
Pre-Existing Diabetes Mellitus, Hypertension and KidneyDisease as Risk Factors of Pre-Eclampsia: A Disease of Theories and Its Association with Genetic Polymorphism
 ,
,  ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Clinical Manifestations of Pre-Eclampsia
3. Pathogenesis of Pre-Eclampsia
4. Stage 1 of Pre-Eclampsia Pathogenesis
4.1. Hypoxia
4.2. Oxidative Stress
4.3. Natural Killer Cell
5. Stage 2 of Pre-Eclampsia Pathogenesis
5.1. Elevated Antiangiogenic Proteins
5.2. Response to Proinflammatory Mediators and Immune Cells
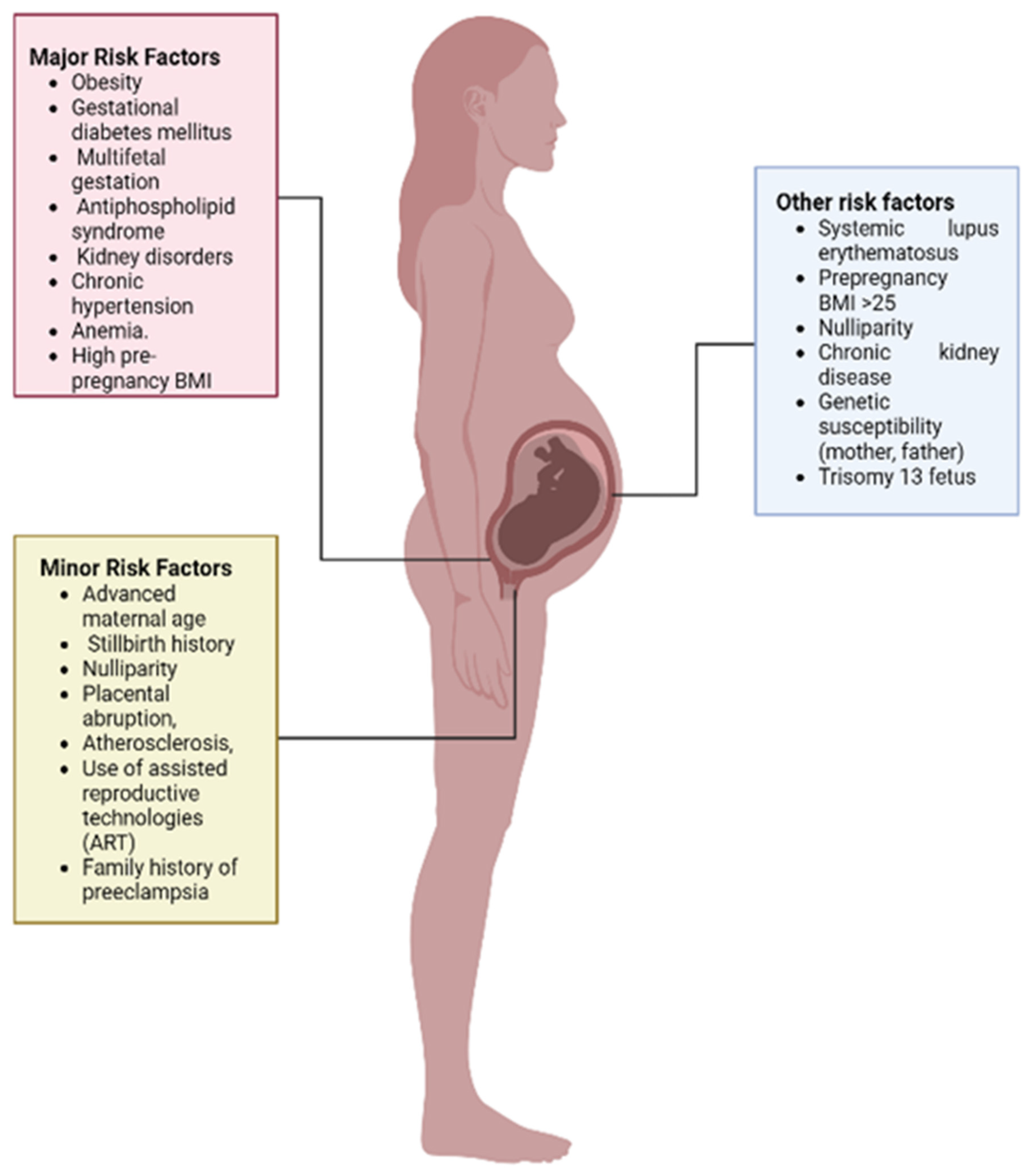
6. Risk Factors Associated with Pre-Eclampsia
6.1. Pre-Existing Disorders
6.2. Multiple Fetal Gestation
6.3. Antiphospholipid Syndrome
6.4. Maternal Age
6.5. Nulliparity
6.6. Family History
6.7. Women with History of PE
6.8. Placental Abruption
6.9. Paternal Risk
7. Genetic Polymorphism and Its Association with Pre-Eclampsia
7.1. FMS-Like Tyrosine Kinase 1
7.2. Endothelial NOS Gene (eNOS)
7.3. Angiotensin-Converting Enzyme Gene
7.4. Interleukin-10 Gene
7.5. Human Leukocyte Antigen-G
7.6. Prostatin Gene
7.7. CXC Chemokine Receptor 2 Gene (CXCR2)
7.8. Forkhead Box P3
7.9. Inhibin Beta B Gene
7.10. Other Candidate Genes

{kind=link}
{kind=link}
| Candidate Gene | Polymorphism | Study Type | Population | Total Participants | Association with PE | Ref. | |
|---|---|---|---|---|---|---|---|
| PE | Control | ||||||
| FLT1 gene | Locus (rs4769613) T/C | Meta-analysis (Data were collected from 13 case control studies) | UK and Iceland | 4380 | 310,238 | Positive association was found | [131] |
| Meta-analysis (Data were collected from 8 case control studies) | Estonia | 96 | 2001 | Positive association was found | [131] | ||
| rs722503 | Case control study | Iranian | 204 | 191 | Positive association found | [132] | |
| eNOS | Glu298Asp | Case control study | Romanian | 69 | 94 | Positive association found | [10] |
| Meta-analysis (Data were collected from 15 case control studies) | Multiethnic group | 1610 | 2975 | Mild association was found | [133] | ||
| Case study | Chinese | 92 | 256 | Positive association was found | [134] | ||
| -786 T/C | Case control study | Serbian | 50 | 50 | Positive association was found | [135] | |
| intron 4 (VNTR4b/a) | Case control study | Serbian | 50 | 50 | Positive association was found | [135] | |
| G894T | Meta-analysis | Multiethnic group | 2450 | 4927 | Positive association was found | [136] | |
| ACE gene | I/D intron (D-allele) | Case control study | Iran | 165 | 131 | Positive association was found | [107] |
| Case control study | Mexican | 66 | 37 | Positive association was found | [137] | ||
| Case control study | Pakistan | 200 | 200 | Positive association was found | [138] | ||
| Case control study | Korean | 104 | 114 | No association was found | [139] | ||
| Meta-analysis (data were collected from 30 case studies) | Asians and Caucasian | 3184 | 3912 | No association was found for Asians and positive association was found in Caucasians | [140] | ||
| Meta-analysis | Columbian | 665 | 1046 | Positive association was found | [141] | ||
| Meta-analysis (data were collected from 11 case studies) | Chinese | 800 | 949 | Positive association was found | [142] | ||
| IL10 (Interleukin 10) | -592A/C | Case control study | Chinese | 155 | 201 | Positive association was found | |
| Meta-analysis (Data were collected from 11 case control studies) | Asian and South American | 1861 | 3632 | Positive association was found | [143] | ||
| Case control study | Indian | 120 | 120 | Positive association was found | [144] | ||
| 819T/C | Case control | Chinese | 177 | 182 | Positive association was found | [145] | |
| Case control study | Indian | 120 | 120 | Positive association was found | [144] | ||
| Meta-analysis (Data were collected from 11 case control studies) | Multiethnic | 1861 | 3632 | Positive association was found in Asian and South American populations | [143] | ||
| 1082-G/G | Case control study | Brazilian (white and nonwhite women) | 151 | 189 | Mild association was found in white women | [109] | |
| Case control study | Indian | 120 | 120 | No significance was found | [144] | ||
| HLA-G | -201AA | Case control study | Multiethnic | 116 | 130 | Positive association was found | [146] |
|
14 BP (I/D) (rs66554220) | Meta-analysis (11 studies) | European Caucasian | 240 | 158 | Positive association was found | [147] | |
| 14 BP (I/D) (rs1704, +2961 − 2974) | Case control study | Norway, Netherlands, and UK | 83 | 83 | Positive association was found | [148] | |
| rs29799440 (CT/TT) | Case control study | Chinese Han | 51 | 48 | Positive association was found | [115] | |
| G*1060 | Case control study | Southeast Asian | 83 | 240 | Positive association was found | [114] | |
| Prostatin gene | rs12597511 (TC/CC) | Case control study | Chinese | 179 | 222 | Positive association was found | [116] |
| Case control study | Pakistani | 76 | 74 | Positive association was found | [118] | ||
