
Autosomal Dominant Hypophosphatemic Rickets: A Case Report and Review of the Literature

 ,
, 

Abstract
:1. Introduction
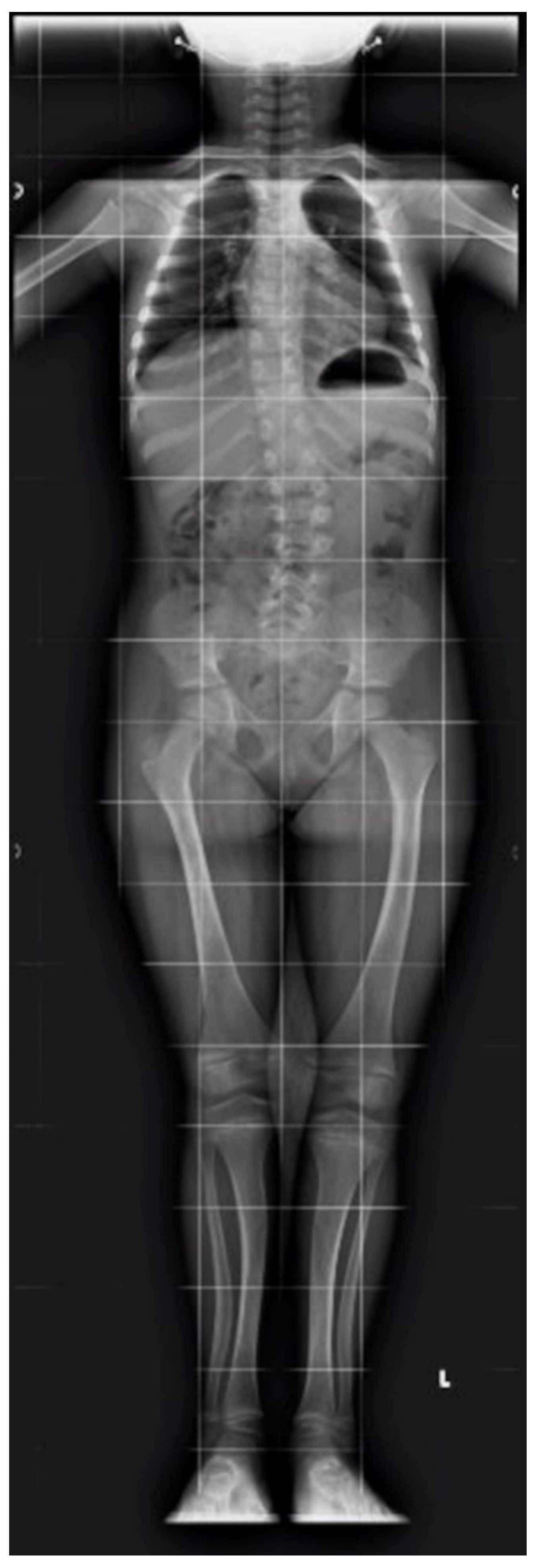
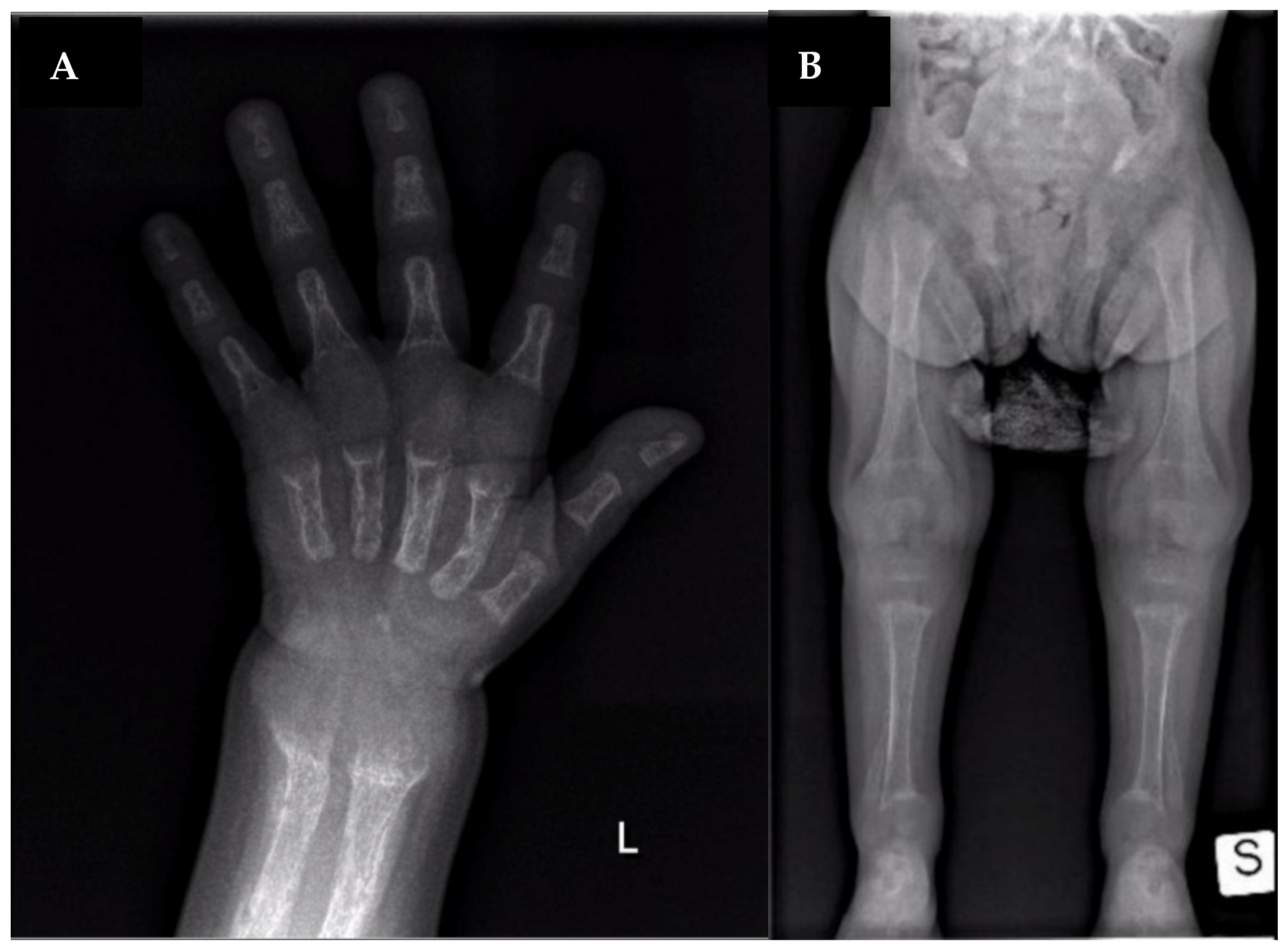
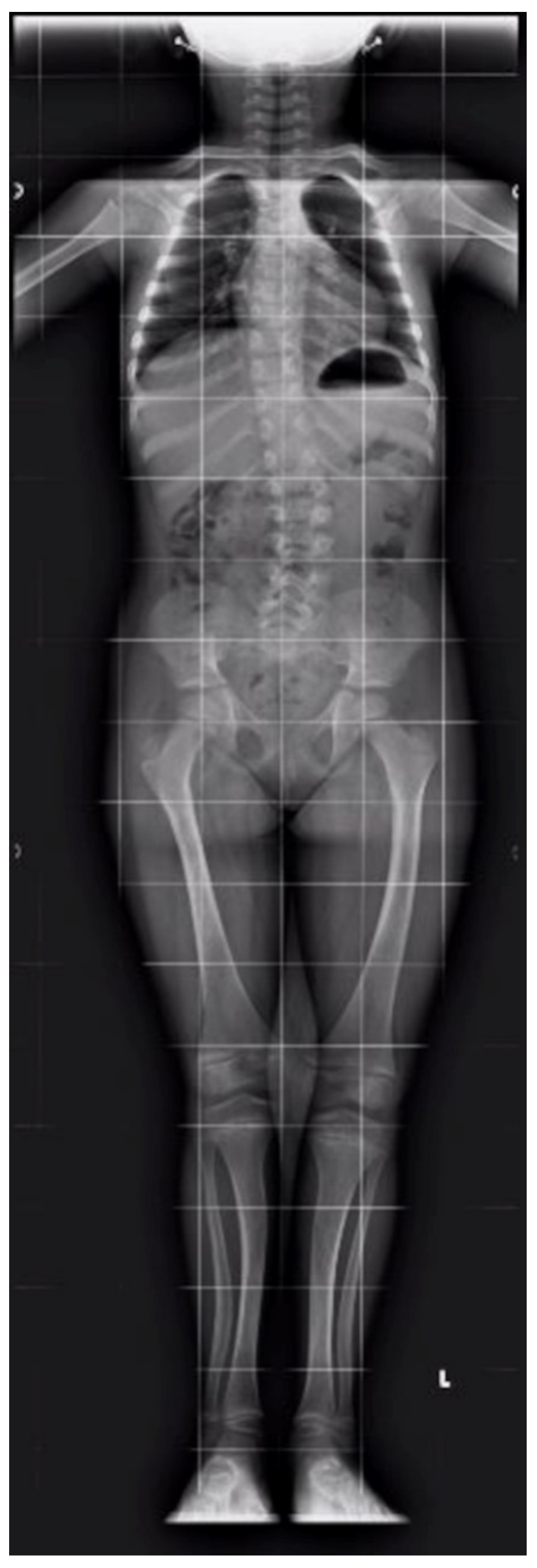
2. Case Report
3. Narrative Review of Literature and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bitzan, M.; Goodyer, P.R. Hypophosphatemic Rickets. Pediatr. Clin. North Am. 2018, 66, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Shaw, N.J.; Portale, A.A.; Ward, L.M.; Abrams, S.A.; Pettifor, J. Rickets. Nat. Rev. Dis. Prim. 2017, 3, 17101. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Evans, W.E.; O’Riordan, J.L.; Speer, M.C.; Econs, M.J.; Lorenz-Depiereux, B.; Grabowski, M.; Meitinger, T.; Strom, T.M. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perwad, F.; Zhang, M.Y.H.; Tenenhouse, H.S.; Portale, A.A. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1α-hydroxylase expression in vitro. Am. J. Physiol. Physiol. 2007, 293, F1577–F1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.-Y.; Miao, D.; Goltzman, D.; Karaplis, A.C. The Autosomal Dominant Hypophosphatemic Rickets R176Q Mutation in Fibroblast Growth Factor 23 Resists Proteolytic Cleavage and Enhances in Vivo Biological Potency. J. Biol. Chem. 2003, 278, 9843–9849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapelari, K.; Köhle, J.; Kotzot, D.; Hoegler, W. Iron Supplementation Associated with Loss of Phenotype in Autosomal Dominant Hypophosphatemic Rickets. J. Clin. Endocrinol. Metab. 2015, 100, 3388–3392. [Google Scholar] [CrossRef] [PubMed]
