
Iron Deficiency and Deranged Myocardial Energetics in Heart Failure

 ,
, {kind=link}
{kind=link}
Abstract
1. Introduction
2. Physiology of Cardiac Energetics
3. Deranged Energy Metabolism in HF
3.1. Adverse Metabolic Myocardial Remodeling in HF
3.2. Mitochondrial Dysfunction in the Failing Heart
4. Iron and Energy Metabolism in the Heart
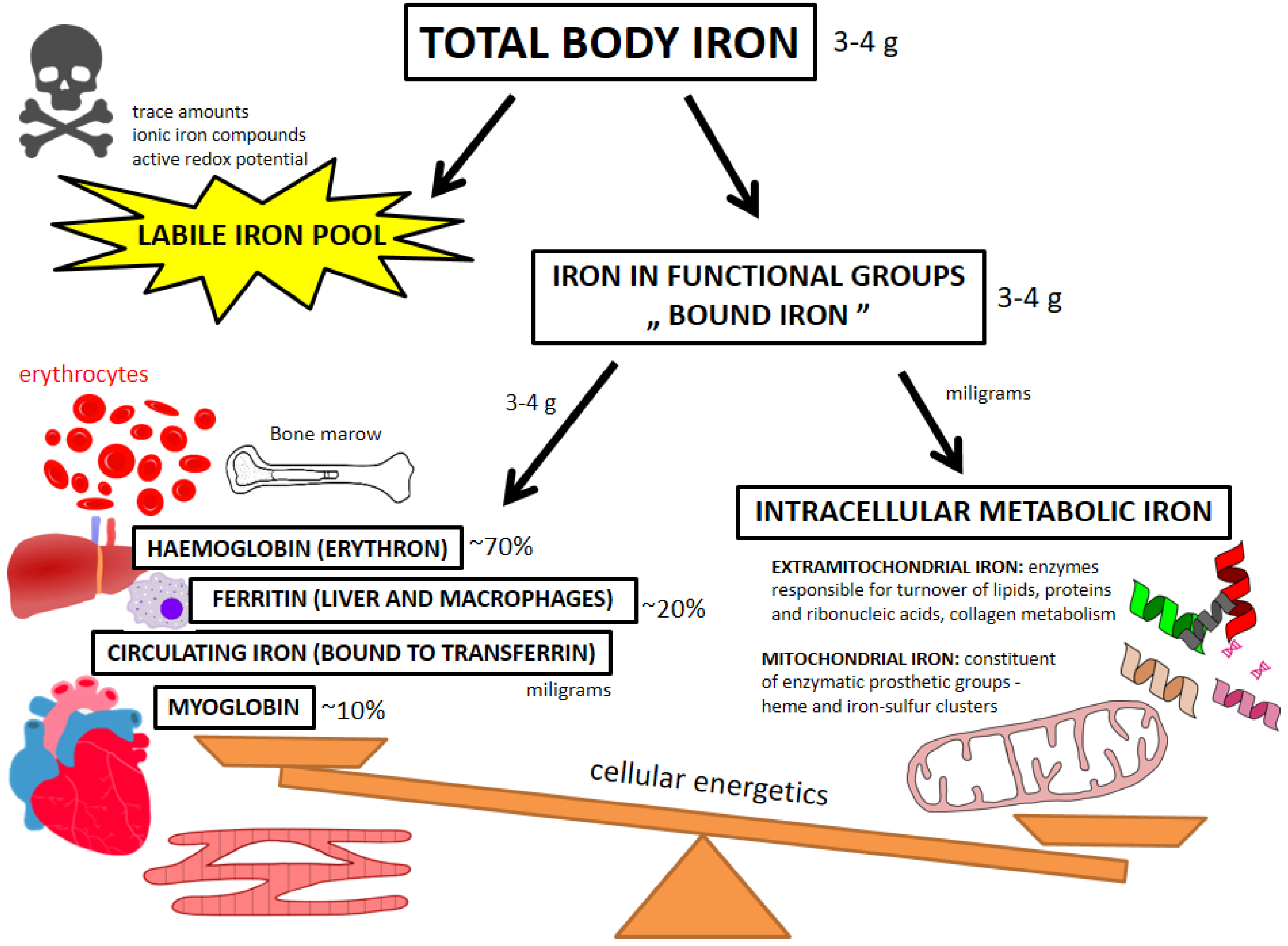
4.1. Beyond Circulating Biomarkers—Pools of Iron
4.2. ID in the Heart—Clinical and Experimental Evidence
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| ATP | adenosine triphosphate |
| CAC | citric acid cycle |
| CK | creatine kinase |
| FAs | fatty acids |
| FCM | ferric carboxymaltose |
| HAMP gene | hepcidin antimicrobial peptide gene (encoding hepcidin) |
| HF | heart failure |
| ID | iron deficiency |
| IRP | iron regulatory proteins |
| LIP | labile iron pool |
| PCr | phosphocreatine |
| ROS | reactive oxygen species |
| sTfR | soluble transferrin receptor |
| TSAT | transferrin saturation (index) |
References
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 2009, 81, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Mettauer, B.; Zoll, J.; Garnier, A.; Ventura-Clapier, R. Heart failure: A model of cardiac and skeletal muscle energetic failure. Pflug. Arch. 2006, 452, 653–666. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Lopez, R.; Marzban, B.; Gao, X.; Lauinger, E.; Van den Bergh, F.; Whitesall, S.E.; Converso-Baran, K.; Burant, C.F.; Michele, D.E.; Beard, D.A. Impaired Myocardial Energetics Causes Mechanical Dysfunction in Decompensated Failing Hearts. Function 2020, 1, zqaa018. [Google Scholar] [CrossRef]
- Tewari, S.G.; Bugenhagen, S.M.; Vinnakota, K.C.; Rice, J.J.; Janssen, P.M.L.; Beard, D.A. Influence of metabolic dysfunction on cardiac mechanics in decompensated hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2016, 94, 162–175. [Google Scholar] [CrossRef]
- Lin, D.; Hollander, Z.; Meredith, A.; Stadnick, E.; Sasaki, M.; Cohen Freue, G.; Qasimi, P.; Mui, A.; Ng, R.T.; Balshaw, R.; et al. Biomarkers in Transplantation Team; & NCE CECR PROOF Centre of Excellence. Molecular signatures of end-stage heart failure. J. Card. Fail. 2011, 17, 867–874. [Google Scholar] [CrossRef]
- van Bilsen, M.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovasc. Res. 2009, 81, 420–428. [Google Scholar] [CrossRef]
- Melenovsky, V.; Hlavata, K.; Sedivy, P.; Dezortova, M.; Borlaug, B.A.; Petrak, J.; Kautzner, J.; Hajek, M. Skeletal Muscle Abnormalities and Iron Deficiency in Chronic Heart Failure. An Exercise 31P Magnetic Resonance Spectroscopy Study of Calf Muscle. Circ. Heart Fail. 2018, 11, e004800. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Zhang, X.; Culver, B.; Chew, H.G., Jr.; Kelley, R.O.; Ren, J. Dietary iron deficiency induces ventricular dilation; mitochondrial ultrastructural aberrations and cytochrome c release: Involvement of nitric oxide synthase and protein tyrosine nitration. Clin. Sci. 2005, 109, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Toblli, J.E.; Cao, G.; Rivas, C.; Giani, J.F.; Dominici, F.P. Intravenous iron sucrose reverses anemia-induced cardiac remodeling; prevents myocardial fibrosis, and improves cardiac function by attenuating oxidative/nitrosative stress and inflammation. Int. J. Cardiol. 2016, 212, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Kirwan, B.A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: A multicentre; double-blind; randomized, controlled. Lancet 2020, 396, 1895–1904, Correction in Lancet 2021, 398, 1964. [Google Scholar] [CrossRef]
