


Metagenomic Analysis Reveals the Response of Microbial Communities and Their Functions in Lake Sediment to Environmental Factors

 ,
,
Abstract
1. Introduction
2. Materials and Methods
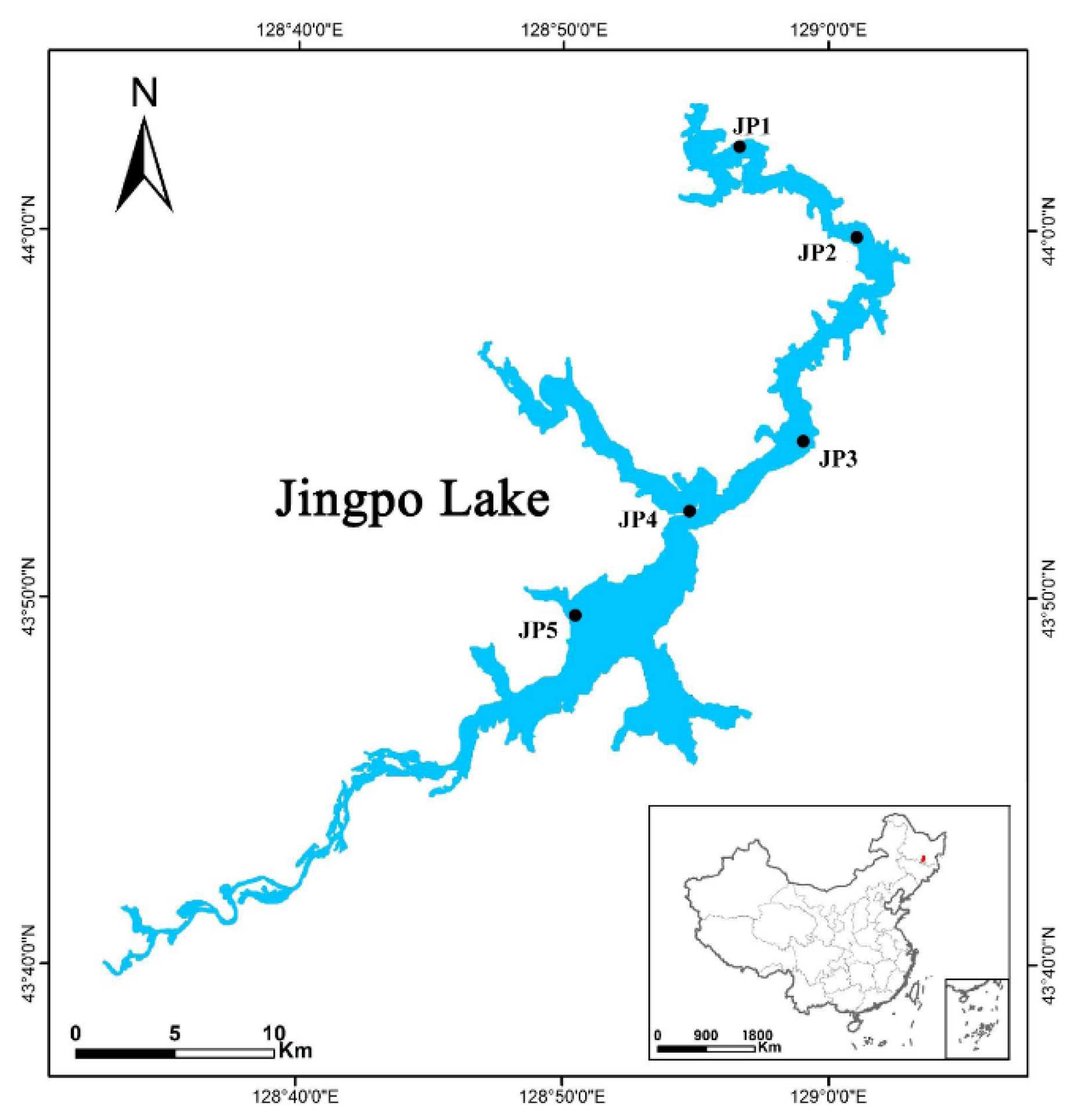
2.1. Study Site Description and Sampling Procedure
2.2. DNA Extraction and Metagenomic Sequencing
2.3. Bioinformatic Analysis
2.4. Statistical Analysis
3. Results
3.1. Physicochemical Characteristics of the Sampling Sites
3.2. Microbial Community Composition
3.3. Differences in Microbial Communities of Sampling Sites
3.4. Differences in the KEGG Function of Sampling Sites
3.5. Correlations between Physicochemical Factors, Microbial Community and KEGG Metabolism Function
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | JP1 | JP2 | JP3 | JP4 |
|---|---|---|---|---|
| Eigenvalues | 0.3721 | 0.1083 | 0.0114 | 0.0362 |
| Decorana values | 0.4139 | 0.0354 | 0.0038 | 0.0016 |
| Axis lengths | 2.3403 | 1.1780 | 0.8193 | 0.7493 |

References
- Davis, J.; O’Grady, A.P.; Dale, A.; Arthington, A.H.; Gell, P.A.; Driver, P.D.; Bond, N.; Casanova, M.; Finlayson, M.; Watts, R.J.; et al. When trends intersect: The challenge of protecting freshwater ecosystems under multiple land use and hydrological intensification scenarios. Sci. Total Environ. 2015, 534, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Feng, K.; Du, X.; Yuan, J.; Liu, J.; Li, Z. Effects of land use and environmental gradients on the taxonomic and functional diversity of rotifer assemblages in lakes along the Yangtze River, China. Ecol. Indic. 2022, 142, 109199. [Google Scholar] [CrossRef]
- Walther, G.-R.; Post, E.; Convey, P.; Menzel, A.; Parmesan, C.; Beebee, T.J.C.; Fromentin, J.-M.; Hoegh-Guldberg, O.; Bairlein, F. Ecological responses to recent climate change. Nature 2002, 416, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Parmesan, C.; Yohe, G. A globally coherent fingerprint of climate change impacts across natural systems. Nature 2003, 421, 37–72. [Google Scholar] [CrossRef]
- Cross, W.F.; Hood, J.M.; Benstead, J.P.; Huryn, A.D.; Nelson, D. Interactions between temperature and nutrients across levels of ecological organization. Glob. Change Biol. 2015, 21, 1025–1040. [Google Scholar] [CrossRef]
- IPCC. Summary for Policymakers. In Climate Change 2013: The Physical Science Basis; Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK, 2013; pp. 3–29. [Google Scholar]
- Ficke, A.D.; Myrick, C.A.; Hansen, L.J. Potential impacts of global climate change on freshwater fisheries. Rev. Fish Biol. Fish. 2007, 17, 581–613. [Google Scholar] [CrossRef]
- Bai, Y.; Ochuodho, T.O.; Yang, J. Impact of land use and climate change on water-related ecosystem services in Kentucky, USA. Ecol. Indic. 2019, 102, 51–64. [Google Scholar] [CrossRef]
- Crain, C.M.; Kroeker, K.; Halpern, B.S. Interactive and cumulative effects of multiple human stressors in marine systems. Ecol. Lett. 2008, 11, 1304–1315. [Google Scholar] [CrossRef]
- Zaharescu, D.G.; Burghelea, C.I.; Hooda, P.S.; Lester, R.N.; Palanca-Soler, A. Small lakes in big landscape: Multi-scale drivers of littoral ecosystem in alpine lakes. Sci. Total Environ. 2016, 551–552, 496–505. [Google Scholar] [CrossRef]