- Econs, M.J.; McEnery, P.T. Autosomal Dominant Hypophosphatemic Rickets/Osteomalacia: Clinical Characterization of a Novel Renal Phosphate-Wasting Disorder. J. Clin. Endocrinol. Metab. 1997, 82, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.; Woelfel, D.; Strom, T.M. Loss of Renal Phosphate Wasting in a Child with Autosomal Dominant Hypophosphatemic Rickets Caused by a FGF23 Mutation. Horm. Res. Paediatr. 2001, 55, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.L.; Negrotti, T.; Alonso, G.; Pasqualini, T. Distintasformas de presentacion clinica de un raquitismohipofosfatemico autosomico dominante por mutacion del factor de crecimiento fibroblastico 23 en una familia. Medicina (B Aires) 2004, 64, 103–106. [Google Scholar] [PubMed]
- Gribaa, M.; Younes, M.; Bouyacoub, Y.; Korbaa, W.; Ben Charfeddine, I.; Touzi, M.; Adala, L.; Mamay, O.; Bergaoui, N.; Saâd, A. An autosomal dominant hypophosphatemic rickets phenotype in a Tunisian family caused by a new FGF23 missense mutation. J. Bone Miner. Metab. 2009, 28, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Sandal, S.; Arora, V.; Verma, L.C. Hypophosphatemic Rickets with R179W Mutation in FGFR23 Gene-A Rare But Treatable Cause of Refractory Rickets. Indian J. Pediatr. 2021, 88, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, Z.; Wang, O.; Li, M.; Xing, X.; Hsieh, E.; Fukumoto, S.; Jiang, Y.; Xia, W. Earlier Onset in Autosomal Dominant Hypophosphatemic Rickets of R179 than R176 Mutations in Fibroblast Growth Factor 23: Report of 20 Chinese Cases and Review of the Literature. Calcif. Tissue Int. 2019, 105, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Seton, M.; Jüppner, H. Autosomal dominant hypophosphatemic rickets in an 85 year old woman: Characterization of her disease from infancy through adulthood. Bone 2012, 52, 640–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournis, S.; Koromila, T.; Chatzistamatas, N.; Droggaris, M.; Zafeiris, C.; Makris, K.; Marketou, H.; Papaioannou, N.; Kollia, P.; Gazi, S. Hip fracture leading to the diagnosis of autosomal dominant hypophosphatemic rickets. A case report. J. Musculoskelet. Neuronal. Interact. 2013, 13, 391–394. [Google Scholar] [PubMed]
