
Prenatal Effects of Nicotine on Obesity Risks: A Narrative Review


 ,
, 
Abstract
1. Nicotine
1.1. Epidemiology of Nicotine Usage and Clinical Significance
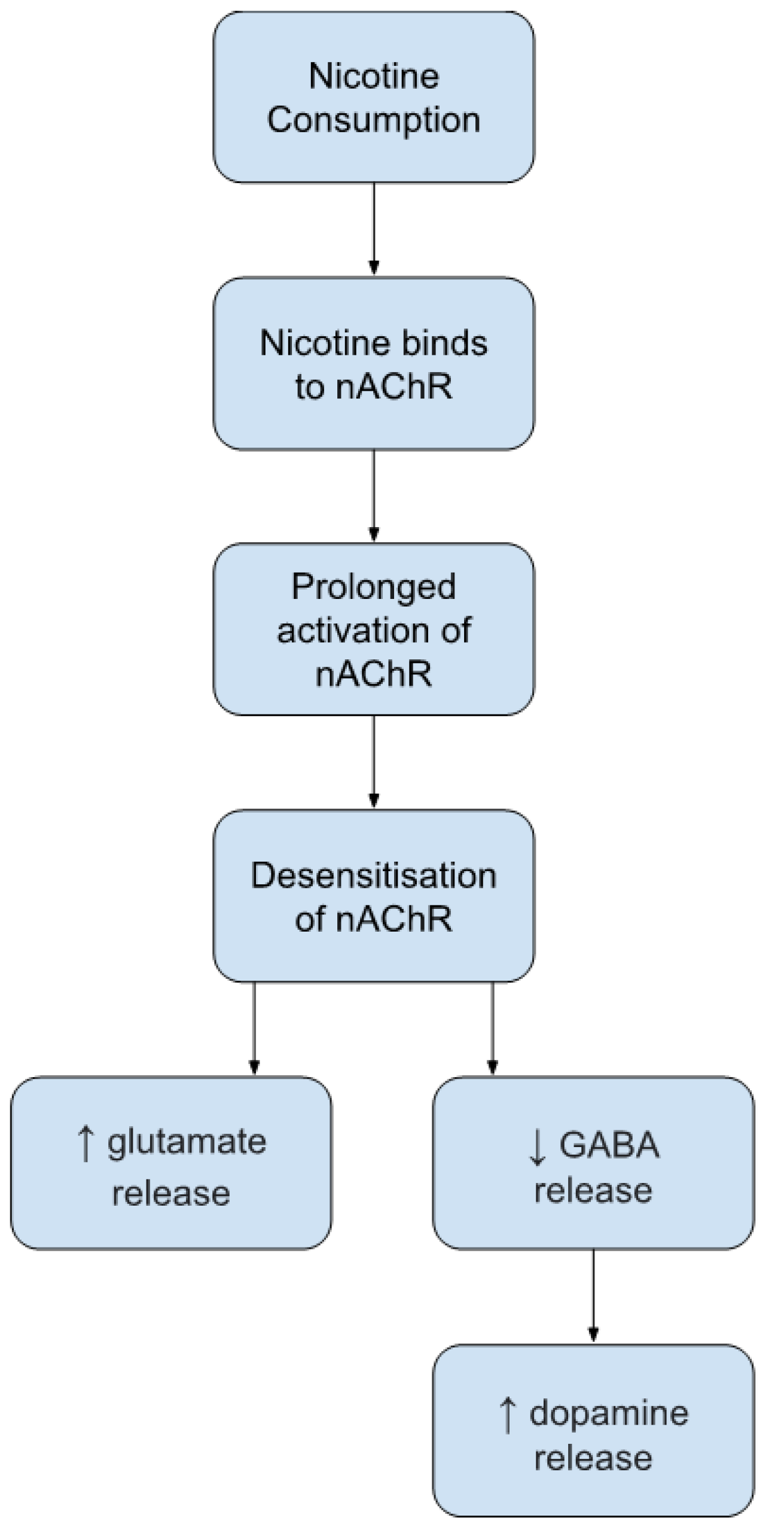
1.2. Nicotine: Mechanisms of Action in Adulthood
2. Obesity
2.1. Epidemiology of Obesity and Clinical Significance
2.2. Obesity and Food Addiction: Mechanisms of Action
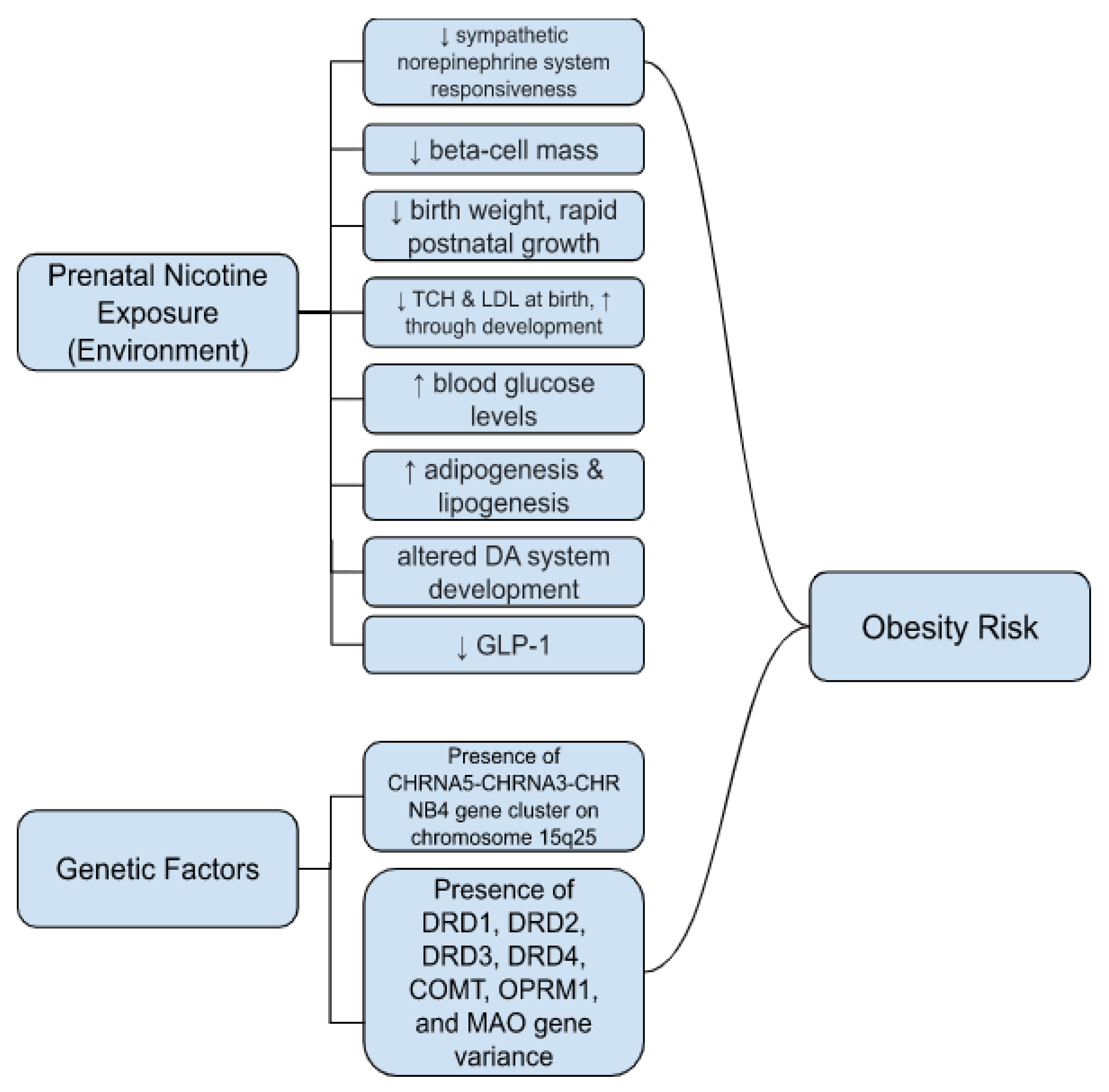
3. Genetic Factors of Nicotine Use and Obesity Risk
Prenatal Nicotine Exposure and Genetic Risk Factors for Obesity
4. Other Hypothesized Mechanisms of Nicotine and Obesity
4.1. Prenatal Nicotine Exposure and Obesity
4.2. The Barker Hypothesis
4.3. Catch-Up Growth Theory
4.4. Thrifty Phenotype Theory
4.5. Neurotransmitter or Endocrine Imbalances Theory
4.6. Adipogenesis, Lipogenesis, and Glucose Metabolism
4.7. Prenatal Nicotine Exposure and Metabolic Disease
4.7.1. Gestational Diabetes Mellitus
4.7.2. Type 1 Diabetes
4.7.3. Type 2 Diabetes
4.7.4. Cholesterol
4.7.5. Liver Disease
5. Prenatal Nicotine Exposure and Obesity Risks through Development
5.1. Leading Mechanism Hypotheses on Prenatal Nicotine Exposure and Obesity
5.2. Prenatal Nicotine Exposure Effects at Early Childhood and Adolescence on Obesity Risks
5.3. Prenatal Nicotine Exposure Effects at Adulthood on Obesity Risks
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valentine, G.; Sofuoglu, M. Cognitive Effects of Nicotine: Recent Progress. Curr. Neuropharmacol. 2018, 16, 403–414. [Google Scholar] [CrossRef]
- Criscitelli, K.; Avena, N.M. The neurobiological and behavioral overlaps of nicotine and food addiction. Prev. Med. 2016, 92, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Lotfipour, S. Nicotine Gateway Effects on Adolescent Substance Use. West J. Emerg. Med. 2019, 20, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Dwoskin, L.P.; Pauly, J.R. Early exposure to nicotine during critical periods of brain development: Mechanisms and consequences. J. Pediatr. Biochem. 2010, 1, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.D. Fetal nicotinic overload, blunted sympathetic responsivity, and obesity. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 481–484. [Google Scholar] [CrossRef]
- Ino, T. Maternal smoking during pregnancy and offspring obesity: Meta-analysis. Pediatr. Int. 2010, 52, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Schwitzgebel, V.M.; Vauthay, D.M.; Aubert, M.L.; Hüppi, P.S. Prenatal nicotine exposure and the programming of metabolic and cardiovascular disorders. Mol. Cell Endocrinol. 2009, 304, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Banderali, G.; Martelli, A.; Landi, M.; Moretti, F.; Betti, F.; Radaelli, G.; Lassandro, C.; Verduci, E. Short and long term health effects of parental tobacco smoking during pregnancy and lactation: A descriptive review. J. Transl. Med. 2015, 13, 327. [Google Scholar] [CrossRef]
- Gaalema, D.E.; Higgins, S.T.; Pepin, C.S.; Heil, S.H.; Bernstein, I.M. Illicit Drug Use Among Pregnant Women Enrolled in Treatment for Cigarette Smoking Cessation. Nicotine Tob. Res. 2013, 15, 987–991. [Google Scholar] [CrossRef]
- Acquavita, S.P.; Talks, A.; Fiser, K. Facilitators and Barriers to Cigarette Smoking While Pregnant for Women With Substance Use Disorders. Nicotine Tob. Res. 2017, 19, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zhang, W.X.; Rao, Y.S.; Xue, J.L.; Wang, F.F.; Zhang, L.; Yan, Y.E. Perinatal Nicotine Exposure Increases Obesity Susceptibility in Adult Male Rat Offspring by Altering Early Adipogenesis. Endocrinology 2016, 157, 4276–4286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kandel, E.R.; Kandel, D.B. A Molecular Basis for Nicotine as a Gateway Drug. N. Engl. J. Med. 2014, 371, 932–943. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Meyerhoff, D.J.; Nixon, S.J. A comprehensive assessment of neurocognition in middle-aged chronic cigarette smokers. Drug Alcohol Depend. 2012, 122, 105–111. [Google Scholar] [CrossRef]
- Tiesler, C.M.T.; Heinrich, J. Prenatal nicotine exposure and child behavioural problems. Eur. Child Adolesc. Psychiatry 2014, 23, 913–929. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, J.; Hinney, A. Environmental and genetic risk factors in obesity. Child Adolesc. Psychiatr. Clin. N. Am. 2009, 18, 83–94. [Google Scholar] [CrossRef]
- Møller, S.E.; Ajslev, T.A.; Andersen, C.S.; Dalgård, C.; Sørensen, T.I. Risk of childhood overweight after exposure to tobacco smoking in prenatal and early postnatal life. PLoS ONE 2014, 9, e109184. [Google Scholar] [CrossRef] [PubMed]
- Rayfield, S.; Plugge, E. Systematic review and meta-analysis of the association between maternal smoking in pregnancy and childhood overweight and obesity. J. Epidemiol. Community Health 2017, 71, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Molnar, D.S.; Rancourt, D.; Schlauch, R.; Wen, X.; Huestis, M.A.; Eiden, R.D. Tobacco Exposure and Conditional Weight-for-Length Gain by 2 Years of Age. J. Pediatric Psychol. 2017, 42, 679–688. [Google Scholar] [CrossRef]
- Rogers, J.M. Smoking and pregnancy: Epigenetics and developmental origins of the metabolic syndrome. Birth Defects Res. 2019, 111, 1259–1269. [Google Scholar] [CrossRef]
- Andersen, M.R.; Simonsen, U.; Uldbjerg, N.; Aalkjær, C.; Stender, S. Smoking Cessation Early in Pregnancy and Birth Weight, Length, Head Circumference, and Endothelial Nitric Oxide Synthase Activity in Umbilical and Chorionic Vessels. Circulation 2009, 119, 857–864. [Google Scholar] [CrossRef]
- Subramaniyan, M.; Dani, J.A. Dopaminergic and cholinergic learning mechanisms in nicotine addiction. Ann. N. Y. Acad. Sci. 2015, 1349, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, E.; Gray, K.; Miller, G.; Tyler, R.; Wiers, C.E.; Volkow, N.D.; Wang, G.J. Food addiction: A common neurobiological mechanism with drug abuse. Front. Biosci. 2018, 23, 811–836. [Google Scholar] [CrossRef]
- Wagner, M.; Schulze-Rauschenbach, S.; Petrovsky, N.; Brinkmeyer, J.; von der Goltz, C.; Gründer, G.; Spreckelmeyer, K.N.; Wienker, T.; Diaz-Lacava, A.; Mobascher, A.; et al. Neurocognitive impairments in non-deprived smokers—results from a population-based multi-center study on smoking-related behavior. Addict. Biol. 2013, 18, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Mackay, D.F.; Gray, L.; Pell, J.P. Impact of smoking and smoking cessation on overweight and obesity: Scotland-wide, cross-sectional study on 40,036 participants. BMC Public Health 2013, 13, 348. [Google Scholar] [CrossRef]
- Dare, S.; Mackay, D.F.; Pell, J.P. Relationship between smoking and obesity: A cross-sectional study of 499,504 middle-aged adults in the UK general population. PLoS ONE 2015, 10, e0123579. [Google Scholar] [CrossRef]
- Jeffery, R.W.; Utter, J. The Changing Environment and Population Obesity in the United States. Obes. Res. 2003, 11, 12S–22S. [Google Scholar] [CrossRef]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 2016, 130, 943–986. [Google Scholar] [CrossRef]
- Rada, P.; Avena, N.M.; Hoebel, B.G. Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 2005, 134, 737–744. [Google Scholar] [CrossRef]
- Thanos, P.K.; Taintor, N.B.; Rivera, S.N.; Umegaki, H.; Ikari, H.; Roth, G.; Ingram, D.K.; Hitzemann, R.; Fowler, J.S.; Gatley, S.J.; et al. DRD2 gene transfer into the nucleus accumbens core of the alcohol preferring and nonpreferring rats attenuates alcohol drinking. Alcohol Clin. Exp. Res. 2004, 28, 720–728. [Google Scholar] [CrossRef]
- Langnäse, K.; Mast, M.; Müller, M.J. Social class differences in overweight of prepubertal children in northwest Germany. Int. J. Obes. 2002, 26, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Gerra, G.; Benedetti, E.; Resce, G.; Potente, R.; Cutilli, A.; Molinaro, S. Socioeconomic Status, Parental Education, School Connectedness and Individual Socio-Cultural Resources in Vulnerability for Drug Use among Students. Int. J. Env. Res. Public Health 2020, 17, 1306. [Google Scholar] [CrossRef] [PubMed]
- Hemmingsson, E. Early Childhood Obesity Risk Factors: Socioeconomic Adversity, Family Dysfunction, Offspring Distress, and Junk Food Self-Medication. Curr. Obes. Rep. 2018, 7, 204–209. [Google Scholar] [CrossRef]
- Drewnowski, A. The Economics of Food Choice Behavior: Why Poverty and Obesity are Linked. Obes. Treat. Prev. New Dir. 2012, 73, 95–112. [Google Scholar] [CrossRef]
- Stice, E.; Burger, K.S.; Yokum, S. Relative ability of fat and sugar tastes to activate reward, gustatory, and somatosensory regions. Am. J. Clin. Nutr. 2013, 98, 1377–1384. [Google Scholar] [CrossRef]