| Foxp3 | rs2232365 (A/G) | Case control study | Chinese Han | 203 | 234 | Positive association was found | [128] |
| Meta-analysis (Data were collected from 7 case control studies) | Multiethnic | 784 | 1415 | Positive association was found | [15] | ||
| Case control study | Iranian | 133 | 143 | Positive association was found | [129] | ||
| Meta-analysis (6 studies) | Multiethnic | 1231 | 1384 | Positive association was found | [149] | ||
| rs3761548 | Case control study | Iranian | 133 | 143 | Positive association was found | [129] | |
| Meta-analysis (8 studies) | Multiethnic | 784 | 1631 | Positive association was found | [15] | ||
| Meta-analysis | Multiethnic | 1023 | 987 | Positive association in Asian Population | [150] | ||
| Meta-analysis (7 studies) | Multiethnic | 1513 | 1600 | Positive association was found | [149] | ||
| INHBB | rs7579169 (CC) genotype | Case control study | Chinese | 181 | 203 | Positive association was found | [130] |
8. Conclusions and Future Recommendations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Phipps, E.; Prasanna, D.; Brima, W.; Jim, B. Preeclampsia: Updates in Pathogenesis, Definitions, and Guidelines. J. Am. Soc. Nephrol. 2016, 11, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar] [CrossRef]
- Thakoordeen, S.; Moodley, J.; Naicker, T. Candidate Gene, Genome-Wide Association and Bioinformatic Studies in Pre-eclampsia: A Review. Curr. Hypertens. Rep. 2018, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Kuklina, E.V.; Ayala, C.; Callaghan, W.M. Hypertensive disorders and severe obstetric morbidity in the United States. Obstet. Gynecol. 2009, 113, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.C.; Foreman, K.J.; Naghavi, M.; Ahn, S.Y.; Wang, M.; Makela, S.M.; Lopez, A.D.; Lozano, R.; Murray, C.J. Maternal mortality for 181 countries, 1980–2008: A systematic analysis of progress towards Millennium Development Goal 5. Lancet 2010, 375, 1609–1623. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- Smyth, A.; Oliveira, G.H.; Lahr, B.D.; Bailey, K.R.; Norby, S.M.; Garovic, V.D. A systematic review and meta-analysis of pregnancy outcomes in patients with systemic lupus erythematosus and lupus nephritis. J. Am. Soc. Nephrol. 2010, 5, 2060–2068. [Google Scholar] [CrossRef]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef]
- Odegård, R.A.; Vatten, L.J.; Nilsen, S.T.; Salvesen, K.A.; Austgulen, R. Preeclampsia and fetal growth. Obstet. Gynecol. 2000, 96, 950–955. [Google Scholar] [CrossRef]
- Procopciuc, L.M.; Caracostea, G.; Hazi, G.M.; Nemeti, G.; Zaharie, G.; Stamatian, F. Maternal/fetal eNOS-Glu298Asp genotypes and their influence on the severity, prognosis, and lipid profile of preeclampsia. J. Matern. Neonatal Med. 2018, 31, 1681–1688. [Google Scholar] [CrossRef]
- Williams, P.J.; Broughton Pipkin, F. The genetics of pre-eclampsia and other hypertensive disorders of pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Thurnau, G.R. Pregnancy-induced hypertension without proteinuria: Is it true preeclampsia? South. Med. J. 1988, 81, 210–213. [Google Scholar] [CrossRef]
- Barton, J.R.; O’Brien, J.M.; Bergauer, N.K.; Jacques, D.L.; Sibai, B.M. Mild gestational hypertension remote from term: Progression and outcome. Am. J. Obstet. Gynecol. 2001, 184, 979–983. [Google Scholar] [CrossRef]
- Espinoza, J.; Vidaeff, A.; Pettker, C.M.; Simhan, H. Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237–e260. [Google Scholar] [CrossRef]
- Karimian, M.; Ghazaey Zidanloo, S.; Jahantigh, D. Influence of FOXP3 gene polymorphisms on the risk of preeclampsia: A meta-analysis and a bioinformatic approach. Clin. Exp. Hypertens. 2022, 44, 280–290. [Google Scholar] [CrossRef]
- Palei, A.C.; Spradley, F.T.; Warrington, J.P.; George, E.M.; Granger, J.P. Pathophysiology of hypertension in pre-eclampsia: A lesson in integrative physiology. Acta Physiol. 2013, 208, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Chaiworapongsa, T. Preeclampsia: A link between trophoblast dysregulation and an antiangiogenic state. J. Clin. Investig. 2013, 123, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Taylor, R.N.; Musci, T.J.; Rodgers, G.M.; Hubel, C.A.; McLaughlin, M.K. Preeclampsia: An endothelial cell disorder. Am. J. Obstet. Gynecol. 1989, 161, 1200–1204. [Google Scholar] [CrossRef]
- Brosens, I.; Robertson, W.B.; Dixon, H.G. The physiological response of the vessels of the placental bed to normal pregnancy. J. Pathol. Bacteriol. 1967, 93, 569–579. [Google Scholar] [CrossRef]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Fisher, S.J. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Investig. 1997, 99, 2152–2164. [Google Scholar] [CrossRef]
- Garrido-Gomez, T.; Dominguez, F.; Quiñonero, A.; Diaz-Gimeno, P.; Kapidzic, M.; Gormley, M.; Ona, K.; Padilla-Iserte, P.; McMaster, M.; Genbacev, O.; et al. Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc. Natl. Acad. Sci. USA 2017, 114, E8468–E8477. [Google Scholar] [CrossRef]