- Sun, Y.; Wang, O.; Xia, W.; Jiang, Y.; Li, M.; Xing, X.; Hu, Y.; Liu, H.; Meng, X.; Zhou, X. FGF23 analysis of a Chinese family with autosomal dominant hypophosphatemic rickets. J. Bone Miner. Metab. 2011, 30, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal Anthropometric Charts: The Italian Neonatal Study Compared with Other European Studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linglart, A.; Duplan, M.B.; Briot, K.; Chaussain, C.; Esterle, L.; Guillaume-Czitrom, S.; Kamenicky, P.; Nevoux, J.; Prié, D.; Rothenbuhler, A.; et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr. Connect. 2014, 3, R13–R30. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, T.O.; Imel, E.A.; Holm, I.A.; De Beur, S.M.J.; Insogna, K.L. A clinician’s guide to X-linked hypophosphatemia. J. Bone Miner. Res. 2011, 26, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Paper [Ref]. | Econs 1997 [8]. | Econs 1997 [8]. | Econs 1997 [8]. | Kruse 2001 [9]. | Negri 2004 [10]. | Gribaa 2010 [11]. | Gribaa 2010 [11]. | Kapelari 2015 [7]. | Sandal 2020 [12]. | Liu 2019 [13]. | Index Case |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case report | Case VI-51 | Case VI-26 | Case VI-5 | Case#1 | Case#2 | Case V3 | Case V4 | Case 1 | Case 1 | Case 3-III1 | Case 1 |
| Sex | F | M | M | F | F | F | M | F | F | F | F |
| Genetic mutation | R176Q NM_020638.3:c.527G>A rs104894347 | R176Q NM_020638.3:c.527G>A; rs104894347, | R176Q NM_020638.3:c.527G>A rs104894347 | R179Q NM_020638.3:c.536G>A; rs193922702 | R179Q NM_020638.3:c.536G>A; rs193922702 | R176W NM_020638.3:c.526C>T rs754201217 | R176W NM_020638.3:c.526C>T rs754201217 | R179Q NM_020638.3:c.536G>A; rs193922702 | R179W NM_020638.3:c.535C>T; rs28937882 | R176Q NM_020638.3:c.527G>A rs104894347 | R179Q NM_020638.3:c.536G>A; rs193922702 |