- Rada, P.; Bocarsly, M.E.; Barson, J.R.; Hoebel, B.G.; Leibowitz, S.F. Reduced accumbens dopamine in Sprague–Dawley rats prone to overeating a fat-rich diet. Physiol. Behav. 2010, 101, 394–400. [Google Scholar] [CrossRef]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Kroemer, N.B.; Wuttig, F.; Bidlingmaier, M.; Zimmermann, U.S.; Smolka, M.N. Nicotine enhances modulation of food-cue reactivity by leptin and ghrelin in the ventromedial prefrontal cortex. Addict. Biol. 2015, 20, 832–844. [Google Scholar] [CrossRef]
- Makris, M.C.; Alexandrou, A.; Papatsoutsos, E.G.; Malietzis, G.; Tsilimigras, D.I.; Guerron, A.D.; Moris, D. Ghrelin and Obesity: Identifying Gaps and Dispelling Myths. A Reappraisal. In Vivo 2017, 31, 1047–1050. [Google Scholar] [CrossRef]
- Abizaid, A. Stress and obesity: The ghrelin connection. J. Neuroendocr. 2019, 31, e12693. [Google Scholar] [CrossRef]
- Imperatore, R.; Palomba, L.; Cristino, L. Role of Orexin-A in Hypertension and Obesity. Curr. Hypertens Rep. 2017, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Nixon, J.P.; Mavanji, V.; Butterick, T.A.; Billington, C.J.; Kotz, C.M.; Teske, J.A. Sleep disorders, obesity, and aging: The role of orexin. Ageing Res. Rev. 2015, 20, 63–73. [Google Scholar] [CrossRef]
- Chandra, R.; Liddle, R.A. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 63–67. [Google Scholar] [CrossRef]
- Rehfeld, J.F. Measurement of cholecystokinin in plasma with reference to nutrition related obesity studies. Nutr. Res. 2020, 76, 1–8. [Google Scholar] [CrossRef]
- Wu, Y.; He, H.; Cheng, Z.; Bai, Y.; Ma, X. The Role of Neuropeptide Y and Peptide YY in the Development of Obesity via Gut-brain Axis. Curr. Protein Pept. Sci. 2019, 20, 750–758. [Google Scholar] [CrossRef]
- Lafferty, R.A.; Flatt, P.R.; Irwin, N. Emerging therapeutic potential for peptide YY for obesity-diabetes. Peptides 2018, 100, 269–274. [Google Scholar] [CrossRef]
- Shende, P.; Desai, D. Physiological and Therapeutic Roles of Neuropeptide Y on Biological Functions. Adv. Exp. Med. Biol. 2020, 1237, 37–47. [Google Scholar] [CrossRef]
- Wang, G.-J.; Volkow, N.D.; Thanos, P.K.; Fowler, J.S. Imaging of brain dopamine pathways: Implications for understanding obesity. J. Addict. Med. 2009, 3, 8–18. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Thanos, P.K.; Logan, J.; Alexoff, D.; Ding, Y.S.; Wong, C.; Ma, Y.; et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: Possible contributing factors. Neuroimage 2008, 42, 1537–1543. [Google Scholar] [CrossRef]
- Davis, L.M.; Michaelides, M.; Cheskin, L.J.; Moran, T.H.; Aja, S.; Watkins, P.A.; Pei, Z.; Contoreggi, C.; McCullough, K.; Hope, B.; et al. Bromocriptine administration reduces hyperphagia and adiposity and differentially affects dopamine D2 receptor and transporter binding in leptin-receptor-deficient Zucker rats and rats with diet-induced obesity. Neuroendocrinology 2009, 89, 152–162. [Google Scholar] [CrossRef]
- Thanos, P.K.; Michaelides, M.; Piyis, Y.K.; Wang, G.J.; Volkow, N.D. Food restriction markedly increases dopamine D2 receptor (D2R) in a rat model of obesity as assessed with in-vivo muPET imaging ([11C] raclopride) and in-vitro ([3H] spiperone) autoradiography. Synapse 2008, 62, 50–61. [Google Scholar] [CrossRef]
- Thanos, P.K.; Cho, J.; Kim, R.; Michaelides, M.; Primeaux, S.; Bray, G.; Wang, G.-J.; Volkow, N.D. Bromocriptine increased operant responding for high fat food but decreased chow intake in both obesity-prone and resistant rats. Behav. Brain Res. 2011, 217, 165–170. [Google Scholar] [CrossRef]
- Thanos, P.K.; Michaelides, M.; Gispert, J.D.; Pascau, J.; Soto-Montenegro, M.L.; Desco, M.; Wang, R.; Wang, G.J.; Volkow, N.D. Differences in response to food stimuli in a rat model of obesity: In-vivo assessment of brain glucose metabolism. Int. J. Obes. 2008, 32, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Thanos, P.K.; Wang, G.J.; Febo, M.; Demetrovics, Z.; Modestino, E.J.; Braverman, E.R.; Baron, D.; Badgaiyan, R.D.; Gold, M.S. The Food and Drug Addiction Epidemic: Targeting Dopamine Homeostasis. Curr. Pharm. Des. 2018, 23, 6050–6061. [Google Scholar] [CrossRef]
- Blum, K.; Febo, M.; Thanos, P.K.; Baron, D.; Fratantonio, J.; Gold, M. Clinically Combating Reward Deficiency Syndrome (RDS) with Dopamine Agonist Therapy as a Paradigm Shift: Dopamine for Dinner? Mol. Neurobiol. 2015, 52, 1862–1869. [Google Scholar] [CrossRef]
- Aleixandre, A.; Miguel, M. Ratas Zucker como modelo experimental para el estudio de diferentes enfermedades. Endocrinol. Y Nutr. 2008, 55, 217–222. [Google Scholar] [CrossRef]
- Michaelides, M.; Thanos, P.K.; Kim, R.; Cho, J.; Ananth, M.; Wang, G.J.; Volkow, N.D. PET imaging predicts future body weight and cocaine preference. Neuroimage 2012, 59, 1508–1513. [Google Scholar] [CrossRef]
- Hamilton, J.; Swenson, S.; Hajnal, A.; Thanos, P.K. Roux-en-Y gastric bypass surgery normalizes dopamine D1, D2, and DAT levels. Synapse 2018, 72, e22058. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Kaname, T.; Shimizu-Okabe, C.; Takayama, C.; Tsutsui, M.; Matsushita, M.; Abe, K.; Masuzaki, H. Impact of brown rice-specific γ-oryzanol on epigenetic modulation of dopamine D2 receptors in brain striatum in high-fat-diet-induced obesity in mice. Diabetologia 2017, 60, 1502–1511. [Google Scholar] [CrossRef]
- Michaelides, M.; Thanos, P.K.; Volkow, N.D.; Wang, G.J. Translational neuroimaging in drug addiction and obesity. Ilar J. 2012, 53, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Zald, D.H.; Treadway, M.T. Reward Processing, Neuroeconomics, and Psychopathology. Annu Rev. Clin. Psychol. 2017, 13, 471–495. [Google Scholar] [CrossRef] [PubMed]
- Alvanzo, A.A.; Wand, G.S.; Kuwabara, H.; Wong, D.F.; Xu, X.; McCaul, M.E. Family history of alcoholism is related to increased D(2)/D(3) receptor binding potential: A marker of resilience or risk? Addict. Biol. 2017, 22, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.F.; Benkelfat, C.; Cherkasova, M.V.; Baker, G.B.; Dagher, A.; Leyton, M. Reduced dopamine response to amphetamine in subjects at ultra-high risk for addiction. Biol. Psychiatry 2014, 76, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Pihl, R.O.; Dagher, A.; Schlagintweit, H.; Casey, K.F.; Benkelfat, C.; Leyton, M. Differential striatal dopamine responses following oral alcohol in individuals at varying risk for dependence. Alcohol Clin. Exp. Res. 2014, 38, 126–134. [Google Scholar] [CrossRef]
- Blum, K.; Thanos, P.K.; Gold, M.S. Dopamine and glucose, obesity, and reward deficiency syndrome. Front. Psychol. 2014, 5, 919. [Google Scholar] [CrossRef] [PubMed]
- Febo, M.; Blum, K.; Badgaiyan, R.D.; Baron, D.; Thanos, P.K.; Colon-Perez, L.M.; Demortrovics, Z.; Gold, M.S. Dopamine homeostasis: Brain functional connectivity in reward deficiency syndrome. Front. Biosci. 2017, 22, 669–691. [Google Scholar] [CrossRef]
- Blum, K.; Chen, A.L.C.; Thanos, P.K.; Febo, M.; Demetrovics, Z.; Dushaj, K.; Kovoor, A.; Baron, D.; Smith, D.E.; Roy, A.K., III; et al. Genetic addiction risk score (GARS) ™, a predictor of vulnerability to opioid dependence. Front. Biosci. 2018, 10, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; McLaughlin, T.; Bowirrat, A.; Modestino, E.J.; Baron, D.; Gomez, L.L.; Ceccanti, M.; Braverman, E.R.; Thanos, P.K.; Cadet, J.L.; et al. Reward Deficiency Syndrome (RDS) Surprisingly Is Evolutionary and Found Everywhere: Is It “Blowin’ in the Wind”? J. Pers. Med. 2022, 12, 321. [Google Scholar] [CrossRef]
- Blum, K.; Febo, M.; McLaughlin, T.; Cronjé, F.J.; Han, D.; Gold, S.M. Hatching the behavioral addiction egg: Reward Deficiency Solution System (RDSS)™ as a function of dopaminergic neurogenetics and brain functional connectivity linking all addictions under a common rubric. J. Behav. Addict. 2014, 3, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Bowirrat, A.; Oscar-Berman, M. Relationship between dopaminergic neurotransmission, alcoholism, and Reward Deficiency syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet 2005, 132b, 29–37. [Google Scholar] [CrossRef]