- Soleymanlou, N.; Jurisica, I.; Nevo, O.; Ietta, F.; Zhang, X.; Zamudio, S.; Post, M.; Caniggia, I. Molecular evidence of placental hypoxia in preeclampsia. J. Clin. Endocrinol. Metab. 2005, 90, 4299–4308. [Google Scholar] [CrossRef]
- Jauniaux, E.; Watson, A.L.; Hempstock, J.; Bao, Y.P.; Skepper, J.N.; Burton, G.J. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am. J. Pathol. 2000, 157, 2111–2122. [Google Scholar] [CrossRef]
- Rajakumar, A.; Brandon, H.M.; Daftary, A.; Ness, R.; Conrad, K.P. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 2004, 25, 763–769. [Google Scholar] [CrossRef]
- Tal, R.; Shaish, A.; Barshack, I.; Polak-Charcon, S.; Afek, A.; Volkov, A.; Feldman, B.; Avivi, C.; Harats, D. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: Possible implications for preeclampsia and intrauterine growth restriction. Am. J. Pathol. 2010, 177, 2950–2962. [Google Scholar] [CrossRef]
- Caniggia, I.; Mostachfi, H.; Winter, J.; Gassmann, M.; Lye, S.J.; Kuliszewski, M.; Post, M. Hypoxia-inducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3). J. Clin. Investig. 2000, 105, 577–587. [Google Scholar] [CrossRef]
- Many, A.; Hubel, C.A.; Fisher, S.J.; Roberts, J.M.; Zhou, Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am. J. Pathol. 2000, 156, 321–331. [Google Scholar] [CrossRef]
- Huang, Q.T.; Wang, S.S.; Zhang, M.; Huang, L.P.; Tian, J.W.; Yu, Y.H.; Wang, Z.J.; Zhong, M. Advanced oxidation protein products enhances soluble Fms-like tyrosine kinase 1 expression in trophoblasts: A possible link between oxidative stress and preeclampsia. Placenta 2013, 34, 949–952. [Google Scholar] [CrossRef]
- Vaughan, J.E.; Walsh, S.W. Oxidative stress reproduces placental abnormalities of preeclampsia. Hypertens. Pregnancy 2002, 21, 205–223. [Google Scholar] [CrossRef]
- Zsengellér, Z.K.; Rajakumar, A.; Hunter, J.T.; Salahuddin, S.; Rana, S.; Stillman, I.E.; Karumanchi, S.A. Trophoblast mitochondrial function is impaired in preeclampsia and correlates negatively with the expression of soluble fms-like tyrosine kinase 1. Pregnancy Hypertens. 2016, 6, 313–319. [Google Scholar] [CrossRef]
- Kai, S.; Tanaka, T.; Daijo, H.; Harada, H.; Kishimoto, S.; Suzuki, T.S.; Takenaga, K.; Fukuda, K.; Hirota, K. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid. Redox Signal. 2012, 16, 203–216. [Google Scholar] [CrossRef]
- Covarrubias, A.E.; Lecarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengellér, Z.K. AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts: Relevance for Preeclampsia Pathogenesis. Am. J. Pathol. 2019, 189, 104–114. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Yung, H.W.; Korolchuk, S.; Tolkovsky, A.M.; Charnock-Jones, D.S.; Burton, G.J. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J. 2007, 21, 872–884. [Google Scholar] [CrossRef]
- Lian, I.A.; Løset, M.; Mundal, S.B.; Fenstad, M.H.; Johnson, M.P.; Eide, I.P.; Bjørge, L.; Freed, K.A.; Moses, E.K.; Austgulen, R. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta 2011, 32, 823–829. [Google Scholar] [CrossRef]
- Fu, J.; Zhao, L.; Wang, L.; Zhu, X. Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwan. J. Obstet. Gynecol. 2015, 54, 19–23. [Google Scholar] [CrossRef]
- George, E.M.; Granger, J.P. Heme oxygenase in pregnancy and preeclampsia. Curr. Opin. Nephrol. Hypertens. 2013, 22, 156–162. [Google Scholar] [CrossRef]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef]
- George, E.M.; Colson, D.; Dixon, J.; Palei, A.C.; Granger, J.P. Heme Oxygenase-1 Attenuates Hypoxia-Induced sFlt-1 and Oxidative Stress in Placental Villi through Its Metabolic Products CO and Bilirubin. Int. J. Hypertens. 2012, 2012, 486053. [Google Scholar] [CrossRef] [PubMed]
- Origassa, C.S.; Câmara, N.O. Cytoprotective role of heme oxygenase-1 and heme degradation derived end products in liver injury. World J. Hepatol. 2013, 5, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Moffett, A.; Colucci, F. Uterine NK cells: Active regulators at the maternal-fetal interface. J. Clin. Investig. 2014, 124, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Moffett-King, A. Natural killer cells and pregnancy. Nat. Rev. Immunol. 2002, 2, 656–663. [Google Scholar] [CrossRef]
- Lash, G.E.; Schiessl, B.; Kirkley, M.; Innes, B.A.; Cooper, A.; Searle, R.F.; Robson, S.C.; Bulmer, J.N. Expression of angiogenic growth factors by uterine natural killer cells during early pregnancy. J. Leukoc. Biol. 2006, 80, 572–580. [Google Scholar] [CrossRef]
