.C.; Ouyang, Q.; Pablo, J.; Basile, M.; Izenwasser, S.; Lieberman, A.; Perrin, R.J. Cocaine Abusers Have an Overexpression of α-Synuclein in Dopamine Neurons. J. Neurosci. 2003, 23, 2564–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, T.; Tomiyama, M.; Wakabayashi, K. Neuropathology of Lewy body disease: Clinicopathological crosstalk between typical and atypical cases. Neuropathology 2020, 40, 30–39. [Google Scholar] [CrossRef]
- Todd, G.; Noyes, C.; Flavel, S.C.; Della Vedova, C.B.; Spyropoulos, P.; Chatterton, B.; Berg, D.; White, J.M. Illicit Stimulant Use Is Associated with Abnormal Substantia Nigra Morphology in Humans. PLoS ONE 2013, 8, e56438. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Bonci, A.; Mercuri, N.B.; De Siena, A.; De Sarro, G.; Malferrari, G.; Diana, M.; Gallelli, L. Cocaine Dependence and Stroke: Pathogenesis and Management. Curr. Neurovascular Res. 2015, 12, 163–172. [Google Scholar] [CrossRef]
- Tamrazi, B.; Almast, J. Your Brain on Drugs: Imaging of Drug-related Changes in the Central Nervous System. RadioGraphics 2012, 32, 701–719. [Google Scholar] [CrossRef] [PubMed]