- Zhang, H.; Jamieson, K.L.; Grenier, J.; Nikhanj, A.; Tang, Z.; Wang, F.; Wang, S.; Seidman, J.G.; Seidman, C.E.; Thompson, R.; et al. Myocardial Iron Deficiency and Mitochondrial Dysfunction in Advanced Heart Failure in Humans. J. Am. Heart Assoc. 2022, 7, e022853. [Google Scholar] [CrossRef]
- Charles-Edwards, G.; Amaral, N.; Sleigh, A.; Ayis, S.; Catibog, N.; McDonagh, T.; Monaghan, M.; Amin-Youssef, G.; Kemp, G.J.; Shah, A.M.; et al. Effect of Iron Isomaltoside on Skeletal Muscle Energetics in Patients with Chronic Heart Failure and Iron Deficiency. Circulation 2019, 139, 2386–2398. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Bhatt, K.N.; Butler, J. Myocardial Energetics and Heart Failure: A Review of Recent Therapeutic Trials. Curr. Heart Fail. Rep. 2018, 15, 191–197. [Google Scholar] [CrossRef]
- Ingwall, J.S. ATP and the Heart; Springer Science & Business Media: New York, NY, USA, 2002; Volume 11, ISBN 978-1-4613-5391-1. [Google Scholar]
- Randle, P.J. Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Abel, E.D. Glucose transport in the heart. Front. Biosci. 2004, 9, 201–215. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Bisaccia, G.; Ricci, F.; Gallina, S.; Di Baldassarre, A.; Ghinassi, B. Mitochondrial Dysfunction and Heart Disease: Critical Appraisal of an Overlooked Association. Int. J. Mol. Sci. 2021, 22, 614. [Google Scholar] [CrossRef] [PubMed]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol. 2004, 555 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Kurdi, M.; Booz, G.W. Focus on mitochondria dysfunction and dysregulation in heart failure: Towards new therapeutic strategies to improve heart function. Congest. Heart Fail. 2011, 17, 255–256. [Google Scholar] [CrossRef]
- van Bilsen, M.; Smeets, P.J.; Gilde, A.J.; van der Vusse, G.J. Metabolic remodelling of the failing heart: The cardiac burn-out syndrome? Cardiovasc. Res. 2004, 61, 218–226. [Google Scholar] [CrossRef]
- Beauloye, C.; Bertrand, L.; Horman, S.; Hue, L. AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovasc. Res. 2011, 90, 224–233. [Google Scholar] [CrossRef]
- Ardehali, H.; Sabbah, H.N.; Burke, M.A.; Sarma, S.; Liu, P.P.; Cleland, J.G.; Maggioni, A.; Fonarow, G.C.; Abel, E.D.; Campia, U.; et al. Targeting myocardial substrate metabolism in heart failure: Potential for new therapies. Eur. J. Heart Fail. 2012, 14, 120–129. [Google Scholar] [CrossRef]
- Marino, A.; Hausenloy, D.J.; Andreadou, I.; Horman, S.; Bertrand, L.; Beauloye, C. AMP-activated protein kinase: A remarkable contributor to preserve a healthy heart against ROS injury. Free Radic. Biol. Med. 2021, 166, 238–254. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Minicucci, M.F.; Santos, P.P.; Paiva, S.A.; Zornoff, L.A. Energy metabolism in cardiac remodeling and heart failure. Cardiol. Rev. 2013, 21, 135–140. [Google Scholar] [CrossRef]