- Parham, P.; Moffett, A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat. Rev. Immunol. 2013, 13, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Apps, R.; Murphy, S.P.; Fernando, R.; Gardner, L.; Ahad, T.; Moffett, A. Human leucocyte antigen (HLA) expression of primary trophoblast cells and placental cell lines, determined using single antigen beads to characterize allotype specificities of anti-HLA antibodies. Immunology 2009, 127, 26–39. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmed, A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ. Res. 2004, 95, 884–891. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.S.; Walshe, T.E.; Saint-Geniez, M.; Venkatesha, S.; Maldonado, A.E.; Himes, N.C.; Matharu, K.S.; Karumanchi, S.A.; D’Amore, P.A. VEGF and TGF-beta are required for the maintenance of the choroid plexus and ependyma. J. Exp. Med. 2008, 205, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Harmon, A.; Cornelius, D.; Amaral, L.; Paige, A.; Herse, F.; Ibrahim, T.; Wallukat, G.; Faulkner, J.; Moseley, J.; Dechend, R.; et al. IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens. Pregnancy 2015, 34, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Weel, I.C.; Baergen, R.N.; Romão-Veiga, M.; Borges, V.T.; Ribeiro, V.R.; Witkin, S.S.; Bannwart-Castro, C.; Peraçoli, J.C.; De Oliveira, L.; Peraçoli, M.T. Association between Placental Lesions, Cytokines and Angiogenic Factors in Pregnant Women with Preeclampsia. PLoS ONE 2016, 11, e0157584. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gu, Y.; Sun, J.; Zhu, H.; Lewis, D.F.; Wang, Y. Reduced CD200 expression is associated with altered Th1/Th2 cytokine production in placental trophoblasts from preeclampsia. Am. J. Reprod. Immunol. 2017, 79, e12763. [Google Scholar] [CrossRef]
- Pantham, P.; Askelund, K.J.; Chamley, L.W. Trophoblast deportation part II: A review of the maternal consequences of trophoblast deportation. Placenta 2011, 32, 724–731. [Google Scholar] [CrossRef]
- Attwood, H.D.; Park, W.W. Embolism to the lungs by trophoblast. J. Obstet. Gynaecol. Br. Commonw. 1961, 68, 611–617. [Google Scholar] [CrossRef]
- Germain, S.J.; Sacks, G.P.; Sooranna, S.R.; Sargent, I.L.; Redman, C.W. Systemic inflammatory priming in normal pregnancy and preeclampsia: The role of circulating syncytiotrophoblast microparticles. J. Immunol. 2007, 178, 5949–5956. [Google Scholar] [CrossRef]
- Guller, S.; Tang, Z.; Ma, Y.Y.; Di Santo, S.; Sager, R.; Schneider, H. Protein composition of microparticles shed from human placenta during placental perfusion: Potential role in angiogenesis and fibrinolysis in preeclampsia. Placenta 2011, 32, 63–69. [Google Scholar] [CrossRef]
- Chang, X.; Yao, J.; He, Q.; Liu, M.; Duan, T.; Wang, K. Exosomes From Women With Preeclampsia Induced Vascular Dysfunction by Delivering sFlt (Soluble Fms-Like Tyrosine Kinase)-1 and sEng (Soluble Endoglin) to Endothelial Cells. Hypertension 2018, 72, 1381–1390. [Google Scholar] [CrossRef]
- Zenclussen, A.C. Regulatory T cells in pregnancy. Springer Semin. Immunopathol. 2006, 28, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Regal, J.F.; Burwick, R.M.; Fleming, S.D. The complement system and preeclampsia. Curr. Hypertens. Rep. 2017, 19, 87. [Google Scholar] [CrossRef] [PubMed]
- Burwick, R.M.; Fichorova, R.N.; Dawood, H.Y.; Yamamoto, H.S.; Feinberg, B.B. Urinary excretion of C5b-9 in severe preeclampsia: Tipping the balance of complement activation in pregnancy. Hypertension 2013, 62, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.J.; Fremeaux-Bacchi, V.; Liszewski, M.K.; Pianetti, G.; Noris, M.; Goodship, T.H.; Atkinson, J.P. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood 2008, 111, 624–632. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Fakhouri, F.; Hourmant, M.; Campistol, J.M.; Cataland, S.R.; Espinosa, M.; Gaber, A.O.; Menne, J.; Minetti, E.E.; Provôt, F.; Rondeau, E.; et al. Terminal Complement Inhibitor Eculizumab in Adult Patients With Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am. J. Kidney Dis. 2016, 68, 84–93. [Google Scholar] [CrossRef]
- Burwick, R.M.; Velásquez, J.A.; Valencia, C.M.; Gutiérrez-Marín, J.; Edna-Estrada, F.; Silva, J.L.; Trujillo-Otálvaro, J.; Vargas-Rodríguez, J.; Bernal, Y.; Quintero, A.; et al. Terminal Complement Activation in Preeclampsia. Obstet. Gynecol. 2018, 132, 1477–1485. [Google Scholar] [CrossRef]
- Penning, M.; Chua, J.S.; van Kooten, C.; Zandbergen, M.; Buurma, A.; Schutte, J.; Bruijn, J.A.; Khankin, E.V.; Bloemenkamp, K.; Karumanchi, S.A.; et al. Classical Complement Pathway Activation in the Kidneys of Women With Preeclampsia. Hypertension 2015, 66, 117–125. [Google Scholar] [CrossRef]
- Irani, R.A.; Xia, Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta 2008, 29, 763–771. [Google Scholar] [CrossRef]
- Brown, M.A.; Wang, J.; Whitworth, J.A. The renin-angiotensin-aldosterone system in pre-eclampsia. Clin. Exp. Hypertens. 1997, 19, 713–726. [Google Scholar] [CrossRef]
- LaMarca, B.; Parrish, M.; Ray, L.F.; Murphy, S.R.; Roberts, L.; Glover, P.; Wallukat, G.; Wenzel, K.; Cockrell, K.; Martin, J.N., Jr.; et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: Role of endothelin-1. Hypertension 2009, 54, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, F.; Lau, W.B.; Zhang, S.; Zhang, S.; Liu, H.; Ma, X.L. Autoantibodies isolated from preeclamptic patients induce endothelial dysfunction via interaction with the angiotensin II AT1 receptor. Cardiovasc. Toxicol. 2014, 14, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wen, H.; Bobst, S.; Day, M.C.; Kellems, R.E. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. J. Soc. Gynecol. Investig. 2003, 10, 82–93. [Google Scholar] [CrossRef]
