
Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets

 , ,
, ,  , ,
, , 
 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Gene Expression, miRNA Expression, and DNA Methylation Datasets
2.2. Differential Analyses and Selection of Candidate Genetic and Epigenetic Biomarkers
2.3. Functional Role of the Candidate Genetic and Epigenetic Biomarkers
2.4. Statistical Analyses
3. Results
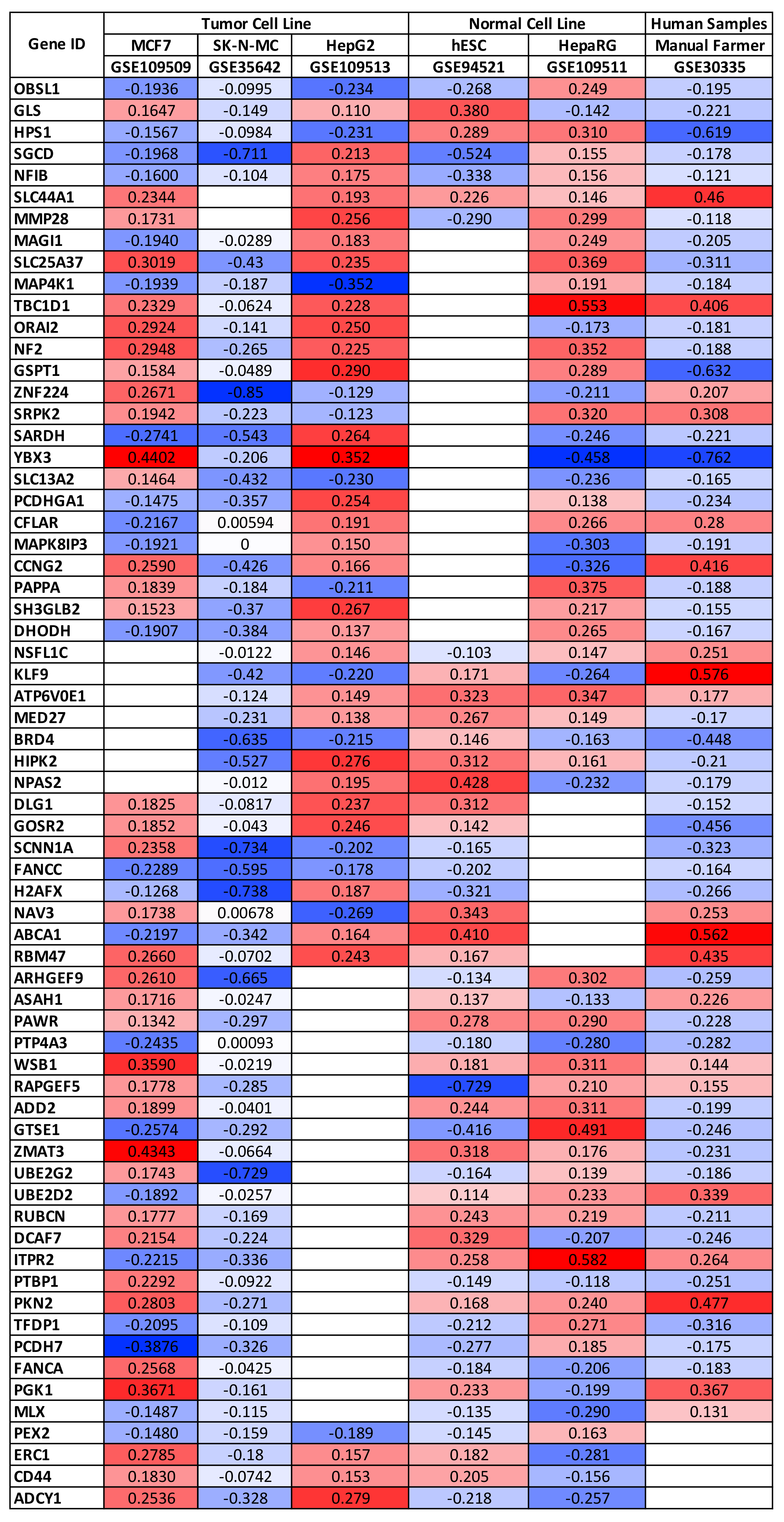
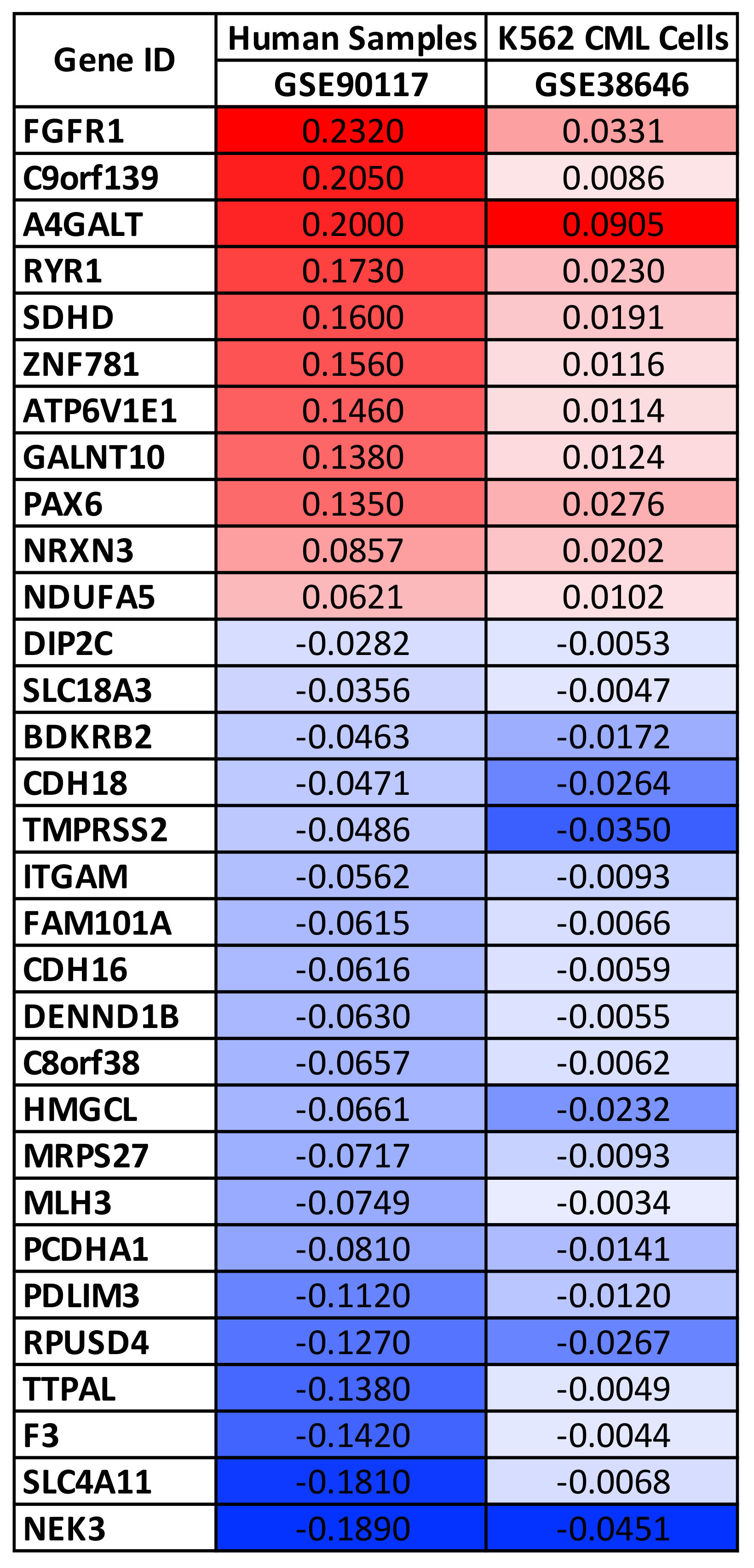
3.1. Selection of Gene Expression, miRNA Expression, and DNA Methylation Datasets and Identification of Potential Genetic and Epigenetic Biomarkers of Pesticide Exposure
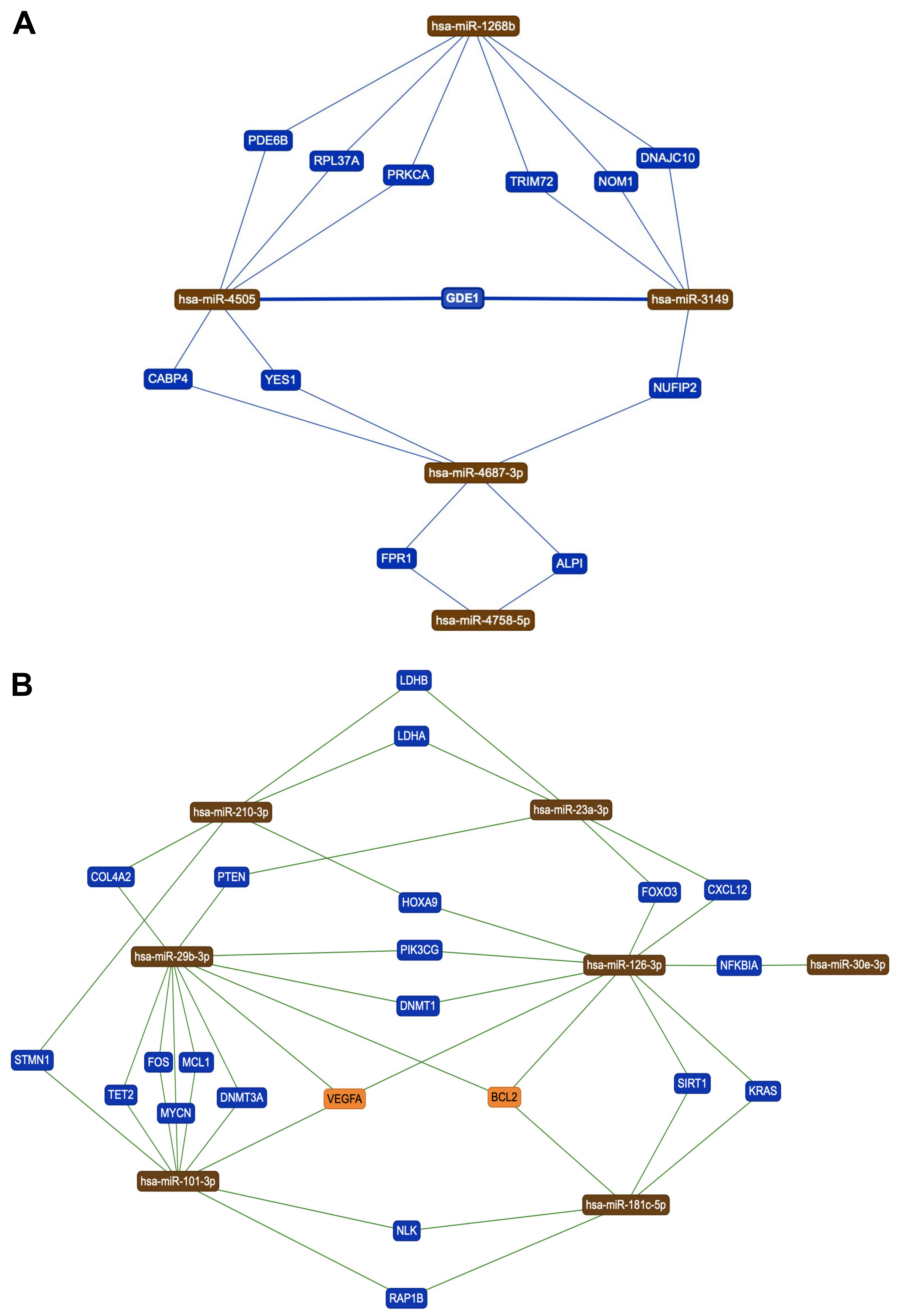
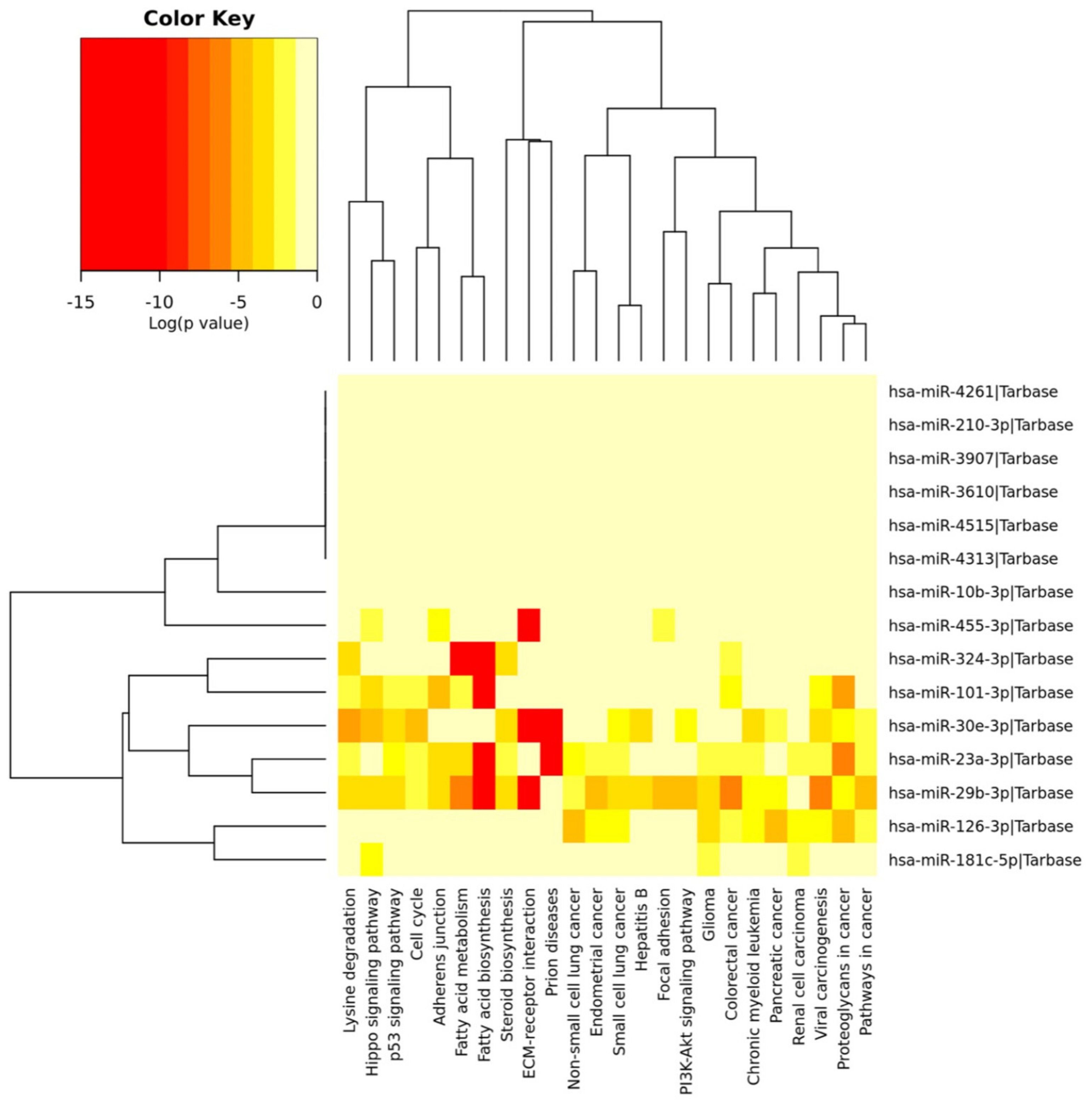
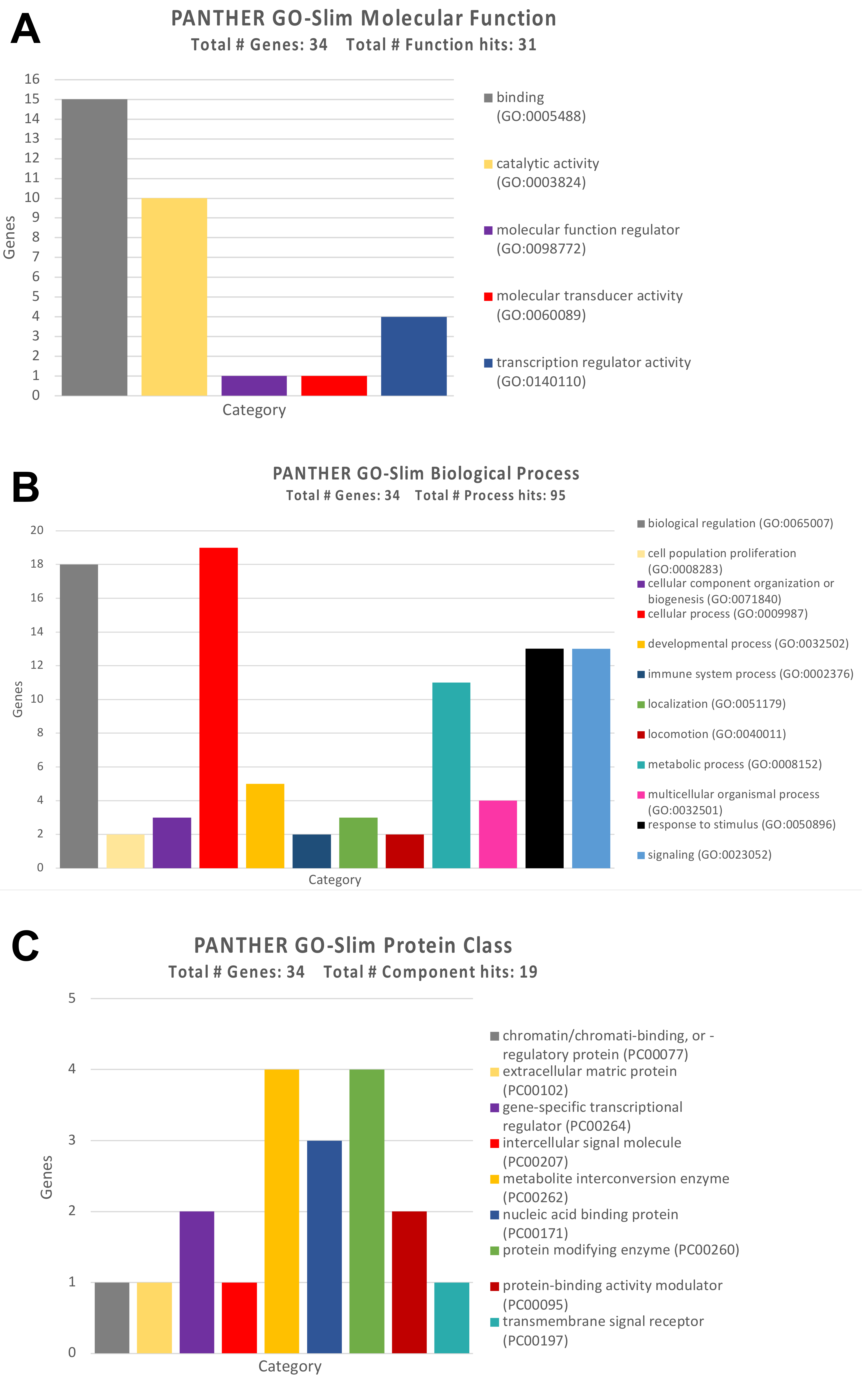
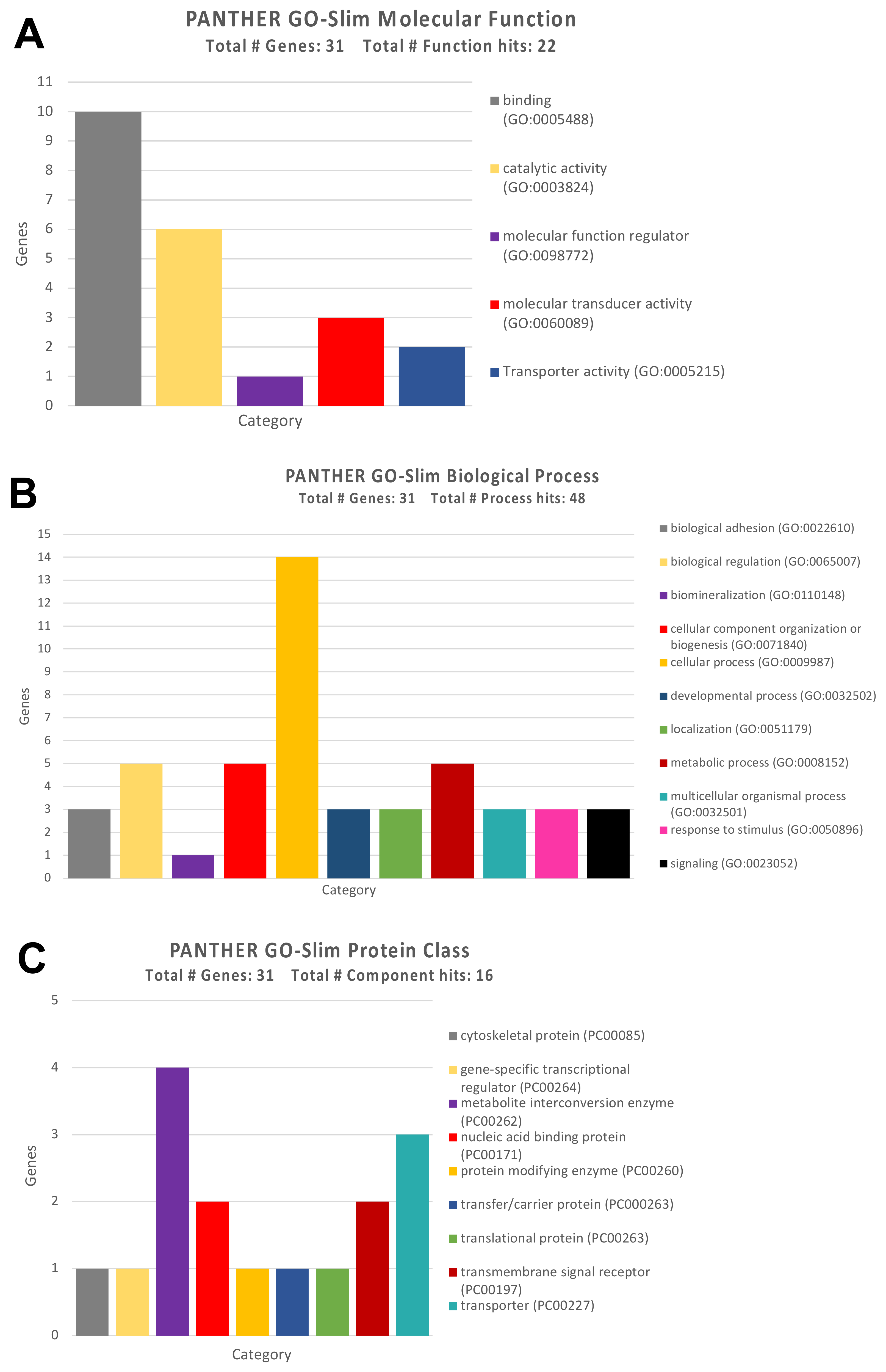
3.2. Identification of Genes and Pathways Modulated by the Selected miRNAs
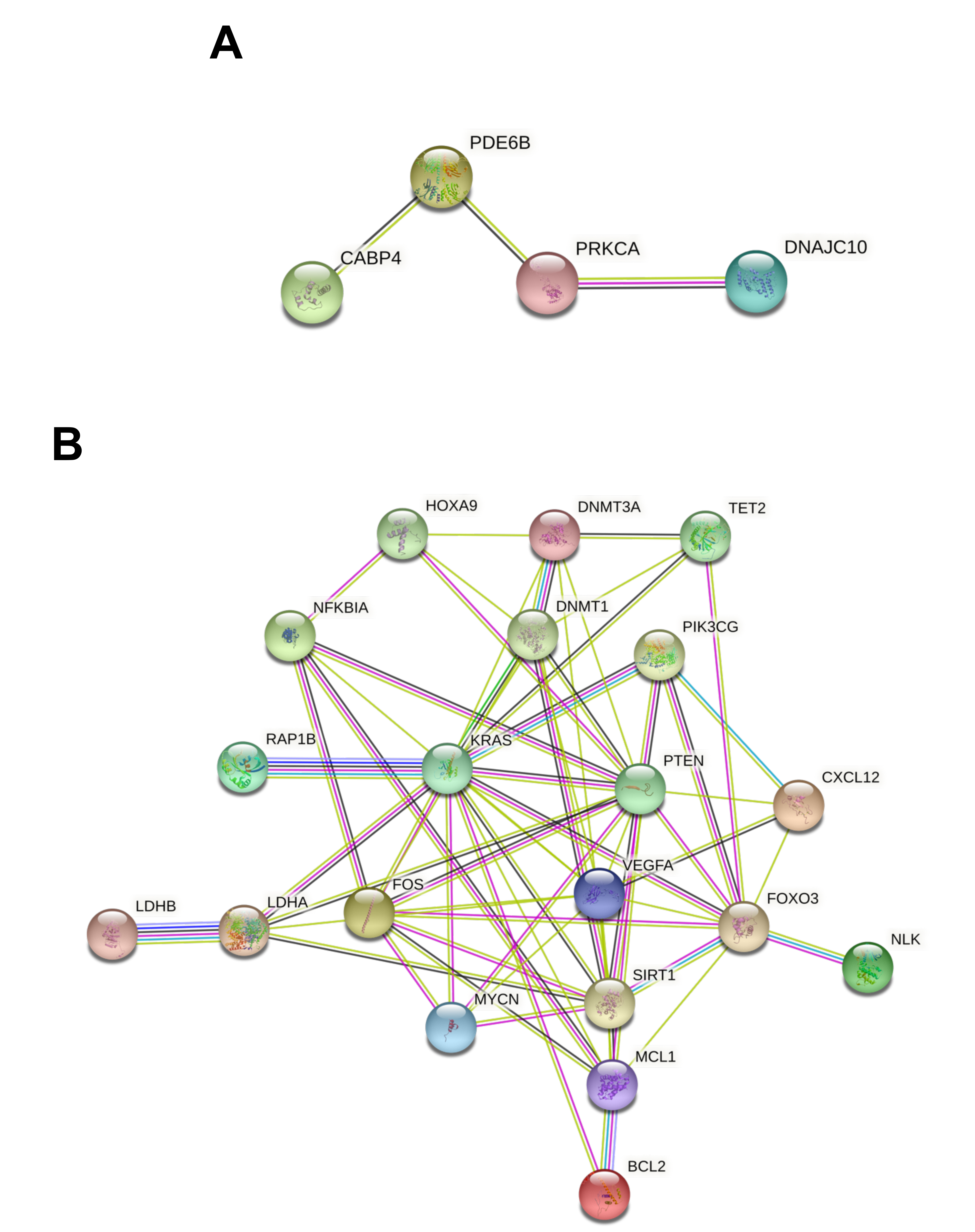
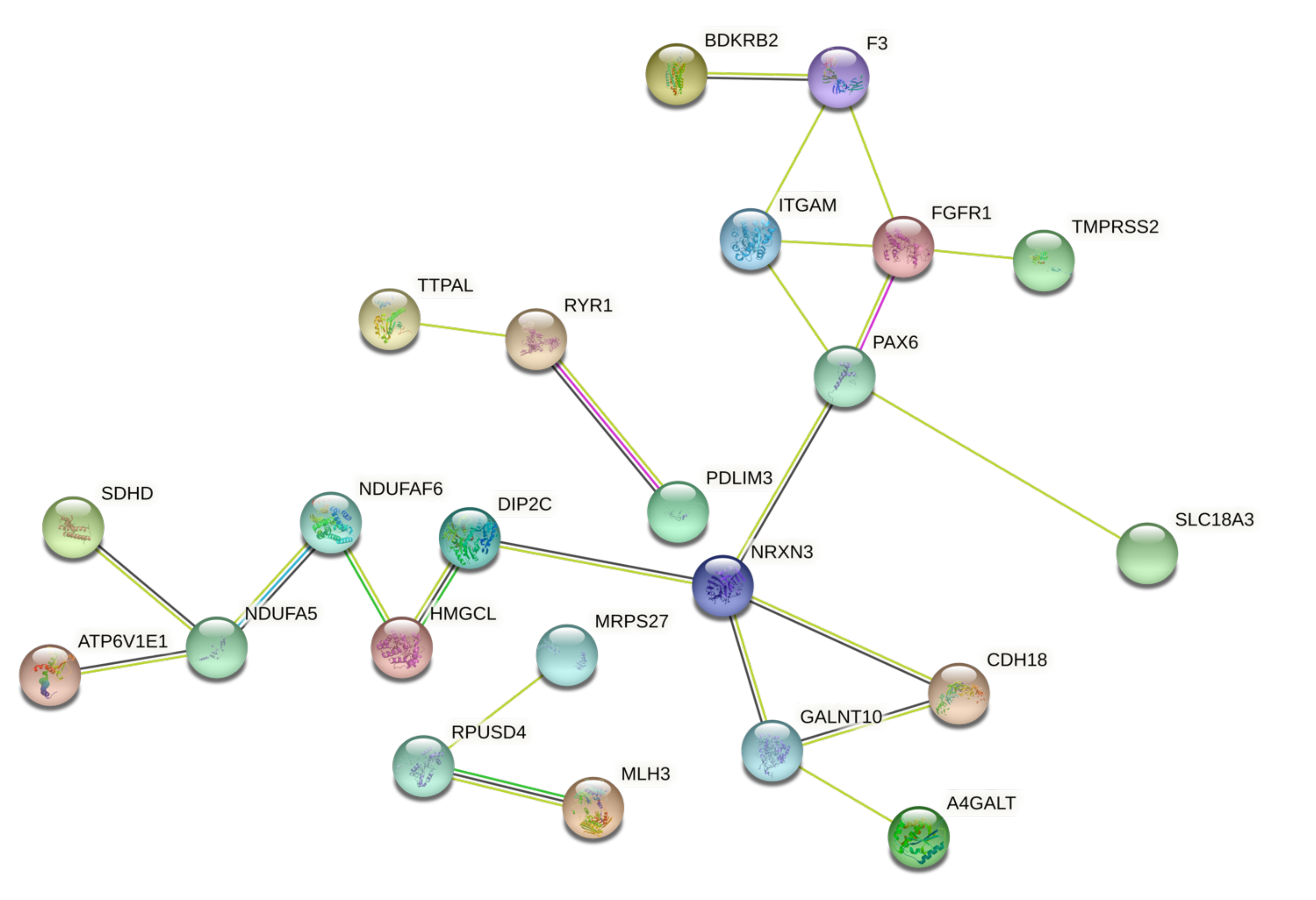
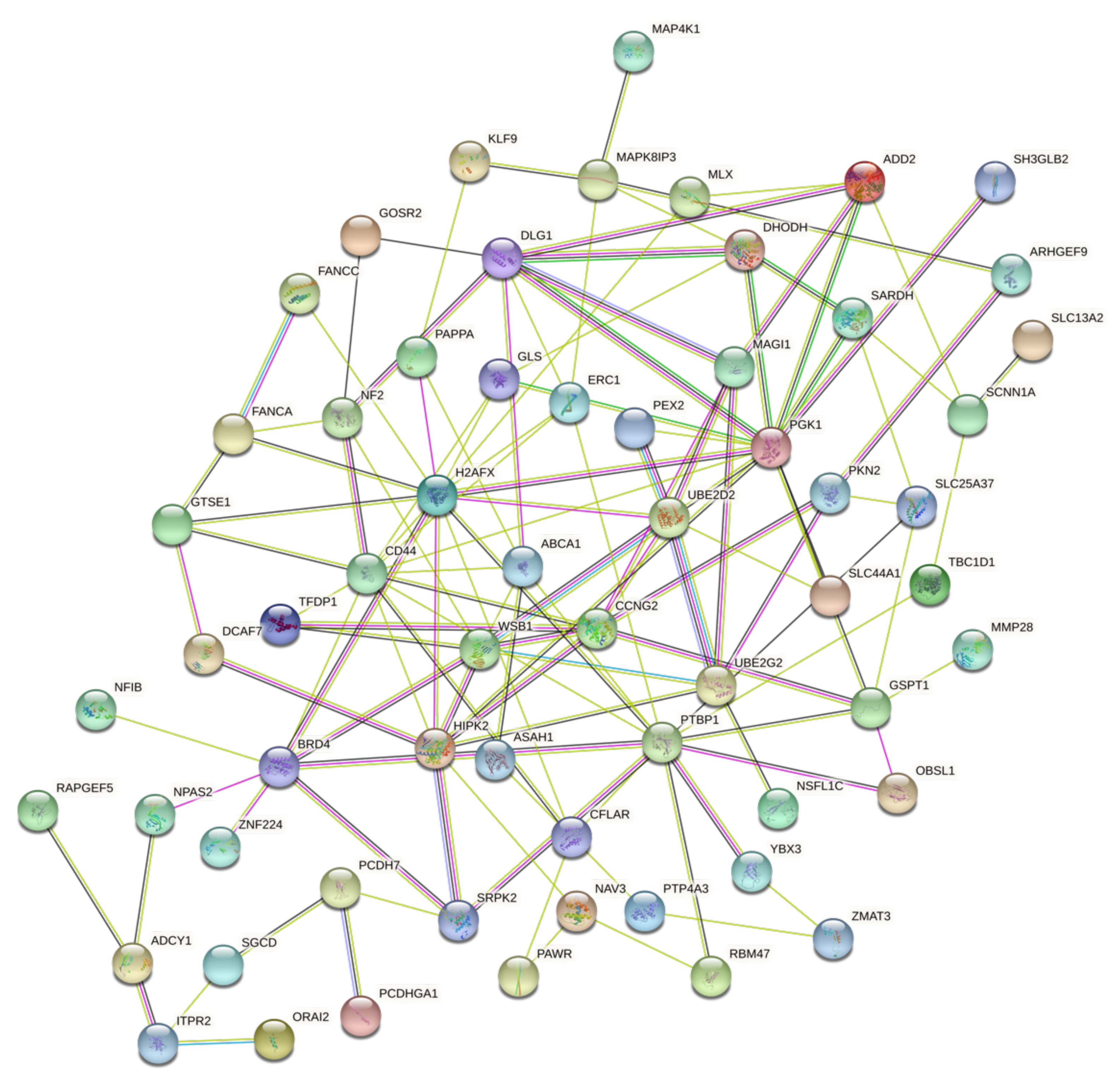
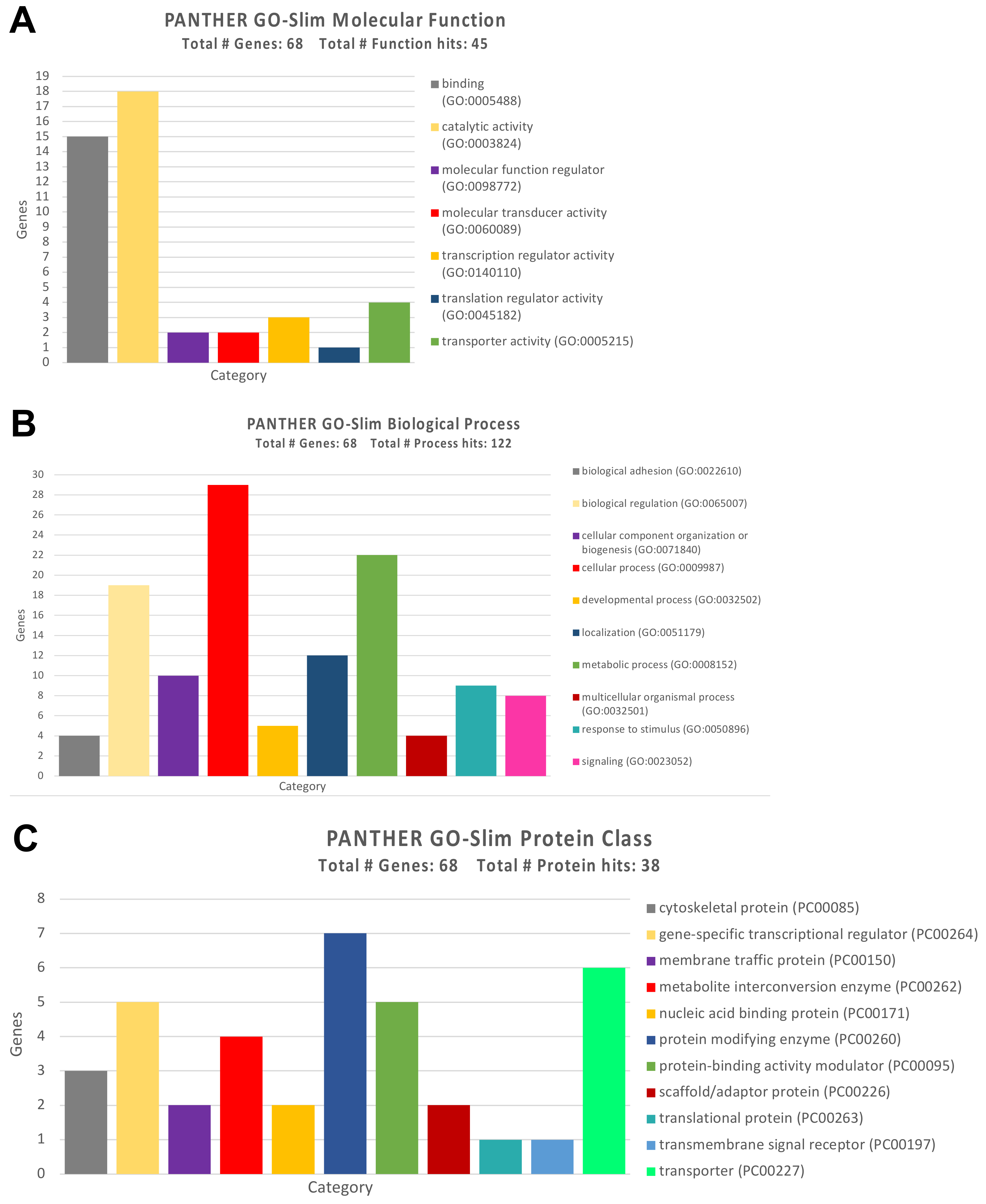
3.3. Protein-Protein Interaction and Functional Role of the Epigenetic Biomarkers and the Differentially Expressed Genes Identified after Pesticide Exposure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Costa, C.; Gangemi, S.; Giambò, F.; Rapisarda, V.; Caccamo, D.; Fenga, C. Oxidative stress biomarkers and paraoxonase 1 polymorphism frequency in farmers occupationally exposed to pesticides. Mol. Med. Rep. 2015, 12, 6353–6357. [Google Scholar] [CrossRef]
- De Oliveira, A.F.B.; de Souza, M.R.; Benedetti, D.; Scotti, A.S.; Piazza, L.S.; Garcia, A.L.H.; Dias, J.F.; Niekraszewicz, L.A.B.; Duarte, A.; Bauer, D.; et al. Investigation of pesticide exposure by genotoxicological, biochemical, genetic polymorphic and in silico analysis. Ecotoxicol. Environ. Saf. 2019, 179, 135–142. [Google Scholar] [CrossRef]
- Damalas, C.A.; Eleftherohorinos, I.G. Pesticide exposure, safety issues, and risk assessment indicators. Int. J. Environ. Res. Public Health 2011, 8, 1402–1419. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, S.; Gofita, E.; Costa, C.; Teodoro, M.; Briguglio, G.; Nikitovic, D.; Tzanakakis, G.; Tsatsakis, A.M.; Wilks, M.F.; Spandidos, D.A.; et al. Occupational and environmental exposure to pesticides and cytokine pathways in chronic diseases (Review). Int. J. Mol. Med. 2016, 38, 1012–1020. [Google Scholar] [CrossRef]
- Aloizou, A.M.; Siokas, V.; Vogiatzi, C.; Peristeri, E.; Docea, A.O.; Petrakis, D.; Provatas, A.; Folia, V.; Chalkia, C.; Vinceti, M.; et al. Pesticides, cognitive functions and dementia: A review. Toxicol. Lett. 2020, 326, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Marconi, A.; Loreto, C.; Franco, S.; Spandidos, D.A.; Libra, M. Occupational exposure to carcinogens: Benzene, pesticides and fibers (Review). Mol. Med. Rep. 2016, 14, 4467–4474. [Google Scholar] [CrossRef] [PubMed]
- Mokarizadeh, A.; Faryabi, M.R.; Rezvanfar, M.A.; Abdollahi, M. A comprehensive review of pesticides and the immune dysregulation: Mechanisms, evidence and consequences. Toxicol. Mech. Methods 2015, 25, 258–278. [Google Scholar] [CrossRef] [PubMed]