- Serafini, G.; Piccinini, G.; Visimberga, S.; Cervetti, A.; Belvederi Murri, M.; Monacelli, F.; Pompili, M.; Amore, M. Aripiprazole-Induced Persistent Hiccup: A Case Report and Review of the Literature. Psychiatr. Danub. 2019, 31, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.G.; Tagliarini, C.; Della Rocca, F.; Flamini, W.; Pagni, G.; Tripodi, B.; Marazziti, D.; Maremmani, I. Protracted Hiccups Induced by Aripiprazole and Regressed after Administration of Gabapentin. Case Rep. Psychiatry 2021, 2021, 556152. [Google Scholar] [CrossRef] [PubMed]
- Loonen, A.J.; Ivanova, S.A. Neurobiological mechanisms associated with antipsychotic drug-induced dystonia. J. Psychopharmacol. 2021, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- APA. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
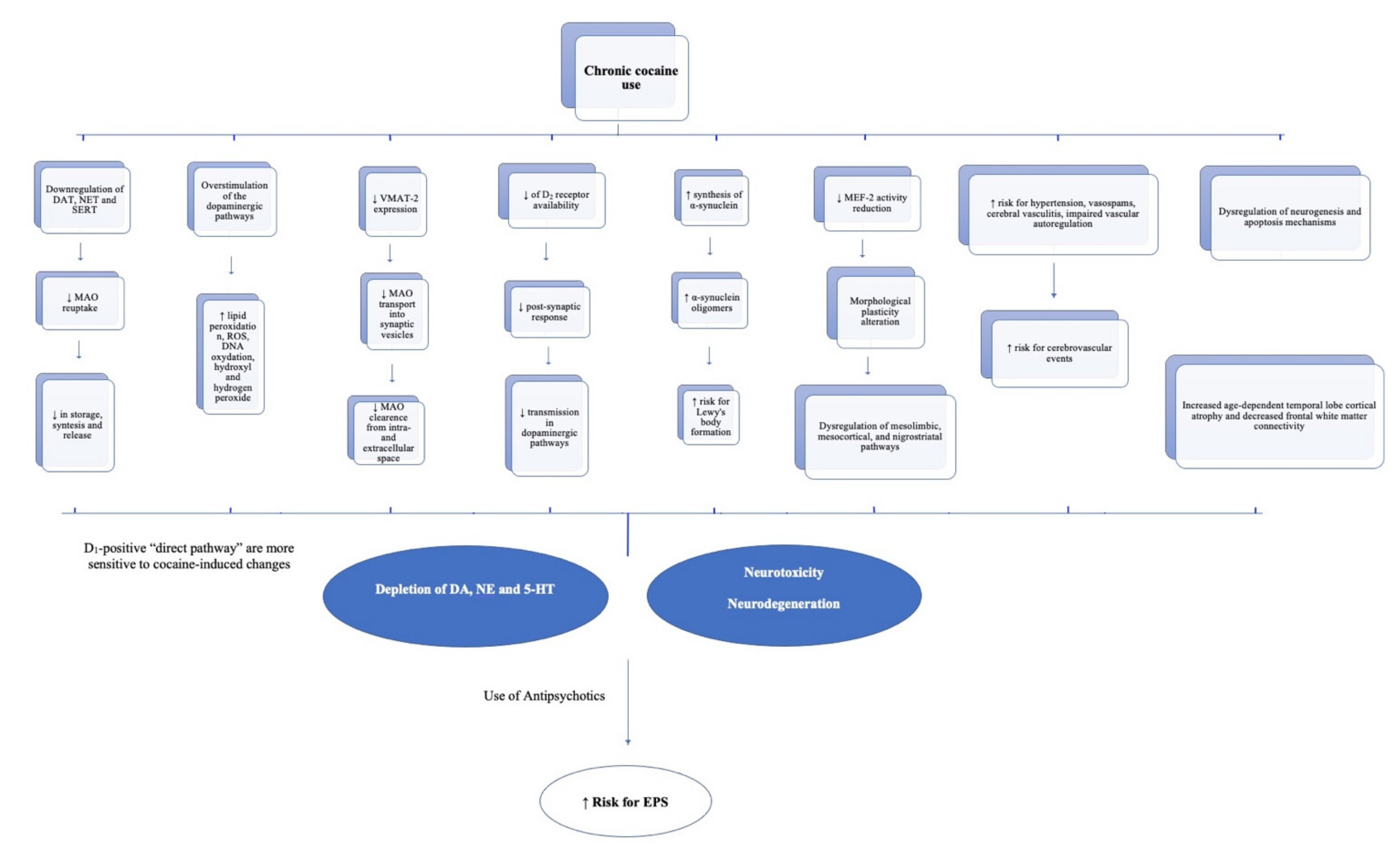

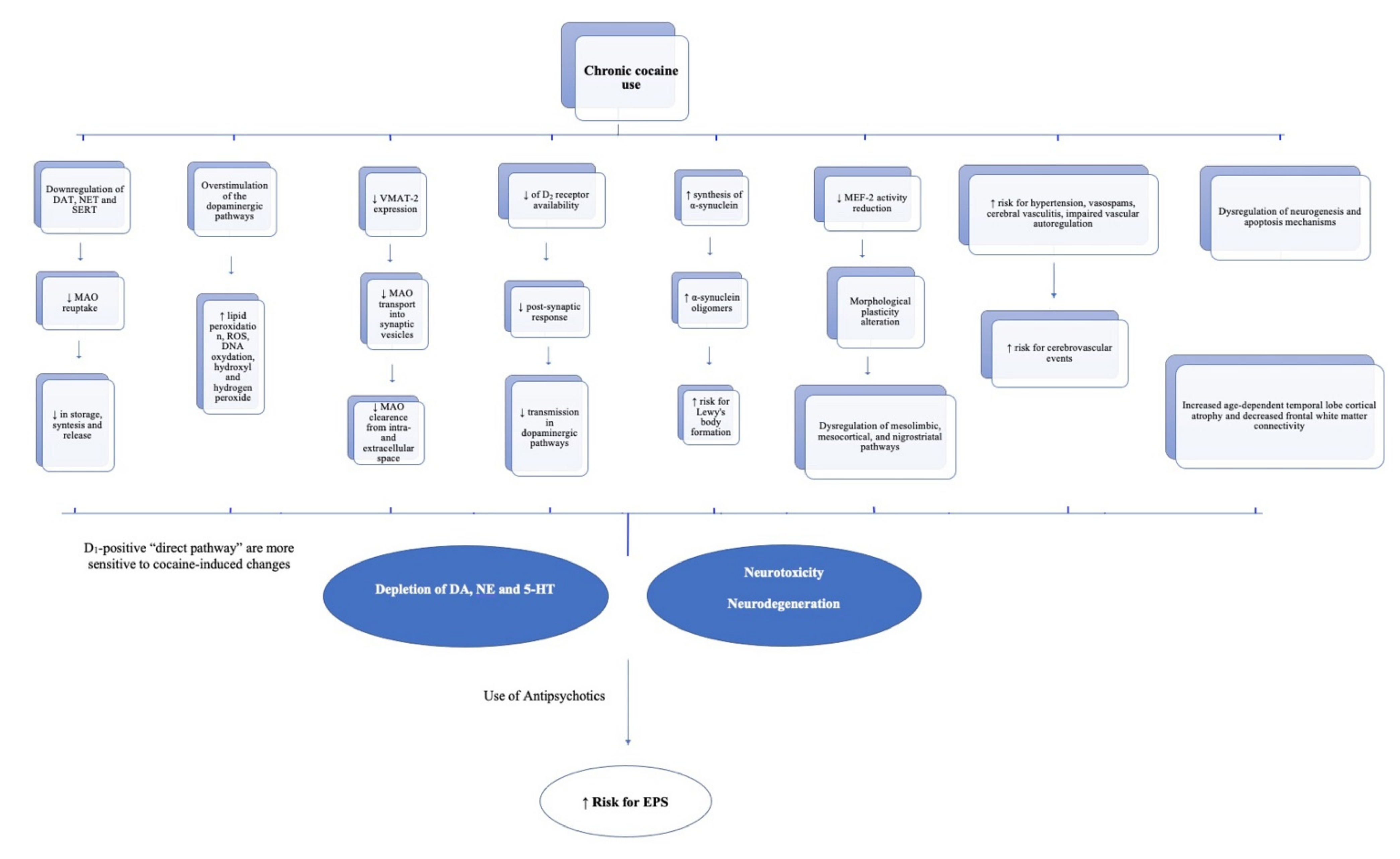{kind=link}
| Drugs | Side Effects | Treatments | Outcomes |
|---|---|---|---|
| Aripiprazole 20 mg/day After the second administration of 10 mg | Protracted hiccups and dyspepsia for 24 h | Chlorpromazine 25 mg and aripiprazole suspension | Gradual resolution in 2–3 h |
| Paliperidone 9 mg/day About 8–10 days after the introduction | Painful dystonia and muscle contractions in the back and neck | Introduction of biperiden and reduction in paliperidone to 6 mg/day | Poor response ↓ paliperidone suspension |