| Age of onset | 14.5 y | 14 m | 19 m | 11 m | 8 m | 3 y | 5 y | 26 m | 2 y | NA | 17 m |
| Age of diagnosis | 14.5 y | 14 m | 19 m | 11 m | 8 m | 3 y | 5 y | 26 m | 13 y | 2 y | 19 m |
| H at diagnosis | N/A | 50° to 5° ple | N/A | 3° ple | 3° ple | N/A | N/A | 79 cm (−2.82 SDS) | 142 cm (−2.68 Z-score) | NA | 68.5 cm (−4.08 SD) |
| W at diagnosis | N/A | N/A | N/A | 10° ple | N/A | N/A | N/A | 9500 g (−2.12 SDS) | 44.2 kg (−0.49 Z-score) | NA | 7.8 kg (−2.2 SD) |
| Ca at diagnosis | 8.9 mg/dL (nv 8.8–10.8) | 9.2 mg/dL (nv 8.8–10.8) | 9.5 mg/dL (nv 8.8–10.8) | 2.53 mol/L (nv 2.44–2.7) | 9.5 mg/dL (nv 8.8–10.8) | 8.82 mg/dL (nv 8.8–10.8) | 9.62 mg/dL (nv 8.8–10.8) | 2.44 mmol/L (nv 2.3–2.7) | 9.65 mg/dL (nv 8.8–10.8) | 2.51 mmol/L (nv 2.3–2.7) | 9.2 mg/dL (nv 8.8–10.8) |
| P at diagnosis | 1.2 mg/dL (nv 4–7) | 2.1–3.3 mg/dL (nv 4–7) | 2.4 mg/dL (nv 4–7) | 1.39 mmol/L (nv 1.64–2.58) | 3 mg/dL (nv 4–7) | 2.79 mg/dL (nv 4–7) | 3.41 mg/dL (nv 4–7) | 0.63 mmol/L (nv 1.1–1.95) | 1.4 mg/dL (nv 4–7) | 2.11 mmol/L | 1.3 mg/dL (nv 4–7) |
| ALP at diagnosis | N/A | 36.5 Bodansky U (nv < 15) | N/A | 860 U/L (nv 200–600) | 1755 U/L (nv < 640) | 190 U/L | N/A | 731 U/L (nv 200–600) | 934 IU/L | 205 U/L | 1173 U/L (nv 140–400) |
| PTH at diagnosis | N/A | 49 µL/Eq-mL (nv < 57) | N/A | 3.8 pmol/L (nv 1.1–5.8) | 45 pg/mL (nv 15–65) | N/A | N/A | 37.3 pg/mL (nv 10–55) | 185 pg/mL (nv 22–84) | NA | 96 pg/mL (nv 15–65) |
| TRP at diagnosis (nv 85–97%) | 85% | N/A | 53% | 47.5% | N/A | N/A | N/A | 75.6% | NA | NA | 69% |