- Chen, A.L.C.; Blum, K.; Chen, T.J.H.; Giordano, J.; Downs, B.W.; Han, D.; Barh, D.; Braverman, E.R. Correlation of the Taq1 dopamine D2 receptor gene and percent body fat in obese and screened control subjects: A preliminary report. Food Funct. 2012, 3, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Crist, R.C.; Reiner, B.C.; Berrettini, W.H. A review of opioid addiction genetics. Curr. Opin. Psychol. 2019, 27, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.C.; Chou, H.W.; Ho, P.S.; Kuo, S.C.; Chen, C.Y.; Huang, C.C.; Liang, C.S.; Lu, R.B.; Huang, S.Y. DRD2 and ANKK1 genes associate with late-onset heroin dependence in men. World J. Biol. Psychiatry 2019, 20, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Uhl, G.; Blum, K.; Noble, E.; Smith, S. Substance abuse vulnerability and D2 receptor genes. Trends Neurosci. 1993, 16, 83–88. [Google Scholar] [CrossRef]
- Lachowicz, M.; Chmielowiec, J.; Chmielowiec, K.; Suchanecka, A.; Masiak, J.; Michałowska-Sawczyn, M.; Mroczek, B.; Mierzecki, A.; Ciechanowicz, I.; Grzywacz, A. Significant association of DRD2 and ANKK1 genes with rural heroin dependence and relapse in men. Ann. Agric. Env. Med. 2020, 27, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Villalba, K.; Devieux, J.G.; Rosenberg, R.; Cadet, J.L. DRD2 and DRD4 genes related to cognitive deficits in HIV-infected adults who abuse alcohol. Behav. Brain Funct. 2015, 11, 25. [Google Scholar] [CrossRef]
- Yang, B.Z.; Kranzler, H.R.; Zhao, H.; Gruen, J.R.; Luo, X.; Gelernter, J. Haplotypic variants in DRD2, ANKK1, TTC12, and NCAM1 are associated with comorbid alcohol and drug dependence. Alcohol Clin. Exp. Res. 2008, 32, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, L.C.; Karoly, H.C.; Thayer, R.E.; Claus, E.D.; Bryan, A.D.; Weiland, B.J.; YorkWilliams, S.; Hutchison, K.E. DRD2 promoter methylation and measures of alcohol reward: Functional activation of reward circuits and clinical severity. Addict. Biol. 2019, 24, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Hagerty, S.L.; YorkWilliams, S.L.; Bidwell, L.C.; Weiland, B.J.; Sabbineni, A.; Blaine, S.K.; Bryan, A.D.; Hutchison, K.E. DRD2 methylation is associated with executive control network connectivity and severity of alcohol problems among a sample of polysubstance users. Addict. Biol. 2020, 25, e12684. [Google Scholar] [CrossRef] [PubMed]
- Stolf, A.R.; Cupertino, R.B.; Müller, D.; Sanvicente-Vieira, B.; Roman, T.; Vitola, E.S.; Grevet, E.H.; von Diemen, L.; Kessler, F.H.P.; Grassi-Oliveira, R.; et al. Effects of DRD2 splicing-regulatory polymorphism and DRD4 48 bp VNTR on crack cocaine addiction. J. Neural. Transm. 2019, 126, 193–199. [Google Scholar] [CrossRef]
- Oyaci, Y.; Aytac, H.M.; Pasin, O.; Cetinay Aydin, P.; Pehlivan, S. Detection of altered methylation of MB-COMT promotor and DRD2 gene in cannabinoid or synthetic cannabinoid use disorder regarding gene variants and clinical parameters. J. Addict. Dis. 2021, 39, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.L.; Nyman, E.; Loukola, A.; Rose, R.J.; Kaprio, J.; Dick, D.M. The association between DRD2/ANKK1 and genetically informed measures of alcohol use and problems. Addict. Biol. 2013, 18, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Voisey, J.; Swagell, C.D.; Hughes, I.P.; van Daal, A.; Noble, E.P.; Lawford, B.R.; Young, R.M.; Morris, C.P. A DRD2 and ANKK1 haplotype is associated with nicotine dependence. Psychiatry Res. 2012, 196, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, F.; Shang, Q.; Song, X.; Miao, X.; Wang, Z. Association between polymorphisms of DRD2 and DRD4 and opioid dependence: Evidence from the current studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156b, 661–670. [Google Scholar] [CrossRef]
- Conner, B.T.; Noble, E.P.; Berman, S.M.; Ozkaragoz, T.; Ritchie, T.; Antolin, T.; Sheen, C. DRD2 genotypes and substance use in adolescent children of alcoholics. Drug Alcohol Depend. 2005, 79, 379–387. [Google Scholar] [CrossRef]
- Esposito-Smythers, C.; Spirito, A.; Rizzo, C.; McGeary, J.E.; Knopik, V.S. Associations of the DRD2 TaqIA polymorphism with impulsivity and substance use: Preliminary results from a clinical sample of adolescents. Pharm. Biochem. Behav. 2009, 93, 306–312. [Google Scholar] [CrossRef][Green Version]
- Picci, G.; Fishbein, D.H.; VanMeter, J.W.; Rose, E.J. Effects of OPRM1 and DRD2 on brain structure in drug-naïve adolescents: Genetic and neural vulnerabilities to substance use. Psychopharmacol. 2022, 239, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Halit, C.; Elif, Y.N.; Hasan, M.A.; Sacide, P.; Yasemin, O.; Deniz, D.; Mehmet, A.U. Methylation of APC2, NR3C1, and DRD2 gene promoters in turkish patients with tobacco use disorder. Niger. J. Clin. Prac. 2022, 25, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Zerdazi, E.H.; Curis, E.; Karsinti, E.; Icick, R.; Fortias, M.; Batel, P.; Cottencin, O.; Orizet, C.; Gay, A.; Coeuru, P.; et al. Occurrence and severity of cocaine-induced hallucinations: Two distinct phenotypes with shared clinical factors but specific genetic risk factors. Drug Alcohol Depend. 2022, 232, 109270. [Google Scholar] [CrossRef] [PubMed]
- Steiger, H.; Thaler, L.; Gauvin, L.; Joober, R.; Labbe, A.; Israel, M.; Kucer, A. Epistatic interactions involving DRD2, DRD4, and COMT polymorphisms and risk of substance abuse in women with binge-purge eating disturbances. J. Psychiatr. Res. 2016, 77, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, L.; Accoto, A.; Couyoumdjian, A.; Conversi, D. A Systematic Review of Genetic Polymorphisms Associated with Binge Eating Disorder. Nutrients 2021, 13, 848. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Levitan, R.D.; Reid, C.; Carter, J.C.; Kaplan, A.S.; Patte, K.A.; King, N.; Curtis, C.; Kennedy, J.L. Dopamine for “wanting” and opioids for “liking”: A comparison of obese adults with and without binge eating. Obesity 2009, 17, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Brunani, A.; Borgomainerio, E.; Tonello, C.; Dioni, L.; Briscini, L.; Redaelli, G.; Molinari, E.; Cavagnini, F.; Carruba, M.O. D2 dopamine receptor (DRD2) gene Taq1A polymorphism and the eating-related psychological traits in eating disorders (anorexia nervosa and bulimia) and obesity. Eat Weight Disord 2007, 12, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Nonino, C.B.; Barato, M.; Ferreira, F.C.; Delfino, H.B.P.; Noronha, N.Y.; Nicoletti, C.F.; Junior, W.S.; Welendorf, C.R.; Souza, D.R.S.; Ferreira-Julio, M.A.; et al. DRD2 and BDNF polymorphisms are associated with binge eating disorder in patients with weight regain after bariatric surgery. Eat Weight Disord 2021, 27, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Levitan, R.D.; Yilmaz, Z.; Kaplan, A.S.; Carter, J.C.; Kennedy, J.L. Binge eating disorder and the dopamine D2 receptor: Genotypes and sub-phenotypes. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 38, 328–335. [Google Scholar] [CrossRef]
- Davis, C.; Levitan, R.D.; Kaplan, A.S.; Carter, J.; Reid, C.; Curtis, C.; Patte, K.; Hwang, R.; Kennedy, J.L. Reward sensitivity and the D2 dopamine receptor gene: A case-control study of binge eating disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 620–628. [Google Scholar] [CrossRef] [PubMed]
- González, L.M.; Mota-Zamorano, S.; García-Herráiz, A.; López-Nevado, E.; Gervasini, G. Genetic variants in dopamine pathways affect personality dimensions displayed by patients with eating disorders. Eat Weight Disord 2021, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, L.; Tuulari, J.; Karlsson, H.; Hirvonen, J.; Helin, S.; Salminen, P.; Parkkola, R.; Hietala, J.; Nuutila, P.; Nummenmaa, L. Aberrant mesolimbic dopamine-opiate interaction in obesity. Neuroimage 2015, 122, 80–86. [Google Scholar] [CrossRef]
- Groleau, P.; Joober, R.; Israel, M.; Zeramdini, N.; DeGuzman, R.; Steiger, H. Methylation of the dopamine D2 receptor (DRD2) gene promoter in women with a bulimia-spectrum disorder: Associations with borderline personality disorder and exposure to childhood abuse. J. Psychiatr. Res. 2014, 48, 121–127. [Google Scholar] [CrossRef]
- Groleau, P.; Steiger, H.; Joober, R.; Bruce, K.R.; Israel, M.; Badawi, G.; Zeramdini, N.; Sycz, L. Dopamine-system genes, childhood abuse, and clinical manifestations in women with Bulimia-Spectrum Disorders. J. Psychiatr. Res. 2012, 46, 1139–1145. [Google Scholar] [CrossRef]
- Frieling, H.; Römer, K.D.; Scholz, S.; Mittelbach, F.; Wilhelm, J.; De Zwaan, M.; Jacoby, G.E.; Kornhuber, J.; Hillemacher, T.; Bleich, S. Epigenetic dysregulation of dopaminergic genes in eating disorders. Int. J. Eat Disord 2010, 43, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Carlin, J.L.; McKee, S.E.; Hill-Smith, T.; Grissom, N.M.; George, R.; Lucki, I.; Reyes, T.M. Removal of high-fat diet after chronic exposure drives binge behavior and dopaminergic dysregulation in female mice. Neuroscience 2016, 326, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Cardel, M.I.; Lemas, D.J.; Lee, A.M.; Miller, D.R.; Huo, T.; Klimentidis, Y.C.; Fernandez, J.R. Taq1a polymorphism (rs1800497) is associated with obesity-related outcomes and dietary intake in a multi-ethnic sample of children. Pediatr. Obes. 2019, 14, e12470. [Google Scholar] [CrossRef] [PubMed]