| Haloperidol 3 mg/day About 4–6 days after the introduction | Aggravating akathisia and psychomotor agitation | Biperiden 5 mg IV and diazepam 10 mg IV | Complete resolution |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cenci, D.; Carbone, M.G.; Callegari, C.; Maremmani, I. Psychomotor Symptoms in Chronic Cocaine Users: An Interpretative Model. Int. J. Environ. Res. Public Health 2022, 19, 1897. https://doi.org/10.3390/ijerph19031897
Cenci D, Carbone MG, Callegari C, Maremmani I. Psychomotor Symptoms in Chronic Cocaine Users: An Interpretative Model. International Journal of Environmental Research and Public Health. 2022; 19(3):1897. https://doi.org/10.3390/ijerph19031897
Chicago/Turabian StyleCenci, Davide, Manuel Glauco Carbone, Camilla Callegari, and Icro Maremmani. 2022. "Psychomotor Symptoms in Chronic Cocaine Users: An Interpretative Model" International Journal of Environmental Research and Public Health 19, no. 3: 1897. https://doi.org/10.3390/ijerph19031897
APA StyleCenci, D., Carbone, M. G., Callegari, C., & Maremmani, I. (2022). Psychomotor Symptoms in Chronic Cocaine Users: An Interpretative Model. International Journal of Environmental Research and Public Health, 19(3), 1897. https://doi.org/10.3390/ijerph19031897