- González, A.; Ravassa, S.; Beaumont, J.; López, B.; Díez, J. New targets to treat the structural remodeling of the myocardium. J. Am. Coll. Cardiol. 2011, 58, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Turer, A.T. Using metabolomics to assess myocardial metabolism and energetics in heart failure. J. Mol. Cell. Cardiol. 2013, 55, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Atherton, H.; Shockcor, J.; Atzori, L. Metabolomics as a tool for cardiac research. Nat. Rev. Cardiol. 2011, 8, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Starling, R.C.; Hammer, D.F.; Altschuld, R.A. Human myocardial ATP content and in vivo contractile function. Mol. Cell. Biochem. 1998, 180, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal; hypertrophied; and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Shen, W.; Asai, K.; Uechi, M.; Mathier, M.A.; Shannon, R.P.; Vatner, S.F.; Ingwall, J.S. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: A compensatory role for the parallel loss of creatine. Circulation 1999, 100, 2113–2118. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Pauletto, P.; Friedrich, J.; Gwathmey, J.K.; Saks, V.; Pessina, A.C.; Allen, P.D. Creatine kinase system in failing and nonfailing human myocardium. Circulation 1996, 94, 1894–1901. [Google Scholar] [CrossRef]
- Ten Hove, M.; Chan, S.; Lygate, C.; Monfared, M.; Boehm, E.; Hulbert, K.; Watkins, H.; Clarke, K.; Neubauer, S. Mechanisms of creatine depletion in chronically failing rat heart. J. Mol. Cell. Cardiol. 2005, 38, 309–313. [Google Scholar] [CrossRef]
- Neubauer, S.; Remkes, H.; Spindler, M.; Horn, M.; Wiesmann, F.; Prestle, J.; Walzel, B.; Ertl, G.; Hasenfuss, G.; Wallimann, T. Downregulation of the Na+-creatine cotransporter in failing human myocardium and in experimental heart failure. Circulation 1999, 100, 1847–1850. [Google Scholar] [CrossRef]
- Sylvén, C.; Lin, L.; Jansson, E.; Sotonyi, P.; Fu, L.X.; Waagstein, F.; Hjalmarsson, A.; Marcus, C.; Brönnegård, M. Ventricular adenine nucleotide translocator mRNA is upregulated in dilated cardiomyopathy. Cardiovasc. Res. 1993, 27, 1295–1299. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Kelly, D.P. The energy substrate switch during development of heart failure: Gene regulatory mechanisms (Review). Int. J. Mol. Med. 1998, 1, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.A.; Givertz, M.M. Modulation of myocardial energetics: Emerging evidence for a therapeutic target in cardiovascular disease. Circulation 2005, 112, 3218–3221. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.P.; Kerner, J.; Huang, H.; Vazquez, E.; Reszko, A.; Martini, W.Z.; Hoppel, C.L.; Imai, M.; Rastogi, S.; Sabbah, H.N.; et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1538–H1543. [Google Scholar] [CrossRef]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef]
- Opie, L.H.; Knuuti, J. The adrenergic-fatty acid load in heart failure. J. Am. Coll. Cardiol. 2009, 54, 1637–1646. [Google Scholar] [CrossRef]
- Kalsi, K.K.; Smolenski, R.T.; Pritchard, R.D.; Khaghani, A.; Seymour, A.M.; Yacoub, M.H. Energetics and function of the failing human heart with dilated or hypertrophic cardiomyopathy. Eur. J. Clin. Investig. 1999, 29, 469–477. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef]
- Taylor, M.; Wallhaus, T.R.; Degrado, T.R.; Russell, D.C.; Stanko, P.; Nickles, R.J.; Stone, C.K. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in Patients with Congestive Heart Failure. J. Nucl. Med. 2001, 42, 55–62. [Google Scholar] [PubMed]