- Bailer, U.F.; Frank, G.K.; Price, J.C.; Meltzer, C.C.; Becker, C.; Mathis, C.A.; Wagner, A.; Barbarich-Marsteller, N.C.; Bloss, C.S.; Putnam, K.; et al. Interaction between serotonin transporter and dopamine D2/D3 receptor radioligand measures is associated with harm avoidant symptoms in anorexia and bulimia nervosa. Psychiatry Res. 2013, 211, 160–168. [Google Scholar] [CrossRef]
- Fried, L.; Modestino, E.J.; Siwicki, D.; Lott, L.; Thanos, P.K.; Baron, D.; Badgaiyan, R.D.; Ponce, J.V.; Giordano, J.; Downs, W.B.; et al. Hypodopaminergia and “Precision Behavioral Management” (PBM): It is a Generational Family Affair. Curr. Pharm. Biotechnol. 2020, 21, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Bowirrat, A.; Gondre Lewis, M.C.; Simpatico, T.A.; Ceccanti, M.; Steinberg, B.; Modestino, E.J.; Thanos, P.K.; Baron, D.; McLaughlin, T.; et al. Exploration of Epigenetic State Hyperdopaminergia (Surfeit) and Genetic Trait Hypodopaminergia (Deficit) During Adolescent Brain Development. Curr. Psychopharmacol. 2021, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gondré-Lewis, M.C.; Bassey, R.; Blum, K. Pre-clinical models of reward deficiency syndrome: A behavioral octopus. Neurosci. Biobehav. Rev. 2020, 115, 164–188. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Cadet, J.L.; Gold, M.S. Psychostimulant use disorder emphasizing methamphetamine and the opioid -dopamine connection: Digging out of a hypodopaminergic ditch. J. Neurol. Sci. 2021, 420, 117252. [Google Scholar] [CrossRef]
- Blum, K.; Gold, M.S.; Llanos-Gomez, L.; Jalali, R.; Thanos, P.K.; Bowirrat, A.; Downs, W.B.; Bagchi, D.; Braverman, E.R.; Baron, D.; et al. Hypothesizing Nutrigenomic-Based Precision Anti-Obesity Treatment and Prophylaxis: Should We Be Targeting Sarcopenia Induced Brain Dysfunction? Int. J. Env. Res. Public Health 2021, 18, 9774. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Morgan, J.; Cadet, J.L.; Baron, D.; Carney, P.R.; Khalsa, J.; Badgaiyan, R.D.; Gold, M.S. Psychoactive Drugs Like Cannabis -Induce Hypodopaminergic Anhedonia and Neuropsychological Dysfunction in Humans: Putative Induction of Dopamine Homeostasis via Coupling of Genetic Addiction Risk Severity (GARS) testing and Precision Pro-dopamine Regulation (KB220). Neurology 2021, 13, 86–92. [Google Scholar]
- Blum, K.; Marcelo, F.; Dushaj, K.; Fried, L.; Badgaiyan, R.D. “Pro-dopamine regulation (KB220Z™)” as a long-term therapeutic modality to overcome reduced resting state dopamine tone in opiate/opioid epidemic in America. J. Syst. Integr. Neurosci. 2016, 2, 162–165. [Google Scholar] [CrossRef]
- Epstein, L.H.; Dearing, K.K.; Erbe, R.W. Parent-child concordance of Taq1 A1 allele predicts similarity of parent-child weight loss in behavioral family-based treatment programs. Appetite 2010, 55, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Comings, D.E.; Gade, R.; MacMurray, J.P.; Muhleman, D.; Peters, W.R. Genetic variants of the human obesity (OB) gene: Association with body mass index in young women, psychiatric symptoms, and interaction with the dopamine D2 receptor (DRD2) gene. Mol. Psychiatry 1996, 1, 325–335. [Google Scholar] [PubMed]
- Thanos, P.K.; Michaelides, M.; Umegaki, H.; Volkow, N.D. D2R DNA transfer into the nucleus accumbens attenuates cocaine self-administration in rats. Synapse 2008, 62, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Thanos, P.K.; Michaelides, M.; Benveniste, H.; Wang, G.J.; Volkow, N.D. Effects of chronic oral methylphenidate on cocaine self-administration and striatal dopamine D2 receptors in rodents. Pharm. Biochem. Behav. 2007, 87, 426–433. [Google Scholar] [CrossRef]
- Thanos, P.K.; Volkow, N.D.; Freimuth, P.; Umegaki, H.; Ikari, H.; Roth, G.; Ingram, D.K.; Hitzemann, R. Overexpression of dopamine D2 receptors reduces alcohol self-administration. J. Neurochem. 2001, 78, 1094–1103. [Google Scholar] [CrossRef]
- Thanos, P.K.; Rivera, S.N.; Weaver, K.; Grandy, D.K.; Rubinstein, M.; Umegaki, H.; Wang, G.J.; Hitzemann, R.; Volkow, N.D. Dopamine D2R DNA transfer in dopamine D2 receptor-deficient mice: Effects on ethanol drinking. Life Sci. 2005, 77, 130–139. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.J.; Fowler, J.S.; Thanos, P.P.; Logan, J.; Gatley, S.J.; Gifford, A.; Ding, Y.S.; Wong, C.; Pappas, N. Brain DA D2 receptors predict reinforcing effects of stimulants in humans: Replication study. Synapse 2002, 46, 79–82. [Google Scholar] [CrossRef]
- Pinheiro, C.R.; Oliveira, E.; Manhães, A.C.; Fraga, M.C.; Claudio-Neto, S.; Younes-Rapozo, V.; Lotufo, B.M.; Moura, E.G.; Lisboa, P.C. Exposure to nicotine increases dopamine receptor content in the mesocorticolimbic pathway of rat dams and offspring during lactation. Pharm. Biochem. Behav. 2015, 136, 87–101. [Google Scholar] [CrossRef]
- Pinheiro, C.R.; Moura, E.G.; Manhães, A.C.; Fraga, M.C.; Claudio-Neto, S.; Younes-Rapozo, V.; Santos-Silva, A.P.; Lotufo, B.M.; Oliveira, E.; Lisboa, P.C. Maternal nicotine exposure during lactation alters food preference, anxiety-like behavior and the brain dopaminergic reward system in the adult rat offspring. Physiol. Behav. 2015, 149, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.R.; Moura, E.G.; Manhães, A.C.; Fraga, M.C.; Claudio-Neto, S.; Abreu-Villaça, Y.; Oliveira, E.; Lisboa, P.C. Concurrent maternal and pup postnatal tobacco smoke exposure in Wistar rats changes food preference and dopaminergic reward system parameters in the adult male offspring. Neuroscience 2015, 301, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Deng, J.; Shi, L.; Wang, Q.; Li, P.; Li, H.; Liu, J.; Que, J.; Chang, S.; Bao, Y.; et al. Neural substrates of smoking and reward cue reactivity in smokers: A meta-analysis of fMRI studies. Transl. Psychiatry 2020, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- David, S.P.; Munafò, M.R.; Johansen-Berg, H.; Smith, S.M.; Rogers, R.D.; Matthews, P.M.; Walton, R.T. Ventral striatum/nucleus accumbens activation to smoking-related pictorial cues in smokers and nonsmokers: A functional magnetic resonance imaging study. Biol. Psychiatry 2005, 58, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Ruzilawati, A.B.; Islam, M.A.; Muhamed, S.K.S.; Ahmad, I. Smoking Genes: A Case-Control Study of Dopamine Transporter Gene (SLC6A3) and Dopamine Receptor Genes (DRD1, DRD2 and DRD3) Polymorphisms and Smoking Behaviour in a Malay Male Cohort. Biomolecules 2020, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Macare, C.; Ducci, F.; Zhang, Y.; Ruggeri, B.; Jia, T.; Kaakinen, M.; Kalsi, G.; Charoen, P.; Casoni, F.; Peters, J.; et al. A neurobiological pathway to smoking in adolescence: TTC12-ANKK1-DRD2 variants and reward response. Eur. Neuropsychopharmacol. 2018, 28, 1103–1114. [Google Scholar] [CrossRef]
- Ducci, F.; Kaakinen, M.; Pouta, A.; Hartikainen, A.L.; Veijola, J.; Isohanni, M.; Charoen, P.; Coin, L.; Hoggart, C.; Ekelund, J.; et al. TTC12-ANKK1-DRD2 and CHRNA5-CHRNA3-CHRNB4 influence different pathways leading to smoking behavior from adolescence to mid-adulthood. Biol. Psychiatry 2011, 69, 650–660. [Google Scholar] [CrossRef]
- Radwan, G.N.; Loffredo, C.A.; Setouhy, E.M.A.; Abdel Hamid, M.; Israel, E.J.; Mohamed, M.K. Waterpipe Smoking and The DRD2/ANKK1 Genotype. J. Egypt Public Health Assoc. 2010, 85, 131–148. [Google Scholar] [PubMed]
- Feistauer, V.; Fisch, J.; da Silva Oliveira, C.K.; Giovenardi, M.; Almeida, S. Restriction and hyperlipidic diets during pregnancy, lactation and adult life modified the expression of dopaminergic system related genes both in female mice and their adult offspring. Brain Res. Bull. 2020, 162, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.S.; Matos, R.J.B.; Cordeiro, G.S.; Perez, G.S.; Santo, D.A.E.; Silva, R.T.; Gonçalves, M.S.; Boaventura, G.T.; Deiró, T.; Barreto-Medeiros, J.M. Perinatal exposure to a high-fat diet alters proopiomelanocortin, neuropeptide Y and dopaminergic receptors gene expression and the food preference in offspring adult rats. Braz. J. Biol. 2021, 82, e234855. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Kaizaki-Mitsumoto, A.; Hattori, N.; Numazawa, S. Fetal methylphenidate exposure induced ADHD-like phenotypes and decreased Drd2 and Slc6a3 expression levels in mouse offspring. Toxicol. Lett. 2021, 344, 1–10. [Google Scholar] [CrossRef]