- Heino, J.; Alahuhta, J.; Bini, L.M.; Cai, Y.; Heiskanen, A.S.; Hellsten, S.; Kortelainen, P.; Kotamäki, N.; Tolonen, K.T.; Vihervaara, P.; et al. Lakes in the era of global change: Moving beyond single-lake thinking in maintaining biodiversity and ecosystem services. Biol. Rev. 2021, 96, 89–106. [Google Scholar] [CrossRef]
- Han, B.; Addo, F.G.; Mu, X.; Zhang, L.; Zhang, S.; Lv, X.; Li, X.; Wang, P.; Wang, C. Epiphytic bacterial community shift drives the nutrient cycle during Potamogeton malaianus decomposition. Chemosphere 2019, 236, 124253. [Google Scholar] [CrossRef] [PubMed]
- Gomez Isaza, D.F.; Cramp, R.L.; Franklin, C.E. Living in polluted waters: A meta-analysis of the effects of nitrate and interactions with other environmental stressors on freshwater taxa. Environ. Pollut. 2020, 261, 114091. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, K.; Asakawa, M.; Anzai, Y.; Sumino, T.; Harada, K. Degradation of microcystins using immobilized microorganism isolated in an eutrophic lake. Chemosphere 2006, 65, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef]
- Han, X.; Schubert, C.J.; Fiskal, A.; Dubois, N.; Lever, M.A. Eutrophication as a driver of microbial community structure in lake sediments. Environ. Microbiol. 2020, 22, 3446–3462. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; Zhao, L.; Li, Y.; Xie, S.; Liu, Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L.; Jiang, H.; Wu, G.; Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016, 6, 25078. [Google Scholar] [CrossRef]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef]
- Zhang, W.; Shen, J.; Wang, J. Linking pollution to biodiversity and ecosystem multi functionality across benthic-pelagic habitats of a large eutrophic lake: A whole ecosystem perspective. Environ. Pollut. 2021, 285, 117501. [Google Scholar] [CrossRef]
- Dai, Y.; Wu, J.; Zhong, F.; Cui, N.; Kong, L.; Liang, W.; Cheng, S. Macrophyte identity shapes water column and sediment bacterial community. Hydrobiologia 2019, 835, 71–82. [Google Scholar] [CrossRef]
- Geng, M.; Zhang, W.; Hu, T.; Wang, R.; Cheng, X.; Wang, J. Eutrophication causes microbial community homogenization via modulating generalist species. Water Res. 2022, 210, 118003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Ge, F.; Peng, X.; Li, X.; Zhang, X.; Zhang, S.; Zhou, Q.; Wu, Z.; Liu, B. Spatial characteristics of nitrogen forms in a large degenerating lake: Its relationship with dissolved organic matter and microbial community. J. Clean. Prod. 2022, 371, 133617. [Google Scholar] [CrossRef]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Poté, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Huang, R.; Zeng, J.; Yan, W.; Wang, J.; Ma, T.; Wang, M.; Wu, Q.L. Diversity analysis of bacterial community compositions in sediments of urban lakes by terminal restriction fragment length polymorphism (T-RFLP). World J. Microbiol. Biotechnol. 2012, 28, 3159–3170. [Google Scholar] [CrossRef] [PubMed]