- Dechend, R.; Viedt, C.; Müller, D.N.; Ugele, B.; Brandes, R.P.; Wallukat, G.; Park, J.K.; Janke, J.; Barta, P.; Theuer, J.; et al. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation 2003, 107, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ 2016, 353, i1753. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, R.B.; Brennecke, S.P. Family history of pre-eclampsia as a predictor for pre-eclampsia in primigravidas. Int. J. Gynaecol. Obstet. 1998, 60, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987-2004. Am. J. Hypertens. 2008, 21, 521–526. [Google Scholar] [CrossRef]
- Lee, C.J.; Hsieh, T.T.; Chiu, T.H.; Chen, K.C.; Lo, L.M.; Hung, T.H. Risk factors for pre-eclampsia in an Asian population. Int. J. Gynaecol. Obstet. 2000, 70, 327–333. [Google Scholar] [CrossRef]
- Duckitt, K.; Harrington, D. Risk factors for pre-eclampsia at antenatal booking: Systematic review of controlled studies. BMJ 2005, 330, 565. [Google Scholar] [CrossRef]
- Santema, J.G.; Koppelaar, I.; Wallenburg, H.C. Hypertensive disorders in twin pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1995, 58, 9–13. [Google Scholar] [CrossRef]
- Canti, V.; Del Rosso, S.; Tonello, M.; Lucianò, R.; Hoxha, A.; Coletto, L.A.; Vaglio Tessitore, I.; Rosa, S.; Manfredi, A.A.; Castiglioni, M.T.; et al. Antiphosphatidylserine/prothrombin Antibodies in Antiphospholipid Syndrome with Intrauterine Growth Restriction and Preeclampsia. J. Rheumatol. 2018, 45, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.A.; Silver, R.M.; Branch, D.W. Do antiphospholipid antibodies cause preeclampsia and HELLP syndrome? Curr. Rheumatol. Rep. 2007, 9, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Frick, A.P. Advanced maternal age and adverse pregnancy outcomes. Best Pract. Res. Clin. Obstet. Gynaecol. 2021, 70, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Stone, J.; Lynch, L.; Lapinski, R.; Berkowitz, G.; Berkowitz, R.L. Pregnancy outcome at age 40 and older. Obstet. Gynecol. 1996, 87, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Syngelaki, A.; Maiz, N.; Zinevich, Y.; Nicolaides, K.H. Maternal age and adverse pregnancy outcome: A cohort study. Ultrasound Obstet. Gynecol. 2013, 42, 634–643. [Google Scholar] [CrossRef]
- Coonrod, D.V.; Hickok, D.E.; Zhu, K.; Easterling, T.R.; Daling, J.R. Risk factors for preeclampsia in twin pregnancies: A population-based cohort study. Obstet. Gynecol. 1995, 85, 645–650. [Google Scholar] [CrossRef]
- Stamilio, D.M.; Sehdev, H.M.; Morgan, M.A.; Propert, K.; Macones, G.A. Can antenatal clinical and biochemical markers predict the development of severe preeclampsia? Am. J. Obstet. Gynecol. 2000, 182, 589–594. [Google Scholar] [CrossRef]
- Shen, M.; Smith, G.N.; Rodger, M.; White, R.R.; Walker, M.C.; Wen, S.W. Comparison of risk factors and outcomes of gestational hypertension and pre-eclampsia. PLoS ONE 2017, 12, e0175914. [Google Scholar] [CrossRef]
- Yang, Y.; Le Ray, I.; Zhu, J.; Zhang, J.; Hua, J.; Reilly, M. Preeclampsia Prevalence, Risk Factors, and Pregnancy Outcomes in Sweden and China. JAMA Netw. Open 2021, 4, e218401. [Google Scholar] [CrossRef]
- Wu, C.T.; Kuo, C.F.; Lin, C.P.; Huang, Y.T.; Chen, S.W.; Wu, H.M.; Chu, P.H. Association of family history with incidence and gestational hypertension outcomes of preeclampsia. Int. J. Cardiol. Hypertens. 2021, 9, 100084. [Google Scholar] [CrossRef]
- Boyd, H.A.; Tahir, H.; Wohlfahrt, J.; Melbye, M. Associations of personal and family preeclampsia history with the risk of early-, intermediate- and late-onset preeclampsia. Am. J. Epidemiol. 2013, 178, 1611–1619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dukler, D.; Porath, A.; Bashiri, A.; Erez, O.; Mazor, M. Remote prognosis of primiparous women with preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 96, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Makkonen, N.; Heinonen, S.; Kirkinen, P. Obstetric prognosis in second pregnancy after preeclampsia in first pregnancy. Hypertens. Pregnancy 2000, 19, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Bellamy, L.; Casas, J.P.; Hingorani, A.D.; Williams, D.J. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: Systematic review and meta-analysis. BMJ 2007, 335, 974. [Google Scholar] [CrossRef]
- Ananth, C.V.; Smulian, J.C.; Vintzileos, A.M. Ischemic placental disease: Maternal versus fetal clinical presentations by gestational age. J. Matern. Fetal Neonatal Med. 2010, 23, 887–893. [Google Scholar] [CrossRef]
- Ananth, C.V.; Getahun, D.; Peltier, M.R.; Smulian, J.C. Placental abruption in term and preterm gestations: Evidence for heterogeneity in clinical pathways. Obstet. Gynecol. 2006, 107, 785–792. [Google Scholar] [CrossRef]