| 25-OH D at diagnosis | N/A | N/A | N/A | N/A | 16 ng/mL (nv > 30) | N/A | N/A | 139.2 nmol/L (nv > 50) | 60 ng/mL (nv > 30) | NA | 17 ng/mL (nv > 30) |
| FGF23 at diagnosis | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | NA | 31.8 pg/mL | NA |
| Ferritin at diagnosis | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 5 μg/L (nv > 30) | NA | NA | NA |
| Clinical features | Bone pain, osteomalacia ankle soreness, low back pain, fatigue, pseudofractures | Rachitic lesions at the wrists and knees, growth retardation | Genu valgum, decreased energy, moderately severe rickets | Rachitic rosary, metaphyseal widening, fraying and cupping of the ulna and the distal femur | Genu varum, growth retardation | Bone deformities, dental hypoplasia, frontal bossing, short stature, pectus carinatum, anterior bowing of both legs, pelvis retroversion | Bone deformities, dental hypoplasia, frontal bossing, short stature, pectus carinatum, anterior bowing of both legs | Genu varu, severe rickets, waddling gait | Rickets, short stature, pain in spine/hips/legs and difficulty in walking, kyphoscoliosis, proximal muscle weakness in all four limbs, and genu varus, deformity of bilateral lower limbs | None | Hypotonia, Rickets, growth retardation |
| Treatment | Vitamin D 50.000–100.000 U/day | Vitamin D 25.000–50.000 U/day, then vitamin D 5–25.000 U/day (vitamin toxicity) | Vitamin D 30.000–50.000 U/day At 11 ys calcitriol 0.25–0.5 mcg twice daily | Calcitriol 0.125 mcg/day Oral phosphate 220–400 mg/day | Oral phosphate + Calcitriol (doses not available) | Oral phosphate (doses not available) | Oral phosphate (doses not available) | Oral phosphate 64 mg/kg/day Alfacalcidol 20 ng/kg/day At 8 ys iron sulfate solution | Vitamin D and calcium till 5 y of age. Since age of 13: Phosphate 60 mg/kg/d in four divided doses and calcitriol 60 ng/kg/d in three divided doses for 6 months | NA | Oral alfacalcidol (0.020 mcg/kg/day) and oral phosphate (30 mg/kg/day, divided in 3 doses) |
| Symptoms Resolution | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | NA | Yes |
| Age at last follow-up visit (H/W if available) | 22 ys H: 163.1 cm W: 65.9 kg | 20 ys | 19.75 ys H 178.8 cm | 8 ys | 3 ys 6 mo | 9 ys H: 138 cm | 12 ys H: 150 cm | 11.5 y H: 125.3 cm (−3.23 SDS) W: 26.2 kg (−2.2 SDS) | 13 y | NA | On going |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mameli, C.; Sangiorgio, A.; Colombo, V.; Gambino, M.; Spaccini, L.; Cattaneo, E.; Zuccotti, G.V. Autosomal Dominant Hypophosphatemic Rickets: A Case Report and Review of the Literature. Int. J. Environ. Res. Public Health 2021, 18, 8771. https://doi.org/10.3390/ijerph18168771
Mameli C, Sangiorgio A, Colombo V, Gambino M, Spaccini L, Cattaneo E, Zuccotti GV. Autosomal Dominant Hypophosphatemic Rickets: A Case Report and Review of the Literature. International Journal of Environmental Research and Public Health. 2021; 18(16):8771. https://doi.org/10.3390/ijerph18168771
Chicago/Turabian StyleMameli, Chiara, Arianna Sangiorgio, Valeria Colombo, Mirko Gambino, Luigina Spaccini, Elisa Cattaneo, and Gian Vincenzo Zuccotti. 2021. "Autosomal Dominant Hypophosphatemic Rickets: A Case Report and Review of the Literature" International Journal of Environmental Research and Public Health 18, no. 16: 8771. https://doi.org/10.3390/ijerph18168771
APA StyleMameli, C., Sangiorgio, A., Colombo, V., Gambino, M., Spaccini, L., Cattaneo, E., & Zuccotti, G. V. (2021). Autosomal Dominant Hypophosphatemic Rickets: A Case Report and Review of the Literature. International Journal of Environmental Research and Public Health, 18(16), 8771. https://doi.org/10.3390/ijerph18168771