- Monteux, S.; Keuper, F.; Fontaine, S.; Gavazov, K.; Hallin, S.; Juhanson, J.; Krab, E.J.; Revaillot, S.; Verbruggen, E.; Walz, J.; et al. Carbon and nitrogen cycling in Yedoma permafrost controlled by microbial functional limitations. Nat. Geosci. 2020, 13, 794–798. [Google Scholar] [CrossRef]
- Jing, Z.; Jiao, Y.; Zhou, Y.; Chen, X. Encyclopedia of Rivers and Lakes in China Heilongjiang River and Liaohe River; China Water Power Press: Beijing, China, 2014. [Google Scholar]
- Du, X.; Song, D.; Ming, K.; Yang, J.; Jin, X.; Wang, H.; Liu, H.; Wang, L.; Zhao, C.; Huo, T. Functional Responses of Phytoplankton Assemblages to Watershed Land Use and Environmental Gradients. Front. Ecol. Evol. 2022, 9, 819252. [Google Scholar] [CrossRef]
- Dai, C.; Wang, S.; Li, Z.; Zhang, Y.; Gao, Y.; Li, C. Review on hydrological geography in Heilongjiang River Basin. Acta Geogr. Sin. 2015, 70, 1823–1834, (In Chinese with English abstract). [Google Scholar]
- Wang, X.; Liu, L.; Li, L.; Zhou, J.; Wang, Y.; Xia, F.; Xia, Y. Correlation analysis of algae composition and environmental factors in Jingpo Lake. China Environ. Sci. 2015, 35, 3403–3413, (In Chinese with English Abstract). [Google Scholar]
- Song, D.; Du, X.; Jin, X.; Liu, H.; Ming, K.; Wang, L.; Wang, H.; Zhao, C.; Huo, T. Hydroacoustic assessment of spatio-temporal distribution and biomass of fish resources in Jingpo Lake. J. Lake Sci. 2022, 34, 2095–2104, (In Chinese with English Abstract). [Google Scholar]
- Zhang, C.; Nie, S.; Liang, J.; Zeng, G.; Wu, H.; Hua, S.; Liu, J.; Yuan, Y.; Xiao, H.; Deng, L.; et al. Effects of heavy metals and soil physicochemical properties on wetland soil microbial biomass and bacterial community structure. Sci. Total Environ. 2016, 557–558, 785–790. [Google Scholar] [CrossRef]
- Ou, Y.; Rousseau, A.N.; Wang, L.; Yan, B.; Gumiere, T.; Zhu, H. Identification of the alteration of riparian wetland on soil properties, enzyme activities and microbial communities following extreme flooding. Geoderma 2019, 337, 825–833. [Google Scholar] [CrossRef]
- Fraterrigo, J.; Downing, J. The Influence of Land Use on Lake Nutrients Varies with Watershed Transport Capacity. Ecosystems 2008, 11, 1021–1034. [Google Scholar] [CrossRef]
- Jeppesen, E.; Moss, B.; Bennion, H.; Carvalho, L.; DeMeester, L.; Feuchtmayr, H.; Friberg, N.; Gessner, M.O.; Hefting, M.; Lauridsen, T.L.; et al. Interaction of Climate Change and Eutrophication. In Climate Change Impacts on Freshwater Ecosystems; Kernan, M., Battarbee, R.W., Moss, B., Eds.; Wiley-Blackwell: Oxford, UK, 2010; pp. 119–151. [Google Scholar]
- Le Moal, M.; Gascuel-Odoux, C.; Ménesguen, A.; Souchon, Y.; Étrillard, C.; Levain, A.; Moatar, F.; Pannard, A.; Souchu, P.; Lefebvre, A.; et al. Eutrophication: A new wine in an old bottle? Sci. Total Environ. 2019, 651, 1–11. [Google Scholar] [CrossRef]
- APHA. Standard Methods for the Examination of Water and Wastewater, 18th ed.; American Public Health Association: Washington, DC, USA, 1992. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 25 October 2022).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.5-7. Available online: https://CRAN.R-project.org/package=vegan.2020 (accessed on 25 October 2022).