- Parker, S.E.; Werler, M.M.; Gissler, M.; Tikkanen, M.; Ananth, C.V. Placental abruption and subsequent risk of pre-eclampsia: A population-based case-control study. Paediatr. Perinat. Epidemiol. 2015, 29, 211–219. [Google Scholar] [CrossRef]
- Dekker, G.; Robillard, P.Y.; Roberts, C. The etiology of preeclampsia: The role of the father. J. Reprod. Immunol. 2011, 89, 126–132. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543. [Google Scholar] [CrossRef]
- Mütze, S.; Rudnik-Schöneborn, S.; Zerres, K.; Rath, W. Genes and the preeclampsia syndrome. J. Perinat. Med. 2008, 36, 38–58. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lim, J.H.; Yang, J.H.; Kim, M.Y.; Han, J.Y.; Ahn, H.K.; Choi, J.S.; Park, S.Y.; Kim, M.J.; Ryu, H.M. Dinucleotide repeat polymorphism in Fms-like tyrosine kinase-1 (Flt-1) gene is not associated with preeclampsia. BMC Med. Genet. 2008, 9, 68. [Google Scholar] [CrossRef]
- McGinnis, R.; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; Sigurdsson, J.K.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260. [Google Scholar] [CrossRef]
- Ozturk, E.; Balat, O.; Pehlivan, S.; Ugur, M.G.; Ozcan, C.; Sever, T.; Kul, S. Endothelial nitric oxide synthase gene polymorphisms in preeclampsia with or without eclampsia in a Turkish population. J. Obstet. Gynaecol. Res. 2011, 37, 1778–1783. [Google Scholar] [CrossRef]
- Fatini, C.; Sticchi, E.; Gensini, F.; Genuardi, M.; Tondi, F.; Gensini, G.F.; Riviello, C.; Parretti, E.; Mello, G.; Abbate, R. Endothelial nitric oxide synthase gene influences the risk of pre-eclampsia, the recurrence of negative pregnancy events, and the maternal-fetal flow. J. Hypertens. 2006, 24, 1823–1829. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, B.H.; Park, H.; Jung, S.C.; Pang, M.G.; Ryu, H.M.; Lee, K.S.; Eom, S.M.; Park, H.Y. No association of the genetic polymorphisms of endothelial nitric oxide synthase, dimethylarginine dimethylaminohydrolase, and vascular endothelial growth factor with preeclampsia in Korean populations. Twin Res. Hum. Genet. 2008, 11, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Sharma, D.; Raghunandan, C.; Bhattacharjee, J. Role of inflammatory cytokines and eNOS gene polymorphism in pathophysiology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Do, A.A.; Esmaeilzadeh, E.; Amin-Beidokhti, M.; Pirjani, R.; Gholami, M.; Mirfakhraie, R. ACE gene rs4343 polymorphism elevates the risk of preeclampsia in pregnant women. J. Hum. Hypertens. 2018, 32, 825–830. [Google Scholar] [CrossRef]
- Zhu, X.; Bouzekri, N.; Southam, L.; Cooper, R.S.; Adeyemo, A.; McKenzie, C.A.; Luke, A.; Chen, G.; Elston, R.C.; Ward, R. Linkage and association analysis of angiotensin I-converting enzyme (ACE)-gene polymorphisms with ACE concentration and blood pressure. Am. J. Hum. Genet. 2001, 68, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Daher, S.; Sass, N.; Oliveira, L.G.; Mattar, R. Cytokine genotyping in preeclampsia. Am. J. Reprod. Immunol. 2006, 55, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhong, M. Association between Interleukin-10 gene polymorphisms and risk of early-onset preeclampsia. Int. J. Clin. Exp. Pathol. 2015, 8, 11659–11664. [Google Scholar] [PubMed]
- Eche, S.; Mackraj, I.; Moodley, J. Circulating fetal and total cell-free DNA, and sHLA-G in black South African women with gestational hypertension and pre-eclampsia. Hypertens. Pregnancy 2017, 36, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Yie, S.M.; Li, L.H.; Li, Y.M.; Librach, C. HLA-G protein concentrations in maternal serum and placental tissue are decreased in preeclampsia. Am. J. Obstet. Gynecol. 2004, 191, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, M.J.; Verloes, A.; Van de Velde, H.; Haentjens, P. Accuracy of soluble human leukocyte antigen-G for predicting pregnancy among women undergoing infertility treatment: Meta-analysis. Hum. Reprod. Update 2008, 14, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.Y.; Ho, J.F.; Chong, Y.S.; Loganath, A.; Chan, Y.H.; Ravichandran, J.; Lee, C.G.; Chong, S.S. Paternal contribution of HLA-G*0106 significantly increases risk for pre-eclampsia in multigravid pregnancies. Mol. Hum. Reprod. 2008, 14, 317–324. [Google Scholar] [CrossRef]
- Ma, C.; Zheng, Y.; Liu, X.; Zhang, W. Association between maternal single-nucleotide polymorphisms in HLA-G gene and risk of preeclampsia. J. Matern. Fetal Neonatal Med. 2021, 35, 1–6. [Google Scholar] [CrossRef]
- Kong, W.; Zhang, Y.; Gong, Y.; Dai, L.; Zhou, R. Association of prostasin gene rs12597511 polymorphism with outcomes of pregnancy with severe preeclampsia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi = Zhonghua Yixue Yichuanxue Zazhi = Chin. J. Med. Genet. 2015, 32, 543–547. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, Y.; Bai, Y.; Liu, X.; Gong, Y.; Zhou, B.; Zhang, L.; Luo, L.; Zhou, R. Prostasin gene polymorphism at rs12597511 is associated with severe preeclampsia in Chinese Han women. Chin. Med. J. 2014, 127, 2048–2052. [Google Scholar]
- Ejaz, S.; Ali, A.; Azim, K.; Mahmood, A.; Khan, A.I.; Almazyad, T.A.; Bilal, B. Association between preeclampsia and prostasin polymorphism in Pakistani females. Saudi Med. J. 2020, 41, 1234–1240. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef]
- Boro, M.; Balaji, K.N. CXCL1 and CXCL2 Regulate NLRP3 Inflammasome Activation via G-Protein-Coupled Receptor CXCR2. J. Immunol. 2017, 199, 1660–1671. [Google Scholar] [CrossRef]
- Shen, X.H.; Xu, S.J.; Jin, C.Y.; Ding, F.; Zhou, Y.C.; Fu, G.S. Interleukin-8 prevents oxidative stress-induced human endothelial cell senescence via telomerase activation. Int. Immunopharmacol. 2013, 16, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Burdick, M.D.; Gomperts, B.N.; Belperio, J.A.; Keane, M.P. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 593–609. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Kang, X.; Zhang, X.; Liu, Z.; Xu, H.; Wang, T.; He, L.; Zhao, A. CXCR2-Mediated Granulocytic Myeloid-Derived Suppressor Cells’ Functional Characterization and Their Role in Maternal Fetal Interface. DNA Cell Biol. 2016, 35, 358–365. [Google Scholar] [CrossRef]
- Pitman, H.; Innes, B.A.; Robson, S.C.; Bulmer, J.N.; Lash, G.E. Altered expression of interleukin-6, interleukin-8 and their receptors in decidua of women with sporadic miscarriage. Hum. Reprod. 2013, 28, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hong, H.; Huang, X.; Huang, L.; He, Z.; Fang, Q.; Luo, Y. CXCR2 is decreased in preeclamptic placentas and promotes human trophoblast invasion through the Akt signaling pathway. Placenta 2016, 43, 17–25. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Dai, L.; Song, Y.; Wang, Y.; Zhou, B.; Zhou, R. Association between polymorphisms in CXCR2 gene and preeclampsia. Mol. Genet. Genom. Med. 2019, 7, e00578. [Google Scholar] [CrossRef]
- Chen, J.; Tan, W.; Wang, D.; Zhao, L.; Gao, H.; Zhang, N.; Wang, C. Association of Foxp3 and TGF-β1 polymorphisms with pre-eclampsia risk in Chinese women. Genet. Test. Mol. Biomark. 2019, 23, 180–187. [Google Scholar] [CrossRef]
- Gholami, M.; Mirfakhraie, R.; Pirjani, R.; Taheripanah, R.; Bayat, S.; Daryabari, S.A.; Noori, M.; Ghaderian, S.M.H. Association study of FOXP3 gene and the risk of 0020 pre-eclampsia. Clin. Exp. Hypertens. 2018, 40, 613–616. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, G.; Guo, C.; Cao, X.; An, L.; Du, M.; Qiu, Y.; Yang, Y.; Wang, Y.; Wang, S.; et al. Single nucleotide polymorphisms near the inhibin beta B gene on 2q14 are associated with pre-eclampsia in Han Chinese women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2015, 193, 127–131. [Google Scholar] [CrossRef]
- Kikas, T.; Inno, R.; Ratnik, K.; Rull, K.; Laan, M. C-allele of rs4769613 Near FLT1 Represents a High-Confidence Placental Risk Factor for Preeclampsia. Hypertension 2020, 76, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Amin-Beidokhti, M.; Gholami, M.; Abedin-Do, A.; Pirjani, R.; Sadeghi, H.; Karamoddin, F.; Yassaee, V.R.; Mirfakhraie, R. An intron variant in the FLT1 gene increases the risk of preeclampsia in Iranian women. Clin. Exp. Hypertens. 2019, 41, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.P.; Sultana, A.; Bammidi, V.K.; Sampathirao, K.; Jamil, K. A meta-analysis of eNOS and ACE gene polymorphisms and risk of pre-eclampsia in women. J. Obstet. Gynaecol. 2011, 31, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.K.; Huang, C.H.; Yeh, H.M.; Lee, C.N.; Shyu, M.K.; Hsieh, F.J.; Lai, L.P.; Sun, W.Z. Polymorphisms in the endothelial nitric oxide synthase gene may be protective against preeclampsia in a Chinese population. Reprod. Sci. 2007, 14, 175–181. [Google Scholar] [CrossRef]
- Sljivancanin Jakovljevic, T.; Kontic-Vucinic, O.; Nikolic, N.; Carkic, J.; Stamenkovic, J.; Soldatovic, I.; Milasin, J. Association Between Endothelial Nitric Oxide Synthase (eNOS) -786 T/C and 27-bp VNTR 4b/a Polymorphisms and Preeclampsia Development. Reprod. Sci. 2021, 28, 3529–3539. [Google Scholar] [CrossRef]
- Qi, H.P.; Fraser, W.D.; Luo, Z.C.; Julien, P.; Audibert, F.; Wei, S.Q. Endothelial nitric oxide synthase gene polymorphisms and risk of preeclampsia. Am. J. Perinatol. 2013, 30, 795–804. [Google Scholar] [CrossRef]
- González-Garrido, J.A.; García-Sánchez, J.R.; Tovar-Rodríguez, J.M.; Olivares-Corichi, I.M. Preeclampsia is associated with ACE I/D polymorphism, obesity and oxidative damage in Mexican women. Pregnancy Hypertens. 2017, 10, 22–27. [Google Scholar] [CrossRef]
- Shaheen, G.; Sajid, S.; Razak, S.; Mazhar, S.B.; Afsar, T.; Almajwal, A.; Alam, I.; Jahan, S. Role of ACE I/D polymorphism in pathological assessment of preeclampsia in Pakistan. Mol. Genet. Genom. Med. 2019, 7, e00799. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, M.H.; Park, H.S.; Lee, K.S.; Ha, E.H.; Pang, M.G. Associations of polymorphisms of the angiotensinogen M235 polymorphism and angiotensin-converting-enzyme intron 16 insertion/deletion polymorphism with preeclampsia in Korean women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 116, 48–53. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P. Meta-analysis of angiotensin-converting enzyme insersion/delection polymorphism and pre-eclampsia susceptibility. J. Obstet. Gynaecol. Res. 2020, 46, 1744–1757. [Google Scholar] [CrossRef]