- Tuunanen, H.; Engblom, E.; Naum, A.; Scheinin, M.; Någren, K.; Airaksinen, J.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Knuuti, J. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: Evidence of relationship with insulin resistance and left ventricular dysfunction. J. Card. Fail. 2006, 12, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Shah, S.; Verma, S.; Oudit, G.Y. Epicardial adipose tissue as a metabolic transducer: Role in heart failure and coronary artery disease. Heart Fail. Rev. 2017, 22, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Jorsal, A.; Iversen, P.; Tolbod, L.; Bouchelouche, K.; Sørensen, J.; Harms, H.J.; Flyvbjerg, A.; Bøtker, H.E.; Wiggers, H. Heart failure patients with prediabetes and newly diagnosed diabetes display abnormalities in myocardial metabolism. J. Nucl. Cardiol. 2018, 25, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Vander Roest, A.S.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Kapadia, S.; Torre-Amione, G.; Durand, J.B.; Bies, R.; Young, J.; Mann, D.L. Differential expression of heat shock proteins in normal and failing human hearts. J. Mol. Cell. Cardiol. 1998, 30, 811–818. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron; an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P. Cellular iron metabolism. Kidney Int. Suppl. 1999, 69, S2–S11. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L. Iron biology in immune function; muscle metabolism and neuronal functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I.; Kakhlon, O.; Epsztejn, S.; Zanninelli, G.; Breuer, W. Intracellular and extracellular labile iron pools. Adv. Exp. Med. Biol. 2002, 509, 55–75. [Google Scholar] [CrossRef]
- Kell, D.B. Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med. Genom. 2009, 2, 2. [Google Scholar] [CrossRef]
- Andrews, N.C. Disorders of iron metabolism. N. Engl. J. Med. 1999, 341, 1986–1995, Erratum in N. Engl. J. Med. 2000, 342, 364. [Google Scholar] [CrossRef]
- Hower, V.; Mendes, P.; Torti, F.M.; Laubenbacher, R.; Akman, S.; Shulaev, V.; Torti, S.V. A general map of iron metabolism and tissue-specific subnetworks. Mol. Biosyst. 2009, 5, 422–443. [Google Scholar] [CrossRef]
- Dunn, L.L.; Suryo Rahmanto, Y.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef]
- Rouault, T.A.; Tong, W.H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef]
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal. 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kölker, S.; Gröne, H.J.; Hentze, M.W. Iron regulatory proteins secure mitochondrial iron sufficiency and function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S. Iron-regulatory proteins: Molecular biology and pathophysiological implications. Expert Rev. Mol. Med. 2007, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Paterek, A.; Mackiewicz, U.; Mączewski, M. Iron and the heart: A paradigm shift from systemic to cardiomyocyte abnormalities. J. Cell. Physiol. 2019, 234, 21613–21629. [Google Scholar] [CrossRef]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of iron metabolism in heart failure: From iron deficiency to iron overload. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef]
- Ghafourian, K.; Shapiro, J.S.; Goodman, L.; Ardehali, H. Iron and Heart Failure: Diagnosis, Therapies, and Future Directions. JACC Basic Transl. Sci. 2020, 5, 300–313. [Google Scholar] [CrossRef]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359, Erratum in N. Engl. J. Med. 2012, 366, 771. [Google Scholar] [CrossRef]