- Collotta, M.; Bertazzi, P.A.; Bollati, V. Epigenetics and pesticides. Toxicology 2013, 307, 35–41. [Google Scholar] [CrossRef]
- Costa, C.; Briguglio, G.; Giambò, F.; Catanoso, R.; Teodoro, M.; Caccamo, D.; Fenga, C. Association between oxidative stress biomarkers and PON and GST polymorphisms as a predictor for susceptibility to the effects of pesticides. Int. J. Mol. Med. 2020, 45, 1951–1959. [Google Scholar] [CrossRef]
- Teodoro, M.; Briguglio, G.; Fenga, C.; Costa, C. Genetic polymorphisms as determinants of pesticide toxicity: Recent advances. Toxicol. Rep. 2019, 6, 564–570. [Google Scholar] [CrossRef]
- Costa, C.; Teodoro, M.; Rugolo, C.A.; Alibrando, C.; Giambò, F.; Briguglio, G.; Fenga, C. MicroRNAs alteration as early biomarkers for cancer and neurodegenerative diseases: New challenges in pesticides exposure. Toxicol. Rep. 2020, 7, 759–767. [Google Scholar] [CrossRef]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, R.G. Early Epigenetic Markers for Precision Medicine. Methods Mol. Biol. 2018, 1856, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The Key Role of Epigenetics in Human Disease Prevention and Mitigation. N. Engl. J. Med. 2018, 378, 1323–1334. [Google Scholar] [CrossRef]
- Piletič, K.; Kunej, T. MicroRNA epigenetic signatures in human disease. Arch. Toxicol. 2016, 90, 2405–2419. [Google Scholar] [CrossRef]
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–944. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, C.; Esteller, M. MicroRNAs and Epigenetics. Adv. Cancer Res. 2017, 135, 189–220. [Google Scholar] [CrossRef]
- Van Cauwenbergh, O.; di Serafino, A.; Tytgat, J.; Soubry, A. Transgenerational epigenetic effects from male exposure to endocrine-disrupting compounds: A systematic review on research in mammals. Clin. Epigenet. 2020, 12, 65. [Google Scholar] [CrossRef]
- Rusiecki, J.A.; Beane Freeman, L.E.; Bonner, M.R.; Alexander, M.; Chen, L.; Andreotti, G.; Barry, K.H.; Moore, L.E.; Byun, H.M.; Kamel, F.; et al. High pesticide exposure events and DNA methylation among pesticide applicators in the agricultural health study. Environ. Mol. Mutagen. 2017, 58, 19–29. [Google Scholar] [CrossRef]
- Falzone, L.; Grimaldi, M.; Celentano, E.; Augustin, L.S.A.; Libra, M. Identification of Modulated MicroRNAs Associated with Breast Cancer, Diet, and Physical Activity. Cancers 2020, 12, 2555. [Google Scholar] [CrossRef]
- Hamberg, M.; Backes, C.; Fehlmann, T.; Hart, M.; Meder, B.; Meese, E.; Keller, A. MiRTargetLink—miRNAs, Genes and Interaction Networks. Int. J. Mol. Sci. 2016, 17, 564. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Black, M.B.; Parks, B.B.; Foley, B.; Wetmore, B.A.; Andersen, M.E.; Clewell, R.A.; Mansouri, K.; McMullen, P.D. A Qualitative Modeling Approach for Whole Genome Prediction Using High-Throughput Toxicogenomics Data and Pathway-Based Validation. Front. Pharmacol. 2018, 9, 1072. [Google Scholar] [CrossRef]