- Moser, K.A.; Baron, J.S.; Brahney, J.; Oleksy, I.A.; Saros, J.E.; Hundey, E.J.; Sadro, S.; Kopáček, J.; Sommaruga, R.; Kainz, M.J.; et al. Mountain lakes: Eyes on global environmental change. Glob. Planet. Change 2019, 178, 77–95. [Google Scholar] [CrossRef]
- Lin, S.S.; Shen, S.L.; Zhou, A.; Lyu, H.M. Assessment and management of lake eutrophication: A case study in lake Erhai, China. Sci. Total Environ. 2020, 751, 141618. [Google Scholar] [CrossRef]
- Liu, M.H.; Fang, R.D.; Zhao, J.P.; Wang, K. Length-weight relationships for five fish species from Jingpo Lake, Heilongjiang, China. J. Appl. Ichthyol. 2016, 32, 151–152. [Google Scholar] [CrossRef]
- Li, C.-H.; Ye, C.; Li, J.-X.; Wei, W.-W.; Zheng, Y.; Kong, M.; Wang, H. Impact of Spring Freshet Flooding and Summer Rainfall Flooding on the Water Quality of an Alpine Barrier Lake. Environ. Sci. Eur. 2019, 32, 57. [Google Scholar] [CrossRef]
- Banda, J.F.; Zhang, Q.; Ma, L.; Pei, L.; Du, Z.; Hao, C.; Dong, H. Both pH and salinity shape the microbial communities of the lakes in Badain Jaran Desert, NW China. Sci. Total Environ. 2021, 791, 148108. [Google Scholar] [CrossRef]
- Yan, H.; Huang, Y.; Wang, G. Water eutrophication evaluation based on rough set and petri nets: A case study in Xiangxi-River, Three Gorges Reservoir. Ecol. Indic. 2016, 69, 463–472. [Google Scholar] [CrossRef]
- Oueriaghli, N.; Castro, D.J.; Llamas, I.; Bejar, V.; Martinez-Checa, F. Study of Bacterial Community Composition and Correlation of Environmental Variables in Rambla Salada, a Hypersaline Environment in South-Eastern Spain. Front. Microbiol. 2018, 9, 1377. [Google Scholar] [CrossRef]
- Lau, N.S.; Zarkasi, K.Z.; Md Sah, A.S.R.; Shu-Chien, A.C. Diversity and Coding Potential of the Microbiota in the Photic and Aphotic Zones of Tropical Man-Made Lake with Intensive Aquaculture Activities: A Case Study on Temengor Lake, Malaysia. Microb. Ecol. 2019, 78, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Yannarell, A.C.; Triplett, E.W. Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 2005, 71, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.J.; Wei, W.W.; Li, C.H.; Ye, C.; Wang, H. Classification of littoral zone and buffer zone in a typical mountain barrier lake and the scheme of ecological restoration: A case study of Jingpo Lake. J. Environ. Eng. Technol. 2021, 11, 1147–1153. [Google Scholar]
- Huang, Y.Y.; Qu, L.Y.; Qu, X.C.; Du, S.Q. Soil microbial community characteristics under different vegetation types at the Holocene-basalt Platform, Jingpo Lake area, Northeast China. Acta Ecol. Sin. 2021, 32, 2827–2836, (In Chinese with English abstract). [Google Scholar] [CrossRef][Green Version]
- Dai, Y.; Yang, Y.; Wu, Z.; Feng, Q.; Xie, S.; Liu, Y. Spatiotemporal variation of planktonic and sediment bacterial assemblages in two plateau freshwater lakes at different trophic status. Appl. Microbiol. Biotechnol. 2016, 100, 4161–4175. [Google Scholar] [CrossRef]
- Yu, S.; He, R.; Song, A.; Huang, Y.; Jin, Z.; Liang, Y.; Li, Q.; Wang, X.; Müller, W.E.G.; Cao, J. Spatial and temporal dynamics of bacterioplankton community composition in a subtropical dammed karst river of southwestern China. Microbiologyopen 2019, 8, e00849. [Google Scholar] [CrossRef] [PubMed]