- Alnuwaysir, R.I.S.; Hoes, M.F.; van Veldhuisen, D.J.; van der Meer, P.; Grote Beverborg, N. Iron Deficiency in Heart Failure: Mechanisms and Pathophysiology. J. Clin. Med. 2021, 11, 125. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.A.; Miller, L.R.; Inamdar, A.R.; Person, R.; Seiler, K.; Mackler, B. Iron deficiency in the rat. Physiological and biochemical studies of muscle dysfunction. J. Clin. Investig. 1976, 58, 447–453. [Google Scholar] [CrossRef] [PubMed]
- McLane, J.A.; Fell, R.D.; McKay, R.H.; Winder, W.W.; Brown, E.B.; Holloszy, J.O. Physiological and biochemical effects of iron deficiency on rat skeletal muscle. Am. J. Physiol. 1981, 241, C47–C54. [Google Scholar] [CrossRef] [PubMed]
- Willis, W.T.; Brooks, G.A.; Henderson, S.A.; Dallman, P.R. Effects of iron deficiency and training on mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 1987, 62, 2442–2446. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Maguire, J.J.; Brooks, G.A.; Dallman, P.R.; Packer, L. Muscle mitochondrial bioenergetics; oxygen supply; and work capacity during dietary iron deficiency and repletion. Am. J. Physiol. 1982, 242, E418–E427. [Google Scholar] [CrossRef]
- Jankowska, E.A.; von Haehling, S.; Anker, S.D.; Macdougall, I.C.; Ponikowski, P. Iron deficiency and heart failure: Diagnostic dilemmas and therapeutic perspectives. Eur. Heart J. 2013, 34, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Manceau, H.; Ausseil, J.; Masson, D.; Feugeas, J.P.; Sablonniere, B.; Guieu, R.; Puy, H.; Peoc’h, K. Neglected Comorbidity of Chronic Heart Failure: Iron Deficiency. Nutrients 2022, 14, 3214. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; von Haehling, S.; Doehner, W.; Banasiak, W.; et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J. Card. Fail. 2011, 17, 899–906. [Google Scholar] [CrossRef]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582. [Google Scholar] [CrossRef]
- Enjuanes, C.; Klip, I.T.; Bruguera, J.; Cladellas, M.; Ponikowski, P.; Banasiak, W.; van Veldhuisen, D.J.; van der Meer, P.; Jankowska, E.A.; Comín-Colet, J. Iron deficiency and health-related quality of life in chronic heart failure: Results from a multicenter European study. Int. J. Cardiol. 2014, 174, 268–275. [Google Scholar] [CrossRef]
- Sindone, A.; Doehner, W.; Manito, N.; McDonagh, T.; Cohen-Solal, A.; Damy, T.; Núñez, J.; Pfister, O.; van der Meer, P.; Comin-Colet, J. Practical Guidance for Diagnosing and Treating Iron Deficiency in Patients with Heart Failure: Why, Who and How? J. Clin. Med. 2022, 11, 2976. [Google Scholar] [CrossRef] [PubMed]
- Beale, A.L.; Warren, J.L.; Roberts, N.; Meyer, P.; Townsend, N.P.; Kaye, D. Iron deficiency in heart failure with preserved ejection fraction: A systematic review and meta-analysis. Open Heart 2019, 6, e001012. [Google Scholar] [CrossRef] [PubMed]
- Alcaide-Aldeano, A.; Garay, A.; Alcoberro, L.; Jiménez-Marrero, S.; Yun, S.; Tajes, M.; García-Romero, E.; Díez-López, C.; González-Costello, J.; Mateus-Porta, G.; et al. Iron Deficiency: Impact on Functional Capacity and Quality of Life in Heart Failure with Preserved Ejection Fraction. J. Clin. Med. 2020, 9, 1199. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Thomas, L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin. Chem. 2002, 48, 1066–1076. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Wojtas, K.; Kasztura, M.; Mazur, G.; Butrym, A.; Kalicinska, E.; Rybinska, I.; Skiba, J.; von Haehling, S.; Doehner, W.; et al. Bone marrow iron depletion is common in patients with coronary artery disease. Int. J. Cardiol. 2015, 182, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Grote Beverborg, N.; Klip, I.T.; Meijers, W.C.; Voors, A.A.; Vegter, E.L.; van der Wal, H.H.; Swinkels, D.W.; van Pelt, J.; Mulder, A.B.; Bulstra, S.K.; et al. Definition of Iron Deficiency Based on the Gold Standard of Bone Marrow Iron Staining in Heart Failure Patients. Circ. Heart Fail. 2018, 11, e004519. [Google Scholar] [CrossRef]