- Pallocca, G.; Grinberg, M.; Henry, M.; Frickey, T.; Hengstler, J.G.; Waldmann, T.; Sachinidis, A.; Rahnenführer, J.; Leist, M. Identification of transcriptome signatures and biomarkers specific for potential developmental toxicants inhibiting human neural crest cell migration. Arch. Toxicol. 2016, 90, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Arvelaiz, Y.; Schiestl, R.H. Transcriptome analysis of a rotenone model of parkinsonism reveals complex I-tied and -untied toxicity mechanisms common to neurodegenerative diseases. PLoS ONE 2012, 7, e44700. [Google Scholar] [CrossRef]
- Declerck, K.; Remy, S.; Wohlfahrt-Veje, C.; Main, K.M.; van Camp, G.; Schoeters, G.; Vanden Berghe, W.; Andersen, H.R. Interaction between prenatal pesticide exposure and a common polymorphism in the PON1 gene on DNA methylation in genes associated with cardio-metabolic disease risk-an exploratory study. Clin. Epigenet. 2017, 9, 35. [Google Scholar] [CrossRef]
- Wirbisky, S.E.; Weber, G.J.; Schlotman, K.E.; Sepúlveda, M.S.; Freeman, J.L. Embryonic atrazine exposure alters zebrafish and human miRNAs associated with angiogenesis, cancer, and neurodevelopment. Food Chem. Toxicol. 2016, 98, 25–33. [Google Scholar] [CrossRef]
- Fenga, C.; Gangemi, S.; di Salvatore, V.; Falzone, L.; Libra, M. Immunological effects of occupational exposure to lead (Review). Mol. Med. Rep. 2017, 15, 3355–3360. [Google Scholar] [CrossRef]
- Polo, A.; Crispo, A.; Cerino, P.; Falzone, L.; Candido, S.; Giudice, A.; de Petro, G.; Ciliberto, G.; Montella, M.; Budillon, A.; et al. Environment and bladder cancer: Molecular analysis by interaction networks. Oncotarget 2017, 8, 65240–65252. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, V.; Ledda, C.; Matera, S.; Fago, L.; Arrabito, G.; Falzone, L.; Marconi, A.; Libra, M.; Loreto, C. Absence of t(14;18) chromosome translocation in agricultural workers after short-term exposure to pesticides. Mol. Med. Rep. 2017, 15, 3379–3382. [Google Scholar] [CrossRef] [PubMed]
- Rapisarda, V.; Salemi, R.; Marconi, A.; Loreto, C.; Graziano, A.C.; Cardile, V.; Basile, M.S.; Candido, S.; Falzone, L.; Spandidos, D.A.; et al. Fluoro-edenite induces fibulin-3 overexpression in non-malignant human mesothelial cells. Oncol. Lett. 2016, 12, 3363–3367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gangemi, S.; Miozzi, E.; Teodoro, M.; Briguglio, G.; de Luca, A.; Alibrando, C.; Polito, I.; Libra, M. Occupational exposure to pesticides as a possible risk factor for the development of chronic diseases in humans (Review). Mol. Med. Rep. 2016, 14, 4475–4488. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, S.; Liu, A.; Wan, J.; Tang, L.; Zheng, N.; Xiong, Y. Inhibition of BDNF production by MPP+ through up-regulation of miR-210-3p contributes to dopaminergic neuron damage in MPTP model. Neurosci. Lett. 2018, 675, 133–139. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. 2013, 4, e588. [Google Scholar] [CrossRef]
- Yuan, H.; Yuan, M.; Tang, Y.; Wang, B.; Zhan, X. MicroRNA expression profiling in human acute organophosphorus poisoning and functional analysis of dysregulated miRNAs. Afr. Health Sci. 2018, 18, 333–342. [Google Scholar] [CrossRef]