- Eisenberg, D.T.; Campbell, B.; Mackillop, J.; Lum, J.K.; Wilson, D.S. Season of birth and dopamine receptor gene associations with impulsivity, sensation seeking and reproductive behaviors. PLoS ONE 2007, 2, e1216. [Google Scholar] [CrossRef] [PubMed]
- Eicher, J.D.; Powers, N.R.; Cho, K.; Miller, L.L.; Mueller, K.L.; Ring, S.M.; Tomblin, J.B.; Gruen, J.R. Associations of prenatal nicotine exposure and the dopamine related genes ANKK1 and DRD2 to verbal language. PLoS ONE 2013, 8, e63762. [Google Scholar] [CrossRef]
- Vrantsidis, D.M.; Clark, C.A.C.; Volk, A.; Wakschlag, L.S.; Andrews Espy, K.; Wiebe, S.A. Exploring the interplay of dopaminergic genotype and parental behavior in relation to executive function in early childhood. Dev. Psychopathol. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, T.C.; Moura, E.G.; Soares, P.N.; Rodrigues, V.S.T.; Claudio-Neto, S.; Oliveira, E.; Manhães, A.C.; Lisboa, P.C. Nicotine exposure during lactation causes disruption of hedonic eating behavior and alters dopaminergic system in adult female rats. Appetite 2021, 160, 105115. [Google Scholar] [CrossRef] [PubMed]
- Oei, J.L.; Xu, H.X.; Abdel-Latif, M.E.; Vunnam, K.; Al-Amry, A.; Clews, S.; Falconer, J.; Feller, J.M.; Lui, K. Dopamine D2 receptor gene polymorphisms in newborn infants of drug-using women. Arch. Dis. Child Fetal Neonatal. Ed. 2012, 97, F193–F198. [Google Scholar] [CrossRef] [PubMed]
- Bilibio, J.P.; Matte, Ú.; de Conto, E.; Cunha-Filho, J.S. Recurrent miscarriage is associated with the dopamine receptor (DRD2) genotype. Gynecol. Endocrinol. 2015, 31, 866–869. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.M.; Morgan, T.J., Jr.; Lowe, S.E.; Williamson, M.J.; Spencer, T.J.; Biederman, J.; Bhide, P.G. Nicotine exposure of male mice produces behavioral impairment in multiple generations of descendants. PLoS Biol. 2018, 16, e2006497. [Google Scholar] [CrossRef] [PubMed]
- DiNieri, J.A.; Wang, X.; Szutorisz, H.; Spano, S.M.; Kaur, J.; Casaccia, P.; Dow-Edwards, D.; Hurd, Y.L. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biol. Psychiatry 2011, 70, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Said, N.; Lakehayli, S.; El Khachibi, M.; El Ouahli, M.; Nadifi, S.; Hakkou, F.; Tazi, A. Prenatal stress induces vulnerability to nicotine addiction and alters D2 receptors’ expression in the nucleus accumbens in adult rats. Neuroscience 2015, 304, 279–285. [Google Scholar] [CrossRef]
- Aliasghari, F.; Nazm, S.A.; Yasari, S.; Mahdavi, R.; Bonyadi, M. Associations of the ANKK1 and DRD2 gene polymorphisms with overweight, obesity and hedonic hunger among women from the Northwest of Iran. Eat Weight Disord 2021, 26, 305–312. [Google Scholar] [CrossRef]
- Sun, X.; Luquet, S.; Small, D.M. DRD2: Bridging the Genome and Ingestive Behavior. Trends Cogn. Sci. 2017, 21, 372–384. [Google Scholar] [CrossRef]
- Ariza, M.; Garolera, M.; Jurado, M.A.; Garcia-Garcia, I.; Hernan, I.; Sánchez-Garre, C.; Vernet-Vernet, M.; Sender-Palacios, M.J.; Marques-Iturria, I.; Pueyo, R.; et al. Dopamine genes (DRD2/ANKK1-TaqA1 and DRD4-7R) and executive function: Their interaction with obesity. PLoS ONE 2012, 7, e41482. [Google Scholar] [CrossRef]
- Lek, F.Y.; Ong, H.H.; Say, Y.H. Association of dopamine receptor D2 gene (DRD2) Taq1 polymorphisms with eating behaviors and obesity among Chinese and Indian Malaysian university students. Asia Pac. J. Clin. Nutr. 2018, 27, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Iñiguez, I.; Panduro, A.; Ramos-Lopez, O.; Villaseñor-Bayardo, S.J.; Roman, S. DRD2/ANKK1 TaqI A1 polymorphism associates with overconsumption of unhealthy foods and biochemical abnormalities in a Mexican population. Eat Weight Disord 2019, 24, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Heni, M.; Kullmann, S.; Ahlqvist, E.; Wagner, R.; Machicao, F.; Staiger, H.; Häring, H.U.; Almgren, P.; Groop, L.C.; Small, D.M.; et al. Interaction between the obesity-risk gene FTO and the dopamine D2 receptor gene ANKK1/TaqIA on insulin sensitivity. Diabetologia 2016, 59, 2622–2631. [Google Scholar] [CrossRef] [PubMed]
- Noble, E.P.; Noble, R.E.; Ritchie, T.; Syndulko, K.; Bohlman, M.C.; Noble, L.A.; Zhang, Y.; Sparkes, R.S.; Grandy, D.K. D2 dopamine receptor gene and obesity. Int. J. Eat Disord 1994, 15, 205–217. [Google Scholar] [CrossRef]
- Blum, K.; Chen, A.L.; Chen, T.J.; Rhoades, P.; Prihoda, T.J.; Downs, B.W.; Waite, R.L.; Williams, L.; Braverman, E.R.; Braverman, D.; et al. LG839: Anti-obesity effects and polymorphic gene correlates of reward deficiency syndrome. Adv. Ther. 2008, 25, 894–913. [Google Scholar] [CrossRef] [PubMed]
- Stice, E.; Spoor, S.; Bohon, C.; Small, D.M. Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science 2008, 322, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Hillemacher, T.; Rhein, M.; Burkert, A.; Heberlein, A.; Wilhelm, J.; Glahn, A.; Muschler, M.A.N.; Kahl, K.G.; Kornhuber, J.; Bleich, S.; et al. DNA-methylation of the dopamin receptor 2 gene is altered during alcohol withdrawal. Eur. Neuropsychopharmacol. 2019, 29, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Kazmi, S.; Modestino, E.J.; Downs, B.W.; Bagchi, D.; Baron, D.; McLaughlin, T.; Green, R.; Jalali, R.; Thanos, P.K.; et al. A Novel Precision Approach to Overcome the “Addiction Pandemic” by Incorporating Genetic Addiction Risk Severity (GARS) and Dopamine Homeostasis Restoration. J. Pers. Med. 2021, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Bowirrat, A.; Baron, D.; Lott, L.; Ponce, J.V.; Brewer, R.; Siwicki, D.; Boyett, B.; Gondre-Lewis, M.C.; Smith, D.E.; et al. Biotechnical development of genetic addiction risk score (GARS) and selective evidence for inclusion of polymorphic allelic risk in substance use disorder (SUD). J. Syst. Integr. Neurosci. 2020, 6, 1–38. [Google Scholar] [CrossRef]
- Blum, K.; Bowirrat, A.; Braverman, E.R.; Baron, D.; Cadet, J.L.; Kazmi, S.; Elman, I.; Thanos, P.K.; Badgaiyan, R.D.; Downs, W.B.; et al. Reward Deficiency Syndrome (RDS): A Cytoarchitectural Common Neurobiological Trait of All Addictions. Int. J. Env. Res. Public Health 2021, 18, 11529. [Google Scholar] [CrossRef]
- Moran, M.; Blum, K.; Ponce, J.V.; Lott, L.; Gondré-Lewis, M.C.; Badgaiyan, S.; Brewer, R.; Downs, B.W.; Fynman, P.; Weingarten, A.; et al. High Genetic Addiction Risk Score (GARS) in Chronically Prescribed Severe Chronic Opioid Probands Attending Multi-pain Clinics: An Open Clinical Pilot Trial. Mol. Neurobiol. 2021, 58, 3335–3346. [Google Scholar] [CrossRef]
- Blum, K.; Modestino, E.J.; Gondre-Lewis, M.; Chapman, E.J.; Neary, J.; Siwicki, D.; Baron, D.; Hauser, M.; Smith, D.E.; Roy, A.K.; et al. The Benefits of Genetic Addiction Risk Score (GARS(™)) Testing in Substance Use Disorder (SUD). Int. J. Genom. Data Min. 2018, 2018, 115. [Google Scholar] [CrossRef]
- Blum, K.; Steinberg, B.; Gondre-Lewis, M.C.; Baron, D.; Modestino, E.J.; Badgaiyan, R.D.; Downs, B.W.; Bagchi, D.; Brewer, R.; McLaughlin, T.; et al. A Review of DNA Risk Alleles to Determine Epigenetic Repair of mRNA Expression to Prove Therapeutic Effectiveness in Reward Deficiency Syndrome (RDS): Embracing “Precision Behavioral Management”. Psychol. Res. Behav. Manag. 2021, 14, 2115–2134. [Google Scholar] [CrossRef]
- Blum, K.; Madigan, M.A.; Fried, L.; Braverman, E.R.; Giordano, J.; Badgaiyan, R.D. Coupling Genetic Addiction Risk Score (GARS) and Pro Dopamine Regulation (KB220) to Combat Substance Use Disorder (SUD). Glob J. Addict. Rehabil. Med. 2017, 1, 555556. [Google Scholar] [CrossRef]
- Blum, K.; Oscar-Berman, M.; Demetrovics, Z.; Barh, D.; Gold, M.S. Genetic Addiction Risk Score (GARS): Molecular neurogenetic evidence for predisposition to Reward Deficiency Syndrome (RDS). Mol. Neurobiol. 2014, 50, 765–796. [Google Scholar] [CrossRef]
- Blum, K.; Oscar-Berman, M.; Femino, J.; Waite, R.L.; Benya, L.; Giordano, J.; Borsten, J.; Downs, W.B.; Braverman, E.R.; Loehmann, R.; et al. Withdrawal from Buprenorphine/Naloxone and Maintenance with a Natural Dopaminergic Agonist: A Cautionary Note. J. Addict. Res. 2013, 4, 1–25. [Google Scholar] [CrossRef]
- Blum, K.; Lott, L.; Siwicki, D.; Fried, L.; Hauser, M.; Simpatico, T.; Baron, D.; Howeedy, A.; Badgaiyan, R.D. Genetic Addiction Risk Score (GARS(™)) as a Predictor of Substance Use Disorder: Identifying Predisposition Not Diagnosis. Curr. Trends Med. Diagn Methods 2018, 1, 1–5. [Google Scholar] [CrossRef]
- Blum, K.; Downs, B.W.; Dushaj, K.; Li, M.; Braverman, E.R.; Fried, L.; Waite, R.; Demotrovics, Z.; Badgaiyan, R.D. The benefits of customized dna directed nutrition to balance the brain reward circuitry and reduce addictive behaviors. Precis. Med. 2016, 1, 18–33. [Google Scholar]
- Blum, K.; Gondré-Lewis, M.C.; Baron, D.; Thanos, P.K.; Braverman, E.R.; Neary, J.; Elman, I.; Badgaiyan, R.D. Introducing Precision Addiction Management of Reward Deficiency Syndrome, the Construct that Underpins All Addictive Behaviors. Front. Psychiatry 2018, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Blum, K.; Oscar-Berman, M.; Dinubile, N.; Giordano, J.; Braverman, E.R.; Truesdell, C.E.; Barh, D.; Badgaiyan, R. Coupling Genetic Addiction Risk Score (GARS) with Electrotherapy: Fighting Iatrogenic Opioid Dependence. J. Addict. Res. 2013, 4, 1000163. [Google Scholar] [CrossRef]
- Huang, W.; Ma, J.Z.; Payne, T.; Beuten, J.; Dupont, R.T.; Li, M.D. Significant association of DRD1 with nicotine dependence. Qual. Life Res. 2007, 123, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Payne, T.J.; Ma, J.Z.; Li, M.D. A functional polymorphism, rs6280, in DRD3 is significantly associated with nicotine dependence in European-American smokers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147b, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rubio, G.; Ramírez-Venegas, A.; Díaz, V.N.; Gómez, L.G.; Fabián, K.E.; Carmona, S.G.; López-Flores, L.A.; Ambrocio-Ortiz, E.; Romero, R.C.; Alcantar-Ayala, N.; et al. Polymorphisms in HTR2A and DRD4 Predispose to Smoking and Smoking Quantity. PLoS ONE 2017, 12, e0170019. [Google Scholar] [CrossRef]
- Schröder, R.; Reuter, M.; Faßbender, K.; Plieger, T.; Poulsen, J.; Lui, S.S.; Chan, R.C.; Ettinger, U. The role of the SLC6A3 3′ UTR VNTR in nicotine effects on cognitive, affective, and motor function. Psychopharmacology 2022, 239, 489–507. [Google Scholar] [CrossRef] [PubMed]
- Beuten, J.; Payne, T.J.; Ma, J.Z.; Li, M.D. Significant Association of Catechol-O-Methyltransferase (COMT) Haplotypes with Nicotine Dependence in Male and Female Smokers of Two Ethnic Populations. Neuropsychopharmacology 2005, 31, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Schwantes-An, T.-H.; Zhang, J.; Chen, L.-S.; Hartz, S.M.; Culverhouse, R.C.; Chen, X.; Coon, H.; Frank, J.; Kamens, H.; Konte, B.; et al. Association of the OPRM1 Variant rs1799971 (A118G) with Non-Specific Liability to Substance Dependence in a Collaborative de novo Meta-Analysis of European-Ancestry Cohorts. Behav. Genet. 2015, 46, 151–169. [Google Scholar] [CrossRef]
- Dick, D.M.; Edenberg, H.; Xuei, X.; Goate, A.; Kuperman, S.; Schuckit, M.; Crowe, R.; Smith, T.L.; Porjesz, B.; Begleiter, H.; et al. Association of GABRG3 With Alcohol Dependence. Alcohol. Clin. Exp. Res. 2004, 28, 4–9. [Google Scholar] [CrossRef]
- Ito, H.; Hamajima, N.; Matsuo, K.; Okuma, K.; Sato, S.; Ueda, R.; Tajima, K. Monoamine oxidase polymorphisms and smoking behaviour in Japanese. Pharmacogenetics 2003, 13, 73–79. [Google Scholar] [CrossRef]
- Sieminska, A.; Buczkowski, K.; Jassem, E.; Tkacz, E. Lack of association between serotonin transporter gene polymorphism 5-HTTLPR and smoking among Polish population: A case-control study. BMC Med Genet. 2008, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Ducci, F.; Goldman, D. The Genetic Basis of Addictive Disorders. Psychiatr. Clin. N. Am. 2012, 35, 495–519. [Google Scholar] [CrossRef] [PubMed]
- Gorwood, P.; Le Strat, Y.; Ramoz, N. Genetics of addictive behavior: The example of nicotine dependence. Dialog- Clin. Neurosci. 2017, 19, 237–245. [Google Scholar] [CrossRef]
- Thorgeirsson, T.E.; Geller, F.; Sulem, P.; Rafnar, T.; Wiste, A.; Magnússon, K.P.; Manolescu, A.; Thorleifsson, G.; Stefansson, H.; Ingason, A.; et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 2008, 452, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.H.C.; Brick, L.; Nugent, N.; Bidwell, L.C.; McGeary, J.E.; Knopik, V.; Keller, M.C. Examining the role of common genetic variants on alcohol, tobacco, cannabis and illicit drug dependence: Genetics of vulnerability to drug dependence. Addiction 2014, 110, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Tiili, E.M.; Mitiushkina, N.; Sukhovskaya, O.A.; Imyanitov, E.N.; Hirvonen, A.P. The genotypes and methylation of MAO genes as factors behind smoking behavior. Pharm. Genom. 2017, 27, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.L.; Ahmed, M.L.; Emmett, P.M.; Preece, M.A.; Dunger, P.D.B. Association between postnatal catch-up growth and obesity in childhood: Prospective cohort study. BMJ 2000, 320, 967–971. [Google Scholar] [CrossRef]
- Harris, H.R.; Willett, W.C.; Michels, K.B. Parental smoking during pregnancy and risk of overweight and obesity in the daughter. Int. J. Obes. 2013, 37, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Gillman, M.W. Fetal Origins of Obesity. Obes. Res. 2003, 11, 496–506. [Google Scholar] [CrossRef]
- Bruin, J.E.; Gerstein, H.C.; Morrison, K.M.; Holloway, A.C. Increased Pancreatic Beta-Cell Apoptosis following Fetal and Neonatal Exposure to Nicotine is Mediated via the Mitochondria. Toxicol. Sci. 2008, 103, 362–370. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.; Walker, M.; Perkins, S.; Beyene, J.; Murphy, K.; Gibb, W.; Ohlsson, A. The effect of tobacco exposure on the fetal hypothalamic? pituitary? adrenal axis. BJOG Int. J. Obstet. Gynaecol. 2006, 113, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.P. Adult Consequences of Fetal Growth Restriction. Clin. Obstet. Gynecol. 2006, 49, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.C.K. The thrifty phenotype as an adaptive maternal effect. Biol. Rev. 2007, 82, 143–172. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.P. Type 2 (non-insulin-dependent) diabetes mellitus: The thrifty phenotype hypothesis. Diabetologia 1992, 35, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Ozanne, S.E.; Hales, C.N. The long-term consequences of intra-uterine protein malnutrition for glucose metabolism. Proc. Nutr. Soc. 1999, 58, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Young, J.B.; Morrison, S.F. Effects of fetal and neonatal environment on sympathetic nervous system development. Diabetes Care 1998, 21, B156. [Google Scholar]
- Holst, J.J. The Physiology of Glucagon-like Peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Eren-Yazicioglu, C.Y.; Yigit, A.; Dogruoz, R.E.; Yapici-Eser, H. Can GLP-1 Be a Target for Reward System Related Disorders? A Qualitative Synthesis and Systematic Review Analysis of Studies on Palatable Food, Drugs of Abuse, and Alcohol. Front. Behav. Neurosci. 2021, 14, 614884. [Google Scholar] [CrossRef] [PubMed]
- Egecioglu, E.; Engel, J.A.; Jerlhag, E. The Glucagon-Like Peptide 1 Analogue Exendin-4 Attenuates the Nicotine-Induced Locomotor Stimulation, Accumbal Dopamine Release, Conditioned Place Preference as well as the Expression of Locomotor Sensitization in Mice. PLoS ONE 2013, 8, e77284. [Google Scholar] [CrossRef]
- Tuesta, L.M.; Chen, Z.; Duncan, A.; Fowler, C.; Ishikawa, M.; Lee, B.R.; Liu, X.-A.; Lu, Q.; Cameron, M.; Hayes, M.R.; et al. GLP-1 acts on habenular avoidance circuits to control nicotine intake. Nat. Neurosci. 2017, 20, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-L.; Ping, J.; Chen, H.-J.; Zhang, W.-X.; Fan, J.; Peng, D.-S.; Zhang, L.; Yan, Y.-E. Maternal nicotine exposure impairs brown adipose tissue via AMPK-SIRT1-PGC-1α signals in male offspring. Life Sci. 2020, 264, 118695. [Google Scholar] [CrossRef] [PubMed]
- Holloway, A.C.; Lim, G.; Petrik, J.J.; Foster, W.G.; Morrison, K.; Gerstein, H. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia 2005, 48, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- Strauss, W.T.; Hales, C.N. Plasma insulin in minor abnormalities of glucose tolerance: A 5 year follow-up. Diabetologia 1974, 10, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Huh, S.Y.; Taveras, E.M.; Rich-Edwards, J.W.; Gillman, M.W. Associations of Maternal Prenatal Smoking with Child Adiposity and Blood Pressure. Obes. Res. 2005, 13, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Högberg, L.; Cnattingius, S.; Lundholm, C.; D’Onofrio, B.M.; Långström, N.; Iliadou, A.N. Effects of maternal smoking during pregnancy on offspring blood pressure in late adolescence. J. Hypertens. 2012, 30, 693–699. [Google Scholar] [CrossRef]
- Casagrande, S.S.; Linder, B.; Cowie, C.C. Prevalence of gestational diabetes and subsequent Type 2 diabetes among U.S. women. Diabetes Res. Clin. Prac. 2018, 141, 200–208. [Google Scholar] [CrossRef]