- Drury, B.; Rosi-Marshall, E.; Kelly, J.J. Wastewater treatment effluent reduces the abundance and diversity of benthic bacterial communities in urban and suburban rivers. Appl. Environ. Microbiol. 2013, 79, 1897–1905. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Niizuma, S.; Kuroda, N.; Sakamoto, M. Occurrence of heterotrophic bacteria causing lysis of cyanobacteria in a eutrophic lake. Jpn. J. Phycol. 1993, 41, 215–220. [Google Scholar]
- Rahman, K.S.; Thahira-Rahman, J. Towards efficient crude oil degradation by a mixed bacterial consortium. Bioresour. Technol. 2002, 85, 257–261. [Google Scholar] [CrossRef]
- Berg, K.; Lyra, C.; Sivonen, K.; Paulin, L.; Suomalainen, S.; Tuomi, P.; Rapala, J. High diversity of cultivable heterotrophic bacteria in association with cyanobacterial water blooms. ISME J. 2009, 3, 314–325. [Google Scholar] [CrossRef]
- Battin, T.J.; Wille, A.; Sattler, B.; Psenner, R. Phylogenetic and functional heterogeneity of sediment biofilms along environmental gradients in a glacial stream. Appl. Environ. Microbiol. 2001, 67, 799. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, J.B.; Roberto, M.D.C.; Thomaz, A.P. Large scale distribution of bacterial communities in the upper Paraná River floodplain. Braz. J. Microbiol. 2014, 45, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Pati, B.R. A comparative study on phyllosphere nitrogen fixation by newly isolated Corynebacterium sp. & Flavobacterium sp. and their potentialities as biofertilizer. Acta Microbiol. Immunol. Hung. 2004, 51, 47–56. [Google Scholar] [PubMed]
- Oluseyi Osunmakinde, C.; Selvarajan, R.; Mamba, B.B.; Msagati, T. Profiling bacterial diversity and potential pathogens in wastewater treatment plants using high-throughput sequencing analysis. Microorganisms 2019, 7, 506. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.J.; Hou, T.T.; Li, X.J.; Feng, L.L.; Wang, Z.Z. Metagenomic insights into comparative study of nitrogen metabolic potential and microbial community between primitive and urban river sediments. Environ. Res. 2022, 212, 113592. [Google Scholar] [CrossRef]
- Rathour, R.; Gupta, J.; Mishra, A.; Rajeev, A.C.; Dupont, C.L.; Thakur, I.S. A comparative metagenomic study reveals microbial diversity and their role in the biogeochemical cycling of Pangong Lake. Sci. Total Environ. 2020, 731, 139074. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H.; Wang, L.; Liu, R.; Fu, L.; Lin, K. Unique bacterial communities and potential function along the vertical gradient in the deepest marine blue hole. Environ. Microbiol. Rep. 2021, 13, 911–927. [Google Scholar] [CrossRef]
- Gavriilidou, A.; Gutleben, J.; Versluis, D.; Forgiarini, F.; Van Passel, M.W.J.; Ingham, C.J.; Smidt, H.; Sipkema, D. Comparative genomic analysis of Flavobacteriaceae: Insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genom. 2020, 21, 569. [Google Scholar] [CrossRef]
- Bernardet, J.F.; Nakagawa, Y.; Holmes, B. Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int. J. Syst. Evol. Microbiol. 2002, 52, 1049–1070. [Google Scholar]