- Su, L.; Wang, C.; Zheng, C.; Wei, H.; Song, X. A meta-analysis of public microarray data identifies biological regulatory networks in Parkinson’s disease. BMC Med. Genom. 2018, 11, 40. [Google Scholar] [CrossRef]
- Fallico, M.; Raciti, G.; Longo, A.; Reibaldi, M.; Bonfiglio, V.; Russo, A.; Caltabiano, R.; Gattuso, G.; Falzone, L.; Avitabile, T. Current molecular and clinical insights into uveal melanoma (Review). Int. J. Oncol. 2021, 58, 10. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Romano, G.L.; Salemi, R.; Bucolo, C.; Tomasello, B.; Lupo, G.; Anfuso, C.D.; Spandidos, D.A.; Libra, M.; Candido, S. Prognostic significance of deregulated microRNAs in uveal melanomas. Mol. Med. Rep. 2019, 19, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [PubMed]
- Candido, S.; Lupo, G.; Pennisi, M.; Basile, M.S.; Anfuso, C.D.; Petralia, M.C.; Gattuso, G.; Vivarelli, S.; Spandidos, D.A.; Libra, M.; et al. The analysis of miRNA expression profiling datasets reveals inverse microRNA patterns in glioblastoma and Alzheimer’s disease. Oncol. Rep. 2019, 42, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Filetti, V.; Falzone, L.; Rapisarda, V.; Caltabiano, R.; Graziano, E.A.C.; Ledda, C.; Loreto, C. Modulation of microRNA expression levels after naturally occurring asbestiform fibers exposure as a diagnostic biomarker of mesothelial neoplastic transformation. Ecotoxicol. Environ. Saf. 2020, 198, 110640. [Google Scholar] [CrossRef]
- Hafsi, S.; Candido, S.; Maestro, R.; Falzone, L.; Soua, Z.; Bonavida, B.; Spandidos, D.A.; Libra, M. Correlation between the overexpression of Yin Yang 1 and the expression levels of miRNAs in Burkitt’s lymphoma: A computational study. Oncol. Lett. 2016, 11, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Seidler, F.J. Protein kinase C is a target for diverse developmental neurotoxicants: Transcriptional responses to chlorpyrifos; diazinon, dieldrin and divalent nickel in PC12 cells. Brain Res. 2009, 1263, 23–32. [Google Scholar] [CrossRef]
- Pontillo, C.; Español, A.; Chiappini, F.; Miret, N.; Cocca, C.; Alvarez, L.; Kleiman de Pisarev, D.; Sales, M.E.; Randi, A.S. Hexachlorobenzene promotes angiogenesis in vivo, in a breast cancer model and neovasculogenesis in vitro, in the human microvascular endothelial cell line HMEC-1. Toxicol. Lett. 2015, 239, 53–64. [Google Scholar] [CrossRef]
- Chang, Z.S.; Xia, J.B.; Wu, H.Y.; Peng, W.T.; Jiang, F.Q.; Li, J.; Liang, C.Q.; Zhao, H.; Park, K.S.; Song, G.H.; et al. Forkhead box O3 protects the heart against paraquat-induced aging-associated phenotypes by upregulating the expression of antioxidant enzymes. Aging Cell 2019, 18, e12990. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Liu, Z.; Zhu, S.; Liu, H.; Fan, W.; Hu, Y.; Hu, T.; Yu, Y.; Li, Y.; et al. Resveratrol Suppresses Rotenone-induced Neurotoxicity Through Activation of SIRT1/Akt1 Signaling Pathway. Anat. Rec. 2018, 301, 1115–1125. [Google Scholar] [CrossRef]
- Hameed, D.A.; Yassa, H.A.; Agban, M.N.; Hanna, R.T.; Elderwy, A.M.; Zwaita, M.A. Genetic aberrations of the K-ras proto-oncogene in bladder cancer in relation to pesticide exposure. Environ. Sci. Pollut. Res. Int. 2018, 25, 21535–21542. [Google Scholar] [CrossRef] [PubMed]
- Ahkin Chin Tai, J.K.; Freeman, J.L. Zebrafish as an integrative vertebrate model to identify miRNA mechanisms regulating toxicity. Toxicol. Rep. 2020, 7, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Jovanović, B.; Palić, D. In silico prediction of MicroRNA role in regulation of Zebrafish (Danio rerio) responses to nanoparticle exposure. Toxicol. In Vitro 2019, 60, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, F.; Chen, W. Exposure to triclosan changes the expression of microRNA in male juvenile zebrafish (Danio rerio). Chemosphere 2019, 214, 651–658. [Google Scholar] [CrossRef]
- Van der Plaat, D.A.; Vonk, J.M.; Terzikhan, N.; de Jong, K.; de Vries, M.; la Bastide-van Gemert, S.; van Diemen, C.C.; Lahousse, L.; Brusselle, G.G.; Nedeljkovic, I.; et al. Occupational exposure to gases/fumes and mineral dust affect DNA methylation levels of genes regulating expression. Hum. Mol. Genet. 2019, 28, 2477–2485. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, P.; Gavriil, M.; Rantakokko, P.; Ladoukakis, E.; Botsivali, M.; Kelly, R.S.; Bergdahl, I.A.; Kiviranta, H.; Vermeulen, R.C.H.; Spaeth, F.; et al. DNA methylation profiling implicates exposure to PCBs in the pathogenesis of B-cell chronic lymphocytic leukemia. Environ. Int. 2019, 126, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Murakami, M.; Ikeda, Y.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y. Role of tumor suppressor molecules in genomic perturbations and damaged DNA repair involved in the pathogenesis of cancer and neurodegeneration (Review). Biomed. Rep. 2020, 13, 10. [Google Scholar] [CrossRef]
- Napoli, S.; Scuderi, C.; Gattuso, G.; Bella, V.D.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef]
- Doukas, S.G.; Vageli, D.P.; Nikolouzakis, T.K.; Falzone, L.; Docea, A.O.; Lazopoulos, G.; Kalbakis, K.; Tsatsakis, A. Role of DNA mismatch repair genes in lung and head and neck cancer (Review). World Acad. Sci. 2019, 1, 184–191. [Google Scholar] [CrossRef]
- Houck, A.L.; Seddighi, S.; Driver, J.A. At the Crossroads Between Neurodegeneration and Cancer: A Review of Overlapping Biology and Its Implications. Curr. Aging Sci. 2018, 11, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Stella, M.; Falzone, L.; Caponnetto, A.; Gattuso, G.; Barbagallo, C.; Battaglia, R.; Mirabella, F.; Broggi, G.; Altieri, R.; Certo, F.; et al. Serum Extracellular Vesicle-Derived circHIPK3 and circSMARCA5 Are Two Novel Diagnostic Biomarkers for Glioblastoma Multiforme. Pharmaceuticals 2021, 14, 618. [Google Scholar] [CrossRef] [PubMed]
- Crimi, S.; Falzone, L.; Gattuso, G.; Grillo, C.M.; Candido, S.; Bianchi, A.; Libra, M. Droplet Digital PCR Analysis of Liquid Biopsy Samples Unveils the Diagnostic Role of hsa-miR-133a-3p and hsa-miR-375-3p in Oral Cancer. Biology 2020, 9, 379. [Google Scholar] [CrossRef]
- Tuaeva, N.O.; Falzone, L.; Porozov, Y.B.; Nosyrev, A.E.; Trukhan, V.M.; Kovatsi, L.; Spandidos, D.A.; Drakoulis, N.; Kalogeraki, A.; Mamoulakis, C.; et al. Translational Application of Circulating DNA in Oncology: Review of the Last Decades Achievements. Cells 2019, 8, 1251. [Google Scholar] [CrossRef]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients with BRAFV600E Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef]
- Falzone, L.; Gattuso, G.; Tsatsakis, A.; Spandidos, D.A.; Libra, M. Current and innovative methods for the diagnosis of COVID-19 infection (Review). Int. J. Mol. Med. 2021, 47, 100. [Google Scholar] [CrossRef]
- Ren, N.; Atyah, M.; Chen, W.Y.; Zhou, C.H. The various aspects of genetic and epigenetic toxicology: Testing methods and clinical applications. J. Transl. Med. 2017, 15, 110. [Google Scholar] [CrossRef]
- Duke, S.O.; Bajsa, J.; Pan, Z. Omics methods for probing the mode of action of natural and synthetic phytotoxins. J. Chem. Ecol. 2013, 39, 333–347. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset ID | Exposed (N) | Unexposed (N) | Platform | Sample Type | Pesticide | Ref |
|---|---|---|---|---|---|---|
| Gene Expression | ||||||
| GSE30335 | 20 | 20 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | Whole blood from exposed workers | Multiple exposure | N/A |
| GSE109513 | 81 | 9 | GPL13158 [HT_HG-U133_Plus_PM] Affymetrix HT HG-U133+ PM Array Plate | HepG2—Liver cancer cells | Imazalil, Fenbuconazole, 2,4-D | [30] |
| GSE109511 | 81 | 9 | GPL13158 [HT_HG-U133_Plus_PM] Affymetrix HT HG-U133+ PM Array Plate | HepaRG—Liver normal cells | Imazalil, Fenbuconazole, 2,4-D | [30] |
| GSE109509 | 81 | 9 | GPL13158 [HT_HG-U133_Plus_PM] Affymetrix HT HG-U133+ PM Array Plate | MCF7—Breast cancer cells | Imazalil, Fenbuconazole, 2,4-D | [30] |
| GSE94521 | 30 | 20 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | (hESC)-derived neural crest cells (NCC)—Normal Cells | Arsenic trioxide, Triadimefon, Cyproconazole, Geldanamycin, PBDE99, Trichostatin A | [31] |
| GSE35642 | 12 | 6 | GPL96 [HG-U133A] Affymetrix Human Genome U133A Array | SK-N-MC—Brain cancer cells | Rotenone | [32] |
| DNA Methylation | ||||||
| GSE90117 | 25 | 23 | GPL13534 Illumina HumanMethylation450 BeadChip (HumanMethylation450_15017482) | Whole blood from child with or without prenatal pesticide exposure | Multiple exposures | [33] |
| GSE38646 | 21 | 6 | GPL8490 Illumina HumanMethylation27 BeadChip (HumanMethylation27_270596_v.1.2) | K562—Hematological cancer cells | Diazinon, Dichlorvos, Chlorpyrifos, Terbufos, Fonofos, Phorate, Prathion | N/A |
| miRNA Expression | ||||||
| GSE78805 | 11 | 4 | GPL21545 Agilent-040157 Zebrafish_Human miRNA v18 [miRNA_ID version] | Zebrafish larvae | Atrazine | [34] |
| miRNA ID | log2FC | p-Value |
|---|---|---|
| Up-Regulated | ||
| hsa-miR-3149 | 2.613 | 0.0281 |
| hsa-miR-4505 | 2.200 | 0.0333 |
| hsa-miR-4687-3p | 0.417 | 0.0039 |
| hsa-miR-4758-5p | 0.259 | 0.0133 |
| hsa-miR-1268b | 0.236 | 0.0484 |
| Down-Regulated | ||
| hsa-miR-455-3p | −0.116 | 0.0349 |
| hsa-miR-23a-3p | −0.124 | 0.0499 |
| hsa-miR-30e-3p | −0.131 | 0.0363 |
| hsa-miR-210 | −0.155 | 0.0471 |
| hsa-miR-4261 | −0.181 | 0.0499 |
| hsa-miR-324-3p | −0.222 | 0.0051 |
| hsa-miR-29b-3p | −0.233 | 0.0246 |
| hsa-miR-181c-5p | −0.363 | 0.0385 |
| hsa-miR-3907 | −0.412 | 0.0003 |
| hsa-miR-101-3p | −0.414 | 0.0058 |
| hsa-miR-3610 | −2.262 | 0.0204 |
| hsa-miR-10b-3p | −2.323 | 0.0220 |
| hsa-miR-4515 | −2.462 | 0.0480 |
| hsa-miR-4313 | −2.545 | 0.0146 |
| hsa-miR-126-3p | −2.945 | 0.0002 |
| N. | KEGG Pathway | p-Value | #Genes | #miRNAs | Genes |
|---|---|---|---|---|---|
| 1 | Fatty acid degradation (hsa00071) | 8.79E-04 | 3 | 3 | ADH4, HADHA, EHHADH |
| 2 | TGF-beta signaling pathway (hsa04350) | 8.79E-04 | 13 | 3 | ID2, ROCK1, SMAD2, THBS1, PPP2CA, SMURF2, SMAD3, BMP8B, SKP1, SMAD4, GDF6, SP1, BMPR1A |
| 3 | Glycosphingolipid biosynthesis-ganglio series (hsa00604) | 1.67E-02 | 4 | 2 | HEXB, ST3GAL1, ST3GAL2, ST6GALNAC6 |
| 4 | Gap junction (hsa04540) | 1.67E-02 | 12 | 4 | PRKCA, SOS2, TJP1, LPAR1, SOS1, GJD2, GUCY1A2, GJA1, MAP3K2, GRB2, PDGFRB, ADCY6 |
| 5 | Cell adhesion molecules (CAMs) (hsa04514) | 3.23E-02 | 21 | 3 | WWC2, NRCAM, CDH2, NEGR1, SPN, SDC3, CD6, CNTNAP2, NRXN1, CLDN22, HLA-F, CD34, CADM1, CD226, NRXN3, VCAN, NLGN2, NLGN1, SDC2, PVRL3, ITGAL |
| 6 | Fatty acid elongation (hsa00062) | 3.71E-02 | 2 | 2 | HADHA, ELOVL7 |
| 7 | Thyroid hormone synthesis (hsa04918) | 3.71E-02 | 8 | 2 | PRKCA, ATP1B2, CREB3L3, TSHR, CREB3L1, TG, SLC5A5, ADCY6 |
| 8 | Amphetamine addiction (hsa05031) | 3.71E-02 | 12 | 4 | PRKCA, CREB3L3, CAMK4, CALM3, SIRT1, SLC18A2, GRIN3B, PDYN, CREB3L1, GRIN2A, GRIA3, GRIN2B |
| N. | KEGG Pathway | p-Value | #Genes | #miRNAs |
|---|---|---|---|---|
| 1 | ECM–receptor interaction (hsa04512) | 3.63E-19 | 38 | 8 |
| 2 | Proteoglycans in cancer (hsa05205) | 4.37E-13 | 92 | 9 |
| 3 | Viral carcinogenesis (hsa05203) | 3.72E-12 | 94 | 9 |
| 4 | Adherens junction (hsa04520) | 3.74E-12 | 46 | 9 |
| 5 | Renal cell carcinoma (hsa05211) | 4.81E-07 | 40 | 8 |
| 6 | Chronic myeloid leukemia (hsa05220) | 6.31E-07 | 43 | 8 |
| 7 | Pancreatic cancer (hsa05212) | 1.40E-06 | 38 | 7 |
| 8 | Hippo signaling pathway (hsa04390) | 1.40E-06 | 58 | 9 |
| 9 | Focal adhesion (hsa04510) | 1.40E-06 | 100 | 9 |
| 10 | p53 signaling pathway (hsa04115) | 4.46E-06 | 41 | 9 |
| 11 | Fatty acid biosynthesis (hsa00061) | 7.57E-06 | 3 | 7 |
| 12 | Colorectal cancer (hsa05210) | 7.57E-06 | 36 | 8 |
| 13 | FoxO signaling pathway (hsa04068) | 2.57E-05 | 67 | 9 |
| 14 | PI3K-Akt signaling pathway (hsa04151) | 2.89E-05 | 141 | 9 |
| 15 | Glioma (hsa05214) | 3.83E-05 | 33 | 8 |
| 16 | Small cell lung cancer (hsa05222) | 5.04E-05 | 46 | 8 |
| 17 | Lysine degradation (hsa00310) | 5.78E-05 | 22 | 7 |
| 18 | Protein processing in endoplasmic reticulum (hsa04141) | 7.18E-05 | 76 | 9 |
| 19 | Nonsmall cell lung cancer (hsa05223) | 1.08E-04 | 29 | 7 |
| 20 | Pathways in cancer (hsa05200) | 1.85E-04 | 157 | 9 |
| 21 | Fatty acid metabolism (hsa01212) | 1.87E-04 | 16 | 8 |
| 22 | Sphingolipid signaling pathway (hsa04071) | 1.87E-04 | 53 | 9 |
| 23 | Cell cycle (hsa04110) | 2.12E-04 | 61 | 9 |
| 24 | Prostate cancer (hsa05215) | 2.53E-04 | 46 | 8 |
| 25 | Estrogen signaling pathway (hsa04915) | 3.85E-04 | 45 | 9 |
| 26 | Regulation of actin cytoskeleton (hsa04810) | 4.42E-04 | 88 | 9 |
| 27 | Endocytosis (hsa04144) | 4.93E-04 | 86 | 9 |
| 28 | Endometrial cancer (hsa05213) | 7.39E-04 | 27 | 8 |
| 29 | Neurotrophin signaling pathway (hsa04722) | 1.07E-03 | 56 | 9 |
| 30 | Ubiquitin mediated proteolysis (hsa04120) | 1.35E-03 | 63 | 8 |
| 31 | mTOR signaling pathway (hsa04150) | 2.96E-03 | 32 | 9 |
| 32 | RNA transport (hsa03013) | 4.16E-03 | 68 | 9 |
| 33 | Thyroid hormone signaling pathway (hsa04919) | 4.16E-03 | 54 | 9 |
| 34 | Signaling pathways regulating pluripotency of stem cells (hsa04550) | 4.58E-03 | 56 | 9 |
| 35 | Melanoma (hsa05218) | 5.40E-03 | 33 | 7 |
| 36 | Progesterone-mediated oocyte maturation (hsa04914) | 5.40E-03 | 41 | 8 |
| 37 | Prolactin signaling pathway (hsa04917) | 5.78E-03 | 33 | 7 |
| 38 | Phosphatidylinositol signaling system (hsa04070) | 5.78E-03 | 35 | 9 |
| 39 | Acute myeloid leukemia (hsa05221) | 7.93E-03 | 27 | 8 |
| 40 | Central carbon metabolism in cancer (hsa05230) | 9.70E-03 | 30 | 9 |
| 41 | Transcriptional misregulation in cancer (hsa05202) | 1.29E-02 | 67 | 8 |
| 42 | Oocyte meiosis (hsa04114) | 1.44E-02 | 49 | 9 |
| 43 | AMPK signaling pathway (hsa04152) | 1.54E-02 | 52 | 9 |
| 44 | TGF-beta signaling pathway (hsa04350) | 1.54E-02 | 33 | 9 |
| 45 | Inositol phosphate metabolism (hsa00562) | 1.90E-02 | 26 | 8 |
| 46 | Insulin signaling pathway (hsa04910) | 2.15E-02 | 58 | 9 |
| 47 | Biotin metabolism (hsa00780) | 2.32E-02 | 1 | 1 |
| 48 | Fc gamma R-mediated phagocytosis (hsa04666) | 2.52E-02 | 39 | 8 |
| 49 | Dorso-ventral axis formation (hsa04320) | 2.66E-02 | 15 | 7 |
| 50 | Axon guidance (hsa04360) | 3.83E-02 | 45 | 9 |
| 51 | Steroid biosynthesis (hsa00100) | 3.93E-02 | 8 | 6 |
| 52 | mRNA surveillance pathway (hsa03015) | 4.71E-02 | 40 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giambò, F.; Leone, G.M.; Gattuso, G.; Rizzo, R.; Cosentino, A.; Cinà, D.; Teodoro, M.; Costa, C.; Tsatsakis, A.; Fenga, C.; et al. Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets. Int. J. Environ. Res. Public Health 2021, 18, 8697. https://doi.org/10.3390/ijerph18168697
Giambò F, Leone GM, Gattuso G, Rizzo R, Cosentino A, Cinà D, Teodoro M, Costa C, Tsatsakis A, Fenga C, et al. Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets. International Journal of Environmental Research and Public Health. 2021; 18(16):8697. https://doi.org/10.3390/ijerph18168697
Chicago/Turabian StyleGiambò, Federica, Gian Marco Leone, Giuseppe Gattuso, Roberta Rizzo, Alessia Cosentino, Diana Cinà, Michele Teodoro, Chiara Costa, Aristides Tsatsakis, Concettina Fenga, and et al. 2021. "Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets" International Journal of Environmental Research and Public Health 18, no. 16: 8697. https://doi.org/10.3390/ijerph18168697
APA StyleGiambò, F., Leone, G. M., Gattuso, G., Rizzo, R., Cosentino, A., Cinà, D., Teodoro, M., Costa, C., Tsatsakis, A., Fenga, C., & Falzone, L. (2021). Genetic and Epigenetic Alterations Induced by Pesticide Exposure: Integrated Analysis of Gene Expression, microRNA Expression, and DNA Methylation Datasets. International Journal of Environmental Research and Public Health, 18(16), 8697. https://doi.org/10.3390/ijerph18168697