- Leszek, P.; Sochanowicz, B.; Szperl, M.; Kolsut, P.; Brzóska, K.; Piotrowski, W.; Rywik, T.M.; Danko, B.; Polkowska-Motrenko, H.; Różański, J.M.; et al. Myocardial iron homeostasis in advanced chronic heart failure patients. Int. J. Cardiol. 2012, 159, 47–52. [Google Scholar] [CrossRef]
- Anker, S.D.; Kirwan, B.A.; van Veldhuisen, D.J.; Filippatos, G.; Comin-Colet, J.; Ruschitzka, F.; Lüscher, T.F.; Arutyunov, G.P.; Motro, M.; Mori, C.; et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: An individual patient data meta-analysis. Eur. J. Heart Fail. 2018, 20, 125–133. [Google Scholar] [CrossRef]
- Haddad, S.; Wang, Y.; Galy, B.; Korf-Klingebiel, M.; Hirsch, V.; Baru, A.M.; Rostami, F.; Reboll, M.R.; Heineke, J.; Flögel, U.; et al. Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur. Heart J. 2017, 38, 362–372. [Google Scholar] [CrossRef]
- Maeder, M.T.; Khammy, O.; dos Remedios, C.; Kaye, D.M. Myocardial and systemic iron depletion in heart failure implications for anemia accompanying heart failure. J. Am. Coll. Cardiol. 2011, 58, 474–480. [Google Scholar] [CrossRef]
- Hirsch, V.G.; Tongers, J.; Bode, J.; Berliner, D.; Widder, J.D.; Escher, F.; Mutsenko, V.; Chung, B.; Rostami, F.; Guba-Quint, A.; et al. Cardiac iron concentration in relation to systemic iron status and disease severity in non-ischaemic heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2020, 22, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Kobak, K.A.; Radwańska, M.; Dzięgała, M.; Kasztura, M.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Structural and functional abnormalities in iron-depleted heart. Heart Fail. Rev. 2019, 24, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Dziegala, M.; Kasztura, M.; Kobak, K.; Bania, J.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Influence of the availability of iron during hypoxia on the genes associated with apoptotic activity and local iron metabolism in rat H9C2 cardiomyocytes and L6G8C5 skeletal myocytes. Mol. Med. Rep. 2016, 14, 3969–3977. [Google Scholar] [CrossRef]
- Kasztura, M.; Dzięgała, M.; Kobak, K.; Bania, J.; Mazur, G.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Both iron excess and iron depletion impair viability of rat H9C2 cardiomyocytes and L6G8C5 myocytes. Kardiol. Pol. 2017, 75, 267–275. [Google Scholar] [CrossRef]
- Dziegala, M.; Kobak, K.A.; Kasztura, M.; Bania, J.; Josiak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Iron Depletion Affects Genes Encoding Mitochondrial Electron Transport Chain and Genes of Non-Oxidative Metabolism, Pyruvate Kinase and Lactate Dehydrogenase, in Primary Human Cardiac Myocytes Cultured upon Mechanical Stretch. Cells 2018, 7, 175. [Google Scholar] [CrossRef]
- Chung, Y.J.; Luo, A.; Park, K.C.; Loonat, A.A.; Lakhal-Littleton, S.; Robbins, P.A.; Swietach, P. Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 2019, 4, e125618. [Google Scholar] [CrossRef] [PubMed]