- La Merrill, M.A.; Cirillo, P.M.; Krigbaum, N.Y.; Cohn, B.A. The impact of prenatal parental tobacco smoking on risk of diabetes mellitus in middle-aged women. J. Dev. Orig. Health Dis. 2015, 6, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, K.; Jönsson, I.; Malmqvist, E.; Larsson, H.E.; Rylander, L. Maternal smoking during pregnancy and offspring type 1 diabetes mellitus risk: Accounting for HLA haplotype. Eur. J. Epidemiol. 2015, 30, 231–238. [Google Scholar] [CrossRef]
- Carrapato, M.R. The offspring of gestational diabetes. J. Périnat. Med. 2003, 31, 5–11. [Google Scholar] [CrossRef]
- Bar-Zeev, Y.; Haile, Z.; Chertok, I.A. Association Between Prenatal Smoking and Gestational Diabetes Mellitus. Obstet. Gynecol. 2019, 135, 91–99. [Google Scholar] [CrossRef] [PubMed]
- DeSisto, C.L.; Kim, S.Y.; Sharma, A.J. Prevalence Estimates of Gestational Diabetes Mellitus in the United States, Pregnancy Risk Assessment Monitoring System (PRAMS), 2007–2010. Prev. Chronic Dis. 2014, 11, E104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-H.; Wen, X.; Qu, W.; Liu, H.-X.; Yan, H.-Y.; Hou, L.-F.; Ping, J. Attenuated Tregs increase susceptibility to type 1 diabetes in prenatal nicotine exposed female offspring mice. Toxicol. Lett. 2019, 315, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.A. Smoking and Type 2 Diabetes Mellitus. Diabetes Metab. J. 2012, 36, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Behl, M.; Rao, D.; Aagaard, K.; Davidson, T.; Levin, E.D.; Slotkin, T.A.; Srinivasan, S.; Wallinga, D.; White, M.F.; Walker, V.R.; et al. Evaluation of the Association between Maternal Smoking, Childhood Obesity, and Metabolic Disorders: A National Toxicology Program Workshop Review. Environ. Health Perspect. 2013, 121, 170–180. [Google Scholar] [CrossRef]
- Hummel, M.; Schenker, M.; Ziegler, A.G. Influence of perinatal factors on the appearance of islet autoantibodies in offspring of parents with type 1 diabetes. Pediatr. Diabetes 2001, 2, 40–42. [Google Scholar]
- Johansson, A.; Hermansson, G.; Ludvigsson, J. Tobacco exposure and diabetes-related autoantibodies in children: Results from the ABIS study. Ann. N. Y. Acad. Sci. 2008, 1150, 197–199. [Google Scholar] [CrossRef]
- Rosenbauer, J.; Herzig, P.; Kaiser, P.; Giani, G. Early Nutrition and Risk of Type 1 Diabetes Mellitus—A Nationwide Case-control Study in Preschool Children. Exp. Clin. Endocrinol. Diabetes 2007, 115, 502–508. [Google Scholar] [CrossRef]
- Stene, L.C.; Barriga, K.; Norris, J.M.; Hoffman, M.; Erlich, H.A.; Eisenbarth, G.S.; McDuffie, R.S.; Rewers, M. Perinatal Factors and Development of Islet Autoimmunity in Early Childhood: The Diabetes Autoimmunity Study in the Young. Am. J. Epidemiol. 2004, 160, 3–10. [Google Scholar] [CrossRef]
- Wadsworth, E.; Hamilton-Shield, J.; Hunt, L.; Baum, J. A Case-control Study of Environmental Factors Associated with Diabetes in the Under 5s. Diabet. Med. 1997, 14, 390–396. [Google Scholar] [CrossRef]
- Toschke, A.M.; Ehlin, A.; Koletzko, B.; Montgomery, S.M. Paternal smoking is associated with a decreased prevalence of type 1 diabetes mellitus among offspring in two national British birth cohort studies (NCDS and BCS70). J. Périnat. Med. 2007, 35, 43–47. [Google Scholar] [CrossRef][Green Version]
- Borowitz, J.L.; Isom, G.E. Nicotine and Type 2 Diabetes. Toxicol. Sci. 2008, 103, 225–227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bruin, E.J.; Kellenberger, L.D.; Gerstein, H.C.; Morrison, K.M.; Holloway, A.C. Fetal and neonatal nicotine exposure and postnatal glucose homeostasis: Identifying critical windows of exposure. J. Endocrinol. 2007, 194, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.D.; Cruickshank, J.K. Glucose, insulin, diabetes and mechanisms of arterial dysfunction. Clin. Exp. Pharmacol. Physiol. 2007, 34, 677–682. [Google Scholar] [CrossRef]
- Yu, B.-L.; Zhao, S.-P.; Hu, J.-R. Cholesterol imbalance in adipocytes: A possible mechanism of adipocytes dysfunction in obesity. Obes. Rev. 2009, 11, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, J.; Huang, W.; Yu, L.; Zhang, Y.; Wang, H. Placental mechanism of prenatal nicotine exposure-reduced blood cholesterol levels in female fetal rats. Toxicol. Lett. 2018, 296, 31–38. [Google Scholar] [CrossRef]
- Lips, K.; Brüggmann, D.; Pfeil, U.; Vollerthun, R.; Grando, S.; Kummer, W. Nicotinic acetylcholine receptors in rat and human placenta. Placenta 2005, 26, 735–746. [Google Scholar] [CrossRef]
- Jaddoe, V.W.; de Ridder, M.A.; Elzen, A.P.V.D.; Hofman, A.; Uiterwaal, C.S.; Witteman, J.C. Maternal smoking in pregnancy is associated with cholesterol development in the offspring: A 27-years follow-up study. Atherosclerosis 2008, 196, 42–48. [Google Scholar] [CrossRef]
- Sarwar, R.; Pierce, N.; Koppe, S. Obesity and nonalcoholic fatty liver disease: Current perspectives. Diabetes, Metab. Syndr. Obes. Targets Ther. 2018, 11, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Crocè, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and Risk Factors for Hepatic Steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Conceição, E.; Peixoto-Silva, N.; Pinheiro, C.; Oliveira, E.; Moura, E.; Lisboa, P. Maternal nicotine exposure leads to higher liver oxidative stress and steatosis in adult rat offspring. Food Chem. Toxicol. 2015, 78, 52–59. [Google Scholar] [CrossRef]
- Valença, S.S.; Gouveia, L.; Pimenta, W.A.; Porto, L.C. Effects of Oral Nicotine on Rat Liver Stereology. Int. J. Morphol. 2008, 26, 1013–1022. [Google Scholar] [CrossRef]
- Micalizzi, L.; Knopik, V.S. Maternal smoking during pregnancy and offspring executive function: What do we know and what are the next steps? Dev. Psychopathol. 2018, 30, 1333–1354. [Google Scholar] [CrossRef] [PubMed]
- Lambers, D.S.; Clark, K.E. The maternal and fetal physiologic effects of nicotine. Semin. Perinatol. 1996, 20, 115–126. [Google Scholar] [CrossRef]
- Ward, C.; Lewis, S.; Coleman, T. Prevalence of maternal smoking and environmental tobacco smoke exposure during pregnancy and impact on birth weight: Retrospective study using Millennium Cohort. BMC Public Health 2007, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, M.; Więckowska, B.; Sztorc, L.; Sajdak, S. Smoking and Smoking Cessation in the Risk for Fetal Growth Restriction and Low Birth Weight and Additive Effect of Maternal Obesity. J. Clin. Med. 2020, 9, 3504. [Google Scholar] [CrossRef]
- Muneoka, K.; Ogawa, T.; Kamei, K.; Muraoka, S.-I.; Tomiyoshi, R.; Mimura, Y.; Kato, H.; Suzuki, M.R.; Takigawa, M. Prenatal nicotine exposure affects the development of the central serotonergic system as well as the dopaminergic system in rat offspring: Involvement of route of drug administrations. Dev. Brain Res. 1997, 102, 117–126. [Google Scholar] [CrossRef]
- Franke, R.M.; Park, M.; Belluzzi, J.D.; Leslie, F.M. Prenatal nicotine exposure changes natural and drug-induced reinforcement in adolescent male rats. Eur. J. Neurosci. 2008, 27, 2952–2961. [Google Scholar] [CrossRef]
- Azam, L.; Chen, Y.; Leslie, F. Developmental regulation of nicotinic acetylcholine receptors within midbrain dopamine neurons. Neuroscience 2007, 144, 1347–1360. [Google Scholar] [CrossRef]
- Dwyer, J.B.; Cardenas, A.; Franke, R.M.; Chen, Y.; Bai, Y.; Belluzzi, J.D.; Lotfipour, S.; Leslie, F.M. Prenatal nicotine sex-dependently alters adolescent dopamine system development. Transl. Psychiatry 2019, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-J.A.; Kelly, R.B. Effect of prenatal or perinatal nicotine exposure on neonatal thyroid status and offspring growth in rats. Life Sci. 2005, 76, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Santiago, S.E.; Huffman, K.J. Postnatal effects of prenatal nicotine exposure on body weight, brain size and cortical connectivity in mice. Neurosci. Res. 2012, 73, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-J.; Holloway, A.C.; Zeng, Z.-H.; Lim, G.; Petrik, J.J.; Foster, W.G.; Lee, R.M. Prenatal Exposure to Nicotine Causes Postnatal Obesity and Altered Perivascular Adipose Tissue Function. Obes. Res. 2005, 13, 687–692. [Google Scholar] [CrossRef]