- Kaushal, G.; Thakur, M.; Rai, A.K.; Singh, S.P. A comprehensive metagenomic analysis framework revealing microbiome profile and potential for hydrocarbon degradation and carbohydrate metabolism in a Himalayan artificial lake. Sustainability 2022, 14, 11455. [Google Scholar] [CrossRef]
- Kolton, M.; Sela, N.; Elad, Y.; Cytryn, E. Comparative Genomic Analysis Indicates that Niche Adaptation of Terrestrial Flavobacteria Is Strongly Linked to Plant Glycan Metabolism. PLoS ONE 2013, 8, e76704. [Google Scholar] [CrossRef] [PubMed]
- Oehme, D.P.; Shafee, T.; Downton, M.T.; Bacic, A.; Doblin, M.S. Differences in protein structural regions that impact functional specificity in GT2 family β-glucan synthases. PLoS ONE 2019, 14, e0224442. [Google Scholar] [CrossRef] [PubMed]
- Yakovlieva, L.; Walvoort, M.T.C. Processivity in Bacterial Glycosyltransferases. ACS Chem. Biol. 2020, 15, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]







| Parameters | JP1 | JP2 | JP3 | JP4 | JP5 |
|---|---|---|---|---|---|
| Water depth (m) | 41.6 | 30.2 | 21.7 | 12.4 | 5.0 |
| Water temperature (°C) | 14.5 | 14.7 | 15.3 | 13.9 | 16.9 |
| Dissolved oxygen (mg/L) | 9.1 | 10.1 | 10.2 | 10.9 | 12.1 |
| Electrical conductivity (μs/cm) | 96.1 | 93 | 91 | 91.6 | 102.8 |
| pH | 7.71 | 7.68 | 7.7 | 7.48 | 8.35 |
| Total phosphorus (mg/L) | 0.047 | 0.043 | 0.036 | 0.055 | 0.081 |
| Total nitrogen (mg/L) | 1.504 | 1.454 | 1.165 | 1.464 | 1.046 |
| Ammonia (mg/L) | 0.084 | 0.093 | 0.076 | 0.102 | 0.091 |
| Nitrite (mg/L) | 0.045 | 0.049 | 0.049 | 0.053 | 0.051 |
| Nitrate (mg/L) | 1.004 | 0.808 | 0.874 | 1.000 | 0.479 |
| Permanganate (mg/L) | 6.04 | 6.26 | 4.36 | 4.93 | 5.02 |
| Chlorophyll a (mg/L) | 13.10 | 12.77 | 11.93 | 9.45 | 48.37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, D.; Huo, T.; Zhang, Z.; Cheng, L.; Wang, L.; Ming, K.; Liu, H.; Li, M.; Du, X. Metagenomic Analysis Reveals the Response of Microbial Communities and Their Functions in Lake Sediment to Environmental Factors. Int. J. Environ. Res. Public Health 2022, 19, 16870. https://doi.org/10.3390/ijerph192416870
Song D, Huo T, Zhang Z, Cheng L, Wang L, Ming K, Liu H, Li M, Du X. Metagenomic Analysis Reveals the Response of Microbial Communities and Their Functions in Lake Sediment to Environmental Factors. International Journal of Environmental Research and Public Health. 2022; 19(24):16870. https://doi.org/10.3390/ijerph192416870
Chicago/Turabian StyleSong, Dan, Tangbin Huo, Zhao Zhang, Lei Cheng, Le Wang, Kun Ming, Hui Liu, Mengsha Li, and Xue Du. 2022. "Metagenomic Analysis Reveals the Response of Microbial Communities and Their Functions in Lake Sediment to Environmental Factors" International Journal of Environmental Research and Public Health 19, no. 24: 16870. https://doi.org/10.3390/ijerph192416870
APA StyleSong, D., Huo, T., Zhang, Z., Cheng, L., Wang, L., Ming, K., Liu, H., Li, M., & Du, X. (2022). Metagenomic Analysis Reveals the Response of Microbial Communities and Their Functions in Lake Sediment to Environmental Factors. International Journal of Environmental Research and Public Health, 19(24), 16870. https://doi.org/10.3390/ijerph192416870