- Serrano, N.C.; Díaz, L.A.; Páez, M.C.; Mesa, C.M.; Cifuentes, R.; Monterrosa, A.; González, A.; Smeeth, L.; Hingorani, A.D.; Casas, J.P. Angiotensin-converting enzyme I/D polymorphism and preeclampsia risk: Evidence of small-study bias. PLoS Med. 2006, 3, e520. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.G.; Wang, Y.; Zhu, H.; Zhao, X. Meta analysis of angiotensin-converting enzyme I/D polymorphism as a risk factor for preeclampsia in Chinese women. Genet. Mol. Res. GMR 2012, 11, 2268–2276. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhu, Z.; Wang, J.; Ye, W.; Ding, Y. Evaluation of association of maternal IL-10 polymorphisms with risk of preeclampsia by A meta-analysis. J. Cell. Mol. Med. 2014, 18, 2466–2477. [Google Scholar] [CrossRef] [PubMed]
- Sowmya, S.; Sri Manjari, K.; Ramaiah, A.; Sunitha, T.; Nallari, P.; Jyothy, A.; Venkateshwari, A. Interleukin 10 gene promoter polymorphisms in women with early-onset pre-eclampsia. Clin. Exp. Immunol. 2014, 178, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Gao, F.Y.; Liu, X.R.; Li, J.; Ji, M.; Dong, J.; Wang, X.T. Investigations into the association between polymorphisms in the interleukin-10 gene and risk of early-onset preeclampsia. Genet. Mol. Res. GMR 2015, 14, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Yan, W.H.; Dai, M.Z.; Chen, X.J.; Li, B.L.; Chen, B.G.; Fan, L.A. Maternal human leukocyte antigen-G polymorphism is not associated with pre-eclampsia in a Chinese Han population. Tissue Antigens 2006, 68, 311–316. [Google Scholar] [CrossRef]
- Pabalan, N.; Jarjanazi, H.; Sun, C.; Iversen, A.C. Meta-analysis of the human leukocyte antigen-G (HLA-G) 14 bp insertion/deletion polymorphism as a risk factor for preeclampsia. Tissue Antigens 2015, 86, 186–194. [Google Scholar] [CrossRef]
- Johnsen, G.M.; Fjeldstad, H.E.S.; Drabbels, J.J.M.; Haasnoot, G.W.; Eikmans, M.; Størvold, G.L.; Alnaes-Katjavivi, P.; Jacobsen, D.P.; Scherjon, S.A.; Redman, C.W.G.; et al. A possible role for HLA-G in development of uteroplacental acute atherosis in preeclampsia. J. Reprod. Immunol. 2021, 144, 103284. [Google Scholar] [CrossRef]
- Liu, J.; Song, G.; Zhao, G.; Meng, T. Gene polymorphism associated with FOXP3, CTLA-4 and susceptibility to pre-eclampsia: A meta-analysis and trial sequential analysis. J. Obstet. Gynaecol. 2022, 42, 1085–1091. [Google Scholar] [CrossRef]
- Fan, Y.X.; Wu, J.H.; Yin, S.J.; Zhou, T.; Huang, Y.H.; Meng, R.; Wang, P.; He, G.H. Associations of FOXP3 gene polymorphisms with susceptibility and severity of preeclampsia: A meta-analysis. Am. J. Reprod. Immunol. 2022, 88, e13554. [Google Scholar] [CrossRef]


| Pre-Eclampsia | |
|---|---|
| Blood pressure |
|
| Proteinuria |
|
| In absence of proteinuria |
|
| Severe Pre-Eclampsia | |
| Blood pressure |
|
| Thrombocytopenia |
|
| Liver dysfunction |
|
| Renal dysfunction |
|
| Other |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alanazi, A.S.; Victor, F.; Rehman, K.; Khan, Y.H.; Yunusa, I.; Alzarea, A.I.; Akash, M.S.H.; Mallhi, T.H. Pre-Existing Diabetes Mellitus, Hypertension and KidneyDisease as Risk Factors of Pre-Eclampsia: A Disease of Theories and Its Association with Genetic Polymorphism. Int. J. Environ. Res. Public Health 2022, 19, 16690. https://doi.org/10.3390/ijerph192416690
Alanazi AS, Victor F, Rehman K, Khan YH, Yunusa I, Alzarea AI, Akash MSH, Mallhi TH. Pre-Existing Diabetes Mellitus, Hypertension and KidneyDisease as Risk Factors of Pre-Eclampsia: A Disease of Theories and Its Association with Genetic Polymorphism. International Journal of Environmental Research and Public Health. 2022; 19(24):16690. https://doi.org/10.3390/ijerph192416690
Chicago/Turabian StyleAlanazi, Abdullah Salah, Francis Victor, Kanwal Rehman, Yusra Habib Khan, Ismaeel Yunusa, Abdulaziz Ibrahim Alzarea, Muhammad Sajid Hamid Akash, and Tauqeer Hussain Mallhi. 2022. "Pre-Existing Diabetes Mellitus, Hypertension and KidneyDisease as Risk Factors of Pre-Eclampsia: A Disease of Theories and Its Association with Genetic Polymorphism" International Journal of Environmental Research and Public Health 19, no. 24: 16690. https://doi.org/10.3390/ijerph192416690
APA StyleAlanazi, A. S., Victor, F., Rehman, K., Khan, Y. H., Yunusa, I., Alzarea, A. I., Akash, M. S. H., & Mallhi, T. H. (2022). Pre-Existing Diabetes Mellitus, Hypertension and KidneyDisease as Risk Factors of Pre-Eclampsia: A Disease of Theories and Its Association with Genetic Polymorphism. International Journal of Environmental Research and Public Health, 19(24), 16690. https://doi.org/10.3390/ijerph192416690