- Petering, D.H.; Stemmer, K.L.; Lyman, S.; Krezoski, S.; Petering, H.G. Iron deficiency in growing male rats: A cause of development of cardiomyopathy. Ann. Nutr. Metab. 1990, 34, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Blayney, L.; Bailey-Wood, R.; Jacobs, A.; Henderson, A.; Muir, J. The effects of iron deficiency on the respiratory function and cytochrome content of rat heart mitochondria. Circ. Res. 1976, 39, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.M.; Beard, J.L. Dietary iron deficiency results in cardiac eccentric hypertrophy in rats. Proc. Soc. Exp. Biol. Med. 1998, 218, 370–375. [Google Scholar] [CrossRef]
- Naito, Y.; Tsujino, T.; Matsumoto, M.; Sakoda, T.; Ohyanagi, M.; Masuyama, T. Adaptive response of the heart to long-term anemia induced by iron deficiency. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H585–H593. [Google Scholar] [CrossRef]
- Rossi, M.A.; Carillo, S.V. Cardiac hypertrophy due to pressure and volume overload: Distinctly different biological phenomena? Int. J. Cardiol. 1991, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Wu, J.; Zhang, Q.; Ye, Y.; Wang, S.; Huang, J.; Liu, H.; Wang, X.; Zhang, W.; Bu, L.; et al. Differential cardiac hypertrophy and signaling pathways in pressure versus volume overload. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, 552–562. [Google Scholar] [CrossRef]
- Kubalova, Z.; Terentyev, D.; Viatchenko-Karpinski, S.; Nishijima, Y.; Györke, I.; Terentyeva, R.; da Cuñha, D.N.; Sridhar, A.; Feldman, D.S.; Hamlin, R.L.; et al. Abnormal intrastore calcium signaling in chronic heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 14104–14109. [Google Scholar] [CrossRef] [PubMed]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife 2016, 5, e19804. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Jairaj Naik, T.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Tkaczyszyn, M.; Suchocki, T.; Drozd, M.; von Haehling, S.; Doehner, W.; Banasiak, W.; Filippatos, G.; Anker, S.D.; Ponikowski, P. Effects of intravenous iron therapy in iron-deficient patients with systolic heart failure: A meta-analysis of randomized controlled trials. Eur. J. Heart Fail. 2016, 18, 786–795. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkaczyszyn, M.; Górniak, K.M.; Lis, W.H.; Ponikowski, P.; Jankowska, E.A. Iron Deficiency and Deranged Myocardial Energetics in Heart Failure. Int. J. Environ. Res. Public Health 2022, 19, 17000. https://doi.org/10.3390/ijerph192417000
Tkaczyszyn M, Górniak KM, Lis WH, Ponikowski P, Jankowska EA. Iron Deficiency and Deranged Myocardial Energetics in Heart Failure. International Journal of Environmental Research and Public Health. 2022; 19(24):17000. https://doi.org/10.3390/ijerph192417000
Chicago/Turabian StyleTkaczyszyn, Michał, Krzysztof Michał Górniak, Weronika Hanna Lis, Piotr Ponikowski, and Ewa Anita Jankowska. 2022. "Iron Deficiency and Deranged Myocardial Energetics in Heart Failure" International Journal of Environmental Research and Public Health 19, no. 24: 17000. https://doi.org/10.3390/ijerph192417000
APA StyleTkaczyszyn, M., Górniak, K. M., Lis, W. H., Ponikowski, P., & Jankowska, E. A. (2022). Iron Deficiency and Deranged Myocardial Energetics in Heart Failure. International Journal of Environmental Research and Public Health, 19(24), 17000. https://doi.org/10.3390/ijerph192417000