- Koçak, N.D.; Eren, A.; Boğa, S.; Aktürk, Ü.A.; Öztürk, Ü.A. Arınç, S.; Şengül, A. Relapse Rate and Factors Related to Relapse in a 1-Year Follow-Up of Subjects Participating in a Smoking Cessation Program. Respir. Care 2015, 60, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.Y.; Shen, S.; Wellman, J.L.; Rickin, E.; Lowers, L. Offspring from families at high risk for alcohol dependence: Increased body mass index in association with prenatal exposure to cigarettes but not alcohol. Psychiatry Res. 2005, 135, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.D.; Smith, G.D.; Rogers, I.S.; Reilly, J.J.; Wells, J.C.; Ness, A.R. Smoking during Pregnancy and Offspring Fat and Lean Mass in Childhood. Obesity 2006, 14, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Dürmuş, B.; Kruithof, C.J.; Gillman, M.H.; Willemsen, S.P.; Hofman, A.; Raat, H.; Eilers, P.H.C.; Steegers, E.A.P.; Jaddoe, V.W.V. Parental smoking during pregnancy, early growth, and risk of obesity in preschool children: The Generation R Study. Am. J. Clin. Nutr. 2011, 94, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Levitan, E.B.; Gillman, M.W. Maternal smoking during pregnancy and child overweight: Systematic review and meta-analysis. Int. J. Obes. 2008, 32, 201–210. [Google Scholar] [CrossRef]
- Fried, A.P.; James, D.S.; Watkinson, B. Growth and pubertal milestones during adolescence in offspring prenatally exposed to cigarettes and marihuana. Neurotoxicol. Teratol. 2001, 23, 431–436. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene/Polymorphism | No. Ref. | Overall Summary |
|---|---|---|
| Dopamine D1 Receptor (DRD1): rs4532—risk allele G | [163] | Several studies supported that genetic variation in dopamine receptors D1 may influence genetic predisposition to substance use disorders. A statistically significant association of DRD1 rs4532 polymorphism with nicotine dependence was found among a pooled sample of European American and African American families (2037 participants). |
| Dopamine D2 Receptor (DRD2): rs1800497—risk allele A1 | [83] | The DRD2 rs1800497 was found associated with greater likelihood of nicotine dependence, with individuals carrying one or two copies of the rs1800497 risk allele being 3.3 times more likely in a study (150 smokers vs. 228 controls). |
| Dopamine D3 Receptor (DRD3): rs6280—risk allele C (Ser9Gly) | [164] | Several case-control studies investigated the association between the DRD3 rs6280 polymorphism with substance use. From a North American study (2037 subjects), the DRD3 rs6280 polymorphism was significantly associated with nicotine dependence in European Americans. |
| Dopamine D4 Receptor (DRD4): rs1800955—risk allele C (48bp repeat VNTR) | [165] | Aa analysis of various case-control studies (total 1157 cases vs. 438 controls) found the DRD4 rs1800955 polymorphism associated with cigarette smoking in both heavy and light smokers compared to non-smokers. |
| Dopamine Transporter Receptor (DAT1): SLC6A3 3′-UTR—risk allele A9 (40bp repeat VNTR) | [166] | DAT1 is a principal regulator of dopaminergic neurotransmission. Lack of literature exists examining the relationship between DAT1 polymorphisms and nicotine dependence. One study saw no association between this particular DAT1 polymorphism and nicotine dependence; however, other studies have seen a strong association with other substances of abuse. |
| Catechol-O-Methyltransferase (COMT): rs4680—risk allele G (Val158Met) | [167] | COMT is a strong candidate gene that contributes to substance use disorder and schizophrenia. An analysis of 602 nuclear families of African American and European American origin showed an association of COMT rs4680 polymorphism with susceptibility to nicotine dependence that is ethnic- and gender-specific. |
| µ-Opioid Receptor (OPRM1): rs1799971—risk allele G (A118G) | [168] | Polymorphisms of the OPRM1 gene expressing µ-opioid receptors could be significantly associated with some features of substance dependence. In a meta-analysis of 25 datasets with over 25,000 subjects from European ancestry, results indicated the OPRM1 risk allele G was associated with general substance dependence, including nicotine dependence. |
| γ-Aminobutyric Acid (GABA) A Receptor, β-3 Subunit (GABRB3): CA repeat—risk allele 181 | [169] | Lack of literature exists examining the relationship between GABRB3 and nicotine dependence. However, many studies have examined an association between the GABRB3 polymorphisms related to other substance use disorders, including alcohol dependence. A family-based association analysis suggests GABRB3 may be involved in the risk for alcohol dependence. |
| Monoamine Oxidase A (MAO-A): 3′ 30bp VNTR—risk allele 4R DNRP | [170] | A clinical study of Japanese outpatients (217 men and 287 women) revealed an association between the MAO-A polymorphisms and nicotine dependence and smoking behavior for both men and women. |
| Serotonin Transporter Receptor (5HTT) Linked Promoter Region (5HTTLPR) in SLC6A4: rs25531—risk allele S′ | [171] | A case-control studied examined the association between the 5-HTTLPR genotype and smoking behavior in Caucasians from Northern Poland (149 cases vs. 158 controls). No significant association was found; however, numerous non-genetic factors may have strongly influenced this genetic susceptibility. |
| Subjects | ROA | Nicotine Dosage | Length of Exposure | Δ Birth Weight Compared to Controls | Δ Adolescent Body Weight Compared to Controls | References |
|---|---|---|---|---|---|---|
| Female Balb/C mice pups | Sc injection | 1.5 mg/kg 2×/day | GD 9–GD 18 |  PND21-42 PND21-42 | N/A | Zhao et al., 2019 [202] |
| Male Wistar rat pups | Sc injection | 1 mg/kg/day | 14 days before mating—PND21 | |  PND49–PND182 PND49–PND182 | Holloway et al., 2005 [192] |
| Male Wistar rat pups | Sc minipump | 6 mg/kg/day | PND2–PND16 | N/A | PND180 | Conceição et al., 2015 [222] |
| Male Wistar rat pups | Sc injection | 1.0 mg/kg 2×/day | GD9–PND28 | No change | PND28PND84–PND182 | Fan et al., 2016 [11] |
| Female and Male Sprague–Dawley rat pups | Implantable nicotine pellet | 0, 15, 25 mg 15mg + 1 and 25mg + 2mg/kg/day | GD0–GD20PND1–PND9 | No change | Females PND35- PND91 Males on PND35 | Chen and Kelly, 2005 [232] |
| CD1 mice pups | Sc injection | 2 mg/kg 2×/day | GD0 to PND0 | | No change | Santiago and Huffman, 2012 [233] |
| Male Wistar rat pups | Sc injection | 1 mg/kg body weight/day | 14 days before mating—PND21 | No change | PND70–PND182 | Gao et al., 2005 [234] |
| Participants | Age (Years) | Length of Exposure (Months) | Dose (Cigarettes/Day) | Δ in Birthweight from Controls | Δ in Weight/BMI from Controls (Childhood/Adolescence) | Δ in Weight/BMI from Controls (Adult) | References |
|---|---|---|---|---|---|---|---|
| 32,747 children | 0–7 | 0–15 mo | 0–20+ | | Weight: BMI: N/A | Weight: N/A BMI: N/A | Møller et al., 2014 [16] |
| 35,370 daughters | 5–18+ | 0–9 mo | 1–25+ | | Weight: N/A BMI: | Weight: N/A BMI: | Harris et al., 2013 [178] |
| 266 children | Newborn | 0–9 mo | 0–29 | | Weight: N/A BMI: N/A | Weight: N/A BMI: N/A | Andersen et al., 2009 [20] |
| 5342 children | 0–4 | 0–9 mo | 0–5+ | | Weight: BMI: | Weight: N/A BMI: N/A | Dürmus et al., 2011 [235] |
| 288 children | 8–18 | 0–9 mo | 0–10+ | N/A | Weight: N/A BMI: | Weight: N/A BMI: | Hill et al., 2005 [236] |
| 5689 children | 9.9 | 0–9 mo | 1–20+ | | Weight: BMI: | Weight: N/A BMI: N/A | Leary et al., 2006 [237] |
| 912 children | Newborn | 0–9 mo | 1–30 | | Weight: N/A BMI: N/A | Weight: N/A BMI: N/A | Lewandowska et al., 2020 [227] |
| 174 children | 0–2 | 0–9 mo | 0–7 | | Weight: BMI: N/A | Weight: N/A BMI: N/A | Molnar et al., 2017 [18] |
| 18,297 children | Newborn | 0–9 mo | 1–20+ daily | | Weight: N/A BMI: N/A | Weight: N/A BMI: N/A | Ward et al., 2007 [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, O.; Roeder, N.; Blum, K.; Eiden, R.D.; Thanos, P.K. Prenatal Effects of Nicotine on Obesity Risks: A Narrative Review. Int. J. Environ. Res. Public Health 2022, 19, 9477. https://doi.org/10.3390/ijerph19159477
White O, Roeder N, Blum K, Eiden RD, Thanos PK. Prenatal Effects of Nicotine on Obesity Risks: A Narrative Review. International Journal of Environmental Research and Public Health. 2022; 19(15):9477. https://doi.org/10.3390/ijerph19159477
Chicago/Turabian StyleWhite, Olivia, Nicole Roeder, Kenneth Blum, Rina D. Eiden, and Panayotis K. Thanos. 2022. "Prenatal Effects of Nicotine on Obesity Risks: A Narrative Review" International Journal of Environmental Research and Public Health 19, no. 15: 9477. https://doi.org/10.3390/ijerph19159477
APA StyleWhite, O., Roeder, N., Blum, K., Eiden, R. D., & Thanos, P. K. (2022). Prenatal Effects of Nicotine on Obesity Risks: A Narrative Review. International Journal of Environmental Research and Public Health, 19(15), 9477. https://doi.org/10.3390/ijerph19159477