
Mechanisms of Ethanol-Induced Cerebellar Ataxia: Underpinnings of Neuronal Death in the Cerebellum




Abstract
1. Introduction
2. Acute Effects of Alcohol
2.1. Clinical Profiles
2.2. Pathophysiology
2.2.1. Deafferentation
2.2.2. Abnormal Activation of Outputs Cells
3. Chronic Effects of Alcohol
3.1. Clinical Profile
3.2. Pathophysiology
3.2.1. Pathology
3.2.2. Malnutrition-Induced Degeneration
3.2.3. Ethanol-Induced Direct Neuronal Degeneration
3.2.4. Genetic Expression Factors
3.2.5. Long-Term Alcohol Exposure
3.3. Therapies and Prognosis
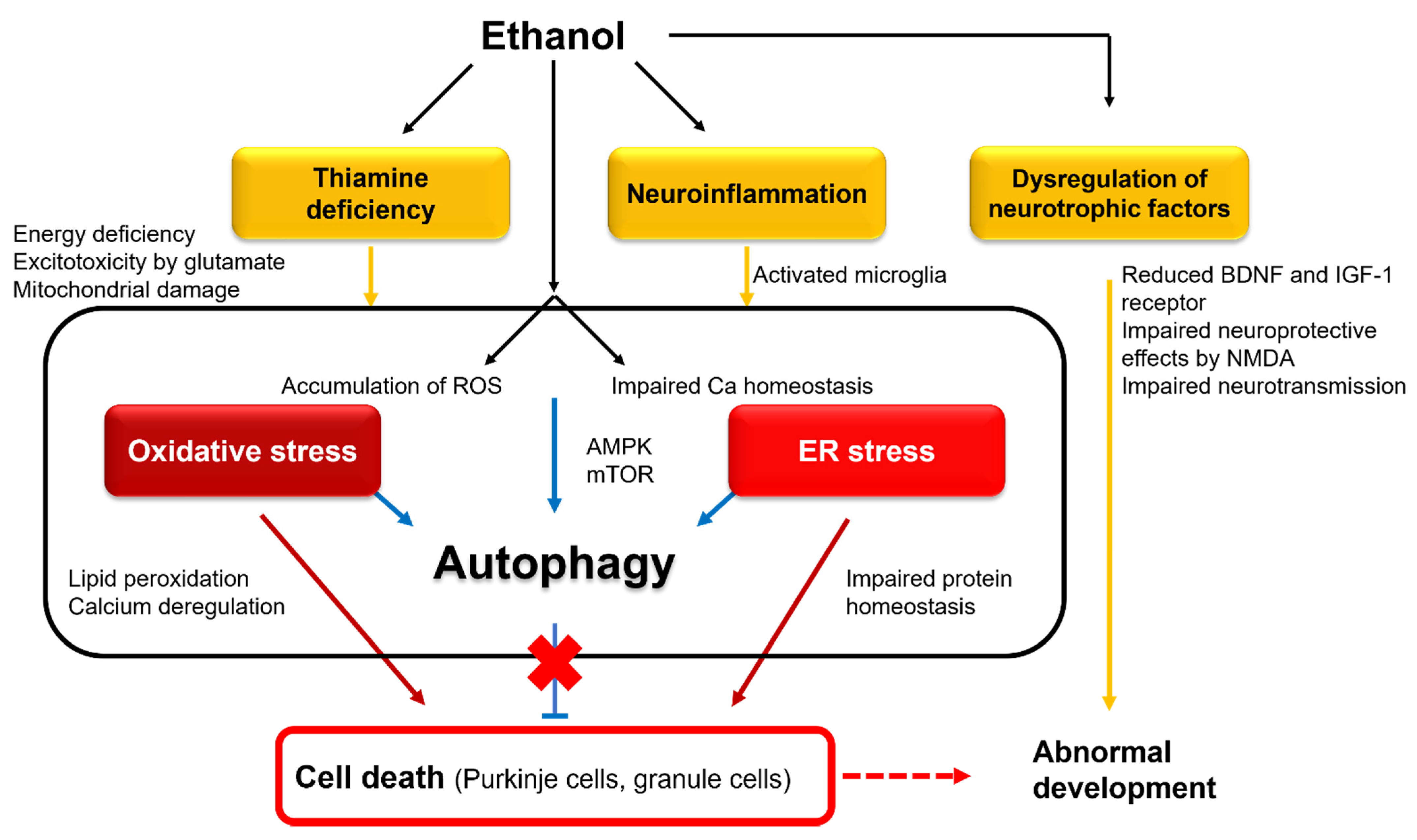
4. Fetal Alcohol Spectrum Disorders
4.1. Clinical Profiles
4.2. Pathophysiology
4.2.1. Interference with Neurotrophic and Retinoic Acid Pathways
4.2.2. Involvement of Oxidative and ER Stresses
4.2.3. Autophagic Protection
4.2.4. Neuroinflammation
4.3. Intervention
5. Conclusions: Crosstalk between Cell Degeneration and Abnormal Development
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Consent for Publication
Code Availability
References
- Manto, M.U.; Jacquy, J. Alcohol toxicity in the cerebellum: Clinical aspects. In The Cerebellum and Its Disorders; Manto, M.U., Pandolfo, M., Eds.; Cambridge University Press: Cambridge, UK, 2002; pp. 336–341. [Google Scholar]
- Laureno, R. Nutritional cerebellar degeneration, with comments on its relationship to Wernicke disease and alcoholism. In Handbook of Clinical Neurology. Vol. 103 (3rd Series) Ataxic Disorders; Subramony, S.H., Dürr, A., Eds.; Elsevier: Amsterdam, The Netherland, 2012; pp. 175–187. [Google Scholar]
- Dar, M.S. Ethanol-induced cerebellar ataxia: Cellular and molecular mechanisms. Cerebellum 2015, 14, 447–465. [Google Scholar] [CrossRef]
- Manto, M.; Perrotta, G. Toxic-induced cerebellar syndrome: From the fetal to the elderly. Handb. Clin. Neurol. 2018, 155, 333–352. [Google Scholar]
- Adamas, R.D. Nutritional cerebellar degeneration. In Handbook of Clinical Neurology; Vinken, P.J., Bruyn, G.W., Eds.; North Holland Publishing Company: Amsterdam, The Netherland, 1976; Volume 28, pp. 271–284. [Google Scholar]
- Victor, M.; Adams, R.D.; Collins, G.H. The Wernicke-Korsakoff Syndrome and Related Neurological Disorders due to Alcoholism and Malnutrition, 2nd ed.; FA Davis: Philadelphia, PA, USA, 1989. [Google Scholar]
- Luo, J. Effects of ethanol on the cerebellum: Advances and prospects. Cerebellum 2015, 14, 383–385. [Google Scholar] [CrossRef]
- Norman, A.L.; Crocker, N.; Mattson, S.N.; Riley, E.P. Neuroimaging and fetal alcohol spectrum disorders. Dev. Disabil. Res. Rev. 2009, 15, 209–217. [Google Scholar] [CrossRef] [PubMed]
- West, J.R. Acute and long-term changes in the cerebellum following developmental exposure to ethanol. Alcohol Alcohol. Suppl. 1993, 2, 199–202. [Google Scholar] [PubMed]
- Marshall, S.; Chen, Y.; Singh, S.; Berrios-Carcamo, P.; Heit, C.; Apostolopoulos, N.; Golla, J.P.; Thompson, D.C.; Vasiliou, V. Engineered Animal Models Designed for Investigating Ethanol Metabolism, Toxicity and Cancer. Adv. Exp. Med. Biol. 2018, 1032, 203–221. [Google Scholar] [PubMed]
- Müller, T.E.; Nunes, M.E.M.; Rodrigues, N.R.; Fontana, B.D.; Hartmann, D.D.; Franco, J.L.; Rosemberg, D.B. Neurochemical mechanisms underlying acute and chronic ethanol-mediated responses in zebrafish: The role of mitochondrial bioenergetics. Neurochem. Int. 2019, 131, 104584. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Autophagy and ethanol neurotoxicity. Autophagy 2014, 10, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Vonghia, L.; Leggio, L.; Ferrulli, A.; Bertini, M.; Gasbarrini, G.; Addolorato, G. Alcoholism Treatment Study Group. Acute alcohol intocication. Eur. J. Intern. Med. 2008, 19, 561–567. [Google Scholar] [CrossRef]
- Ito, M. The Cerebellum and Neural Control; Raven Press: New York, NY, USA, 1984. [Google Scholar]
- Kaplan, J.S.; Mohr, C.; Rossi, D.J. Opposite actions of alcohol on tonic GABA (A) receptor currents mediated by nNOS and PKC activity. Nat. Neurosci. 2013, 16, 1783–1793. [Google Scholar] [CrossRef]
- Valenzuela, C.F.; Jotty, K. Mini-Review: Effects of ethanol on GABAA receptor-mediated neurotransmission in cerebellar cortex—Recent advances. Cerebellum 2015, 14, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Braas, K.M.; Newby, A.C.; Wilson, V.S.; Snyder, S.H. Adenosine contain in neurons in the brain localized by immunocytochemistry. J. Neurosci. 1986, 6, 1952–1961. [Google Scholar] [CrossRef]
- Takahashi, M.; Kovalchuk, Y.; Atwellm, D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J. Neurosci. 1995, 15, 5693–5702. [Google Scholar] [CrossRef]
- Dittman, J.S.; Regehr, W.G. Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J. Neurosci. 1996, 16, 1623–1633. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Diaom, L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J. Pharmacol. Exp. Ther. 1994, 268, 537–545. [Google Scholar]
- Konishi, S.; Mitoma, H. Cyclic AMP-dependent and independent mechanisms for presynaptic modulation of GABAergic transmission in the cerebellar cortex. In Adenosine; Okada, Y., Ed.; Elsevier: Amsterdam, The Netherland, 1997; pp. 89–95. [Google Scholar]
- Geiger, J.D.; Nagy, J.I. Distribution of adenosine deaminase activity in rat brain and spinal cord. J. Neurosci. 1986, 6, 2707–2714. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.A.; Mackey, J.R.; Cass, C.E.; Young, J.D. Nucleoside transporters: Molecular biology and implications for therapeutic development. Mol. Med. Today 1999, 5, 216–224. [Google Scholar] [CrossRef]
- Choi, D.S.; Cascini, M.G.; Mailliardm, W.; Young, H.; Paredes, P.; McMahon, T.; Diamond, I.; Bonci, A.; Messing, R.O. The type 1 equilibrative nucleoside transporter regulates ethanol intoxication and preference. Nat. Neurosci. 2004, 7, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E.; Diamond, I.; Casso, D.J.; Franklin, C.; Gordon, A.S. Ethanol increases extracellular adenosine by inhibiting adenosine uptake via the nucleoside transporter. J. Biol. Chem. 1990, 265, 1946–1951. [Google Scholar] [CrossRef]
- Carta, M.; Mameli, M.; Valenzuela, C.F. Alcohol potently modulates climbing fiber → Purkinje neuron synapses: Role of metabotropic glutamate receptors. J. Neurosci. 2006, 26, 1906–1912. [Google Scholar] [CrossRef]
- Gilman, S.; Adams, K.; Koeppe, R.A.; Berent, S.; Kluin, K.J.; Modell, J.G.; Kroll, P.; Brunberg, J.A. Cerebellar and frontal hypometabolism in alcoholic cerebelalr degeneration studied with positron emission tomography. Ann. Neurol. 1990, 28, 775–785. [Google Scholar] [CrossRef]
- Haubek, A.; Lee, K. Computed tomography in alcoholic cerebellar atrophy. Neuroradiology 1979, 18, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Victor, M.; Adams, R.D.; Mancall, E.L. A restricted form of cerebellar cortical degeneration occurring in alcoholic patients: Cerebellar cortical degenerations. Arch. Neurol. 1959, 1, 579–688. [Google Scholar] [CrossRef]
- Johnson-Greene, D.; Adams, K.M.; Gilman, S.; Kluin, K.J.; Junck, L.; Martorello, S.; Heumann, M. Impaired upper limb coordination in alcoholic cerebellar degeneration. Ann. Neurol. 1997, 54, 436–439. [Google Scholar] [CrossRef]
- Torvik, A.; Lindboe, C.F.; Rogde, S. Brain lesions in alcoholics. A neuropathological study with clinical correlations. J. Neurol. Sci. 1982, 56, 233–248. [Google Scholar] [CrossRef]
- Torvik, A. Brain lesions in alcoholics: Neuropathological observations. Acta. Med. Scand. 1987, 717, 47–54. [Google Scholar] [CrossRef]
- Worner, T.M. Effects of alcohol. In Handbook of Cerebellar Diseases; Lechtenberg, R., Ed.; Marcel Dekker: New York, NY, USA, 1993; pp. 547–566. [Google Scholar]
- Happer, C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J. Neuropathol. Exp. Neurol. 1998, 57, 101–110. [Google Scholar]
- Riethdorf, L.; Warzok, R.; Schwesinger, G. Die Alkoholenzephalopathien im Obdvktionsgut. Zentralbl. Pathol. 1991, 137, 148–156. [Google Scholar]
- Victor, M.; Laurenom, R. Neurologic complication of alcohol abuse: Epidemiologic aspects. Adv. Neurol. 1978, 19, 603–617. [Google Scholar]
- Torvik, A.; Torp, S. The prevalence of alcoholic cerebellar atrophy. A morphometric and histological study of an autopsy material. J. Neurol. Sci. 1986, 75, 43–51. [Google Scholar] [CrossRef]
- Phillips, S.C.; Harper, C.G.; Kril, J.J. The contribution of Wernicke’s disease encephalopathy to alcohol-related cerebellar damage. Drug Alcohol. Rev. 1990, 9, 53–60. [Google Scholar] [CrossRef]
- Nicolás, J.M.; Fernandez-Sola, J.; Robert, J.; Antúnez, E.; Cofán, M.; Cardenal, C.; Sacanella, E.; Estruch, R.; Urbano-Márquez, A. High ethanol intake and malnutrition in alcoholic cerebellar shrinkage. QJM 2000, 93, 449–456. [Google Scholar] [CrossRef][Green Version]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Baker, K.; Harding, A.; Halliday, G.; Kril, J.J.; Harper, C.G. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience 1999, 91, 429–438. [Google Scholar] [CrossRef]
- Suzanne, M.; de la Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2014, 127, 71–90. [Google Scholar]
- Manzo, L.; Locatelli, C.; Candura, S.M.; Costa, L.G. Nutrition and alcohol neurotoxicity. Neurotoxicology 1994, 15, 555–565. [Google Scholar] [PubMed]
- Hazell, A.S.; Todd, K.G.; Butterworth, R.F. Mechanisms of neuronal death in Wernicke’s encephalopathy. Metab. Brain Dis. 1998, 13, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic Roles of Glutamate Decarboxylase 65 Autoantibodies in Cerebellar Ataxias. J. Immunol. Res. 2017, 2017, 2913297. [Google Scholar] [CrossRef]
- Kleinschmidt-DeMasters, B.K.; Norenberg, M.D. Cerebellar degeneration in the rat following rapid correction of hyponatremia. Ann. Neurol. 1981, 10, 561–565. [Google Scholar] [CrossRef]
- Setta, F.; Jacquy, J.; Hildebrand, J.; Manto, M.U. Ataxia indued by small amounts of alcohol. J. Neurol. Neurosurg. Psychiatry 1998, 65, 370–373. [Google Scholar] [CrossRef]
- Estrin, W.J. Alcoholic cerebellar degeneration is not a dose-dependent phenomenon. Alcohol. Clin. Exp. Res. 1987, 11, 372–375. [Google Scholar] [CrossRef]
- Greenwood, J.; Jeyasingham, M.; Pratt, O.E.; Ryle, P.R.; Shaw, G.K.; Thomson, A.D. Heterogeneity of human erythrocyte transketolase: A preliminary report. Alcohol Alcohol. 1984, 19, 123–129. [Google Scholar]
- de la Monte, S.M.; Longato, L.; Tong, M.; DeNucci, S.; Wands, J.R. The liver-brain axis of alcohol-mediated neurodegeneration: Role of toxic lipids. Int. J. Environ. Res. Public Health 2009, 6, 2055–2075. [Google Scholar] [CrossRef]
- Chen, C.H.; Walker, J.; Momenan, R.; Rawlings, R.; Heilig, M.; Hommer, D. Relationship between liver function and brain shrinkage in patients with alcohol dependence. Alcohol. Clin. Exp. Res. 2012, 36, 625–632. [Google Scholar] [CrossRef]
- Dlugos, C.A. Ethanol-induced alterations in Purkinje neuron dendrites in adult and aging rats: A Review. Cerebellum 2015, 14, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.A.; Neafsey, E.J. Ethanol and adult CNS neurodamage: Oxidative stress, but possibly not excitotoxicity. Front. Biosci. 2012, 4, 1358–1367. [Google Scholar] [CrossRef]
- Schöls, L.; Szymanski, S.; Peters, S.; Przuntek, H.; Epplen, J.T.; Hardt, C.; Riess, O. Genetic background of apparently idiopathic sporadic cerebellar ataxia. Hum. Genet. 2000, 107, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, P. Fragile X-associated tremor/ataxia syndrome (FXTAS): Pathology and mechanisms. Acta Neuropathol. 2013, 126, 1–19. [Google Scholar] [CrossRef]
- Dulman, R.S.; Auta, J.; Teppen, T.; Pandey, S.C. Acute Ethanol Produces Ataxia and Induces Fmr1 Expression via Histone Modifications in the Rat Cerebellum. Alcohol. Clin. Exp. Res. 2019, 43, 1191–1198. [Google Scholar] [CrossRef]
- Nagy, J. Alcohol related changes in regulation of NMDA receptor functions. Curr. Neuropharmacol. 2008, 6, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Gilman, S.; Koeppe, R.A.; Adams, K.; Johnson-Greene, D.; Junck, L.; Kluin, K.J.; Brunberg, J.; Martorello, S.; Lohman, M. Positron emission tomographic studies of cerebral benzodiazepine-receptor binding in chronic alcoholics. Ann. Neurol. 1996, 40, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Maini, A.; Rousset, O.G.; Brasić, J.R. Positron emission tomography-a tool for identifying the effects of alcohol dependence on the brain. Alcohol Res. Health 2003, 27, 161–173. [Google Scholar] [PubMed]
- Laureno, R.; Hodics, T. Alcohol intoxication and withdrawal. In Current Therapy in Neurologic Disease, 7th ed.; Johnson, R.T., Griffin, J.W., McArthurm, J.V., Eds.; Mosby: St. Louis, MO, USA, 2002; pp. 345–346. [Google Scholar]
- Jung, M.E. Alcohol Withdrawal and Cerebellar Mitochondria. Cerebellum 2014, 14, 421–437. [Google Scholar] [CrossRef]
- van Overwalle, F.; Manto, M.; Cattaneo, Z.; Clausi, S.; Ferrari, C.; Gabrieli, J.D.E.; Guell, X.; Heleven, E.; Lupo, M.; Ma, Q.; et al. Consensus paper: Cerebellum and social cognition. Cerebellum 2020, 19, 833–868. [Google Scholar] [CrossRef]
- Soyka, M.; Müller, C.A. Pharmacotherapy of alcoholism-an update on approved and off-label medications. Expert Opin. Pharmacother. 2017, 18, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.K.; Balasanova, A.A. Treatment of alcohol use disorder. JAMA 2021, 325, 596. [Google Scholar] [CrossRef]
- Miterko, L.N.; Baker, K.B.; Beckinghausen, J.; Bradnam, L.V.; Cheng, M.Y.; Cooperrider, J.; DeLong, M.R.; Gornati, S.V.; Hallett, M.; Heck, D.H.; et al. Consensus paper: Experimental neurostimulation of the cerebellum. Cerebellum 2019, 18, 1064–1097. [Google Scholar] [CrossRef]
- Luo, J. Mechanisms of ethanol-induced death of cerebellar granule cell. Cerebellum 2012, 11, 145–154. [Google Scholar] [CrossRef]
- Riley, E.P.; McGee, C.L. Fetal alcohol spectrum disorders; an overview with emphasis on changes in brain and behavior. Exp. Biol. Med. 2005, 230, 357–365. [Google Scholar] [CrossRef]
- Mattson, S.N.; Bernes, G.A.; Doyle, L.R. Fetal alcohol spectrum disorders: A review of the neurobehavioral deficits associated with prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 2019, 43, 1046–1062. [Google Scholar] [CrossRef]
- Streissguth, A.P.; Barr, H.M.; Sampson, P.D. Moderate prenatal alcohol exposure: Effects on child IQ and learning problems at age 7½ years. Alcohol. Clin. Exp. Res. 1990, 14, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Coles, C.D.; Platzman, K.A.; Lynch, M.E.; Freides, D. Auditory and visual sustained attention in adolescents prenatally exposed to alcohol. Alcohol. Clin. Exp. Res. 2002, 26, 263–271. [Google Scholar] [CrossRef]
- Adnams, C.M.; Kodituwakku, P.W.; Hay, A.; Moltenom, C.D.; Viljoen, D.; May, P.A. Patterns of cognitive-motor development in children with fetal alcohol syndrome from a community in South Africa. Alcohol. Clin. Exp. Res. 2001, 25, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Connor, P.D.; Sampson, P.D.; Streissguth, A.P.; Bookstein, F.L.; Barr, H.M. Effects of prenatal alcohol exposure on fine motor coordination and balance: A study of two adult samples. Neuropsychologia 2006, 44, 744–751. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Caplan, D. Cognition, emotion and the cerebellum. Brain 2006, 129, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Hoche, F.; Guell, X.; Vangel, M.G.; Sherman, J.C.; Schmahmann, J.D. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain 2018, 141, 248–270. [Google Scholar] [CrossRef]
- Sullivan, E.V.; Moore, E.M.; Lane, B.; Pohl, K.M.; Riley, E.P.; Pfefferbaum, A. Graded cerebellar lobular volume deficits in adolescents and young adults with fetal alcohol spectrum disorders (FASD). Cereb. Cortex 2020, 30, 4729–4746. [Google Scholar] [CrossRef]
- Napper, R.M.; West, J.R. Permanent neuronal cell loss in the cerebellum of rats exposed to continuous low blood alcohol levels during the brain growth spurt: A stereological investigation. J. Comp. Neurol. 1995, 362, 283–292. [Google Scholar] [CrossRef]
- Maier, S.E.; Miller, J.A.; Blackwell, J.M.; West, J.R. Fetal alcohol exposure and temporal vulnerability: Regional differences in cell loss as a function of the timing of binge-like alcohol exposure during brain development. Alcohol. Clin. Exp. Res. 1999, 23, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.E.; West, J.R. Regional differences in cell loss associated with binge-like alcohol exposure during the first two trimesters equivalent in the rat. Alcohol 2001, 23, 49–57. [Google Scholar] [CrossRef]
- Green, J.T. The effects of ethanol on the developing cerebellum and eyeblink classical conditioning. Cerebellum 2004, 3, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Karacay, B.; Li, S.; Bonthius, D.J. Maturation-dependent alcohol resistance in the developing mouse: Cerebellar neuronal loss and gene expression during alcohol-vulnerable and -resistant periods. Alcohol. Clin. Exp. Res. 2008, 32, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Siler-Marsiglio, K.I.; Paiva, M.; Madorsky, I.; Pan, Q.; Shaw, G.; Heaton, M.B. Functional mechanisms of apoptosis-related proteins in neonatal rat cerebellum are differentially influenced by ethanol at postnatal days 4 and 7. J. Neurosci. Res. 2005, 81, 632–643. [Google Scholar] [CrossRef]
- Light, K.E.; Ge, Y.; Belcher, S.M. Early postnatal ethanol exposure selectively decreases BDNF and truncated TrkB-T2 receptor mRNA expression in the rat cerebellum. Brain Res. Mol. Brain Res. 2001, 93, 46–55. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Wands, J.R. Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell Mol. Life Sci. 2002, 59, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Bhave, S.V.; Snell, L.D.; Tabakoff, B.; Hoffman, P.L. Chronic ethanol exposure attenuates the antiapoptotic effect of NMDA in cerebellar granule neurons. J. Neurochem. 2000, 75, 1035–1044. [Google Scholar] [CrossRef]
- Hoffman, P.L. NMDA receptors in alcoholism. Int. Rev. Neurobiol. 2003, 56, 35–82. [Google Scholar]
- Lotfullina, N.; Khazipov, R. Ethanol and the developing brain: Inhibition of neural activity and neuroapoptosis. Neuroscientist 2018, 24, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W. Fetal alcohol syndrome at the cellular level. Addict. Biol. 2004, 9, 137–149. [Google Scholar] [CrossRef]
- Olney, J.W. Focus on apoptosis to decipher how alcohol and many other drugs disrupt brain development. Front. Pediatr. 2014, 4, 81. [Google Scholar] [CrossRef]
- McCaffery, P.J.; Adams, J.; Maden, M.; Rosa-Molinar, E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 2003, 18, 457–472. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, C.K.; DiPette, D.D.; Singh, U.S. Ethanol impairs activation of retinoic acid receptors in cerebellar granule cells in a rodent model of fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2010, 34, 928–937. [Google Scholar] [CrossRef]
- Heaton, M.B.; Paiva, M.; Mayer, J.; Miller, R. Ethanol-mediated generation of reactive oxygen species in developing rat cerebellum. Neurosci. Lett. 2002, 334, 83–86. [Google Scholar] [CrossRef]
- Heaton, M.B.; Moore, D.B.; Paiva, M.; Madorsky, I.; Mayer, J.; Shaw, G. The role of neurotrophic factors, apoptosis-related proteins, and endogenous antioxidants in the differential temporal vulnerability of neonatal cerebellum to ethanol. Alcohol. Clin. Exp. Res. 2003, 27, 657–669. [Google Scholar] [CrossRef]
- Ke, Z.; Wang, X.; Liu, Y.; Fan, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Li, M.; Fang, S.; et al. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcohol. Clin. Exp. Res. 2011, 35, 1574–1583. [Google Scholar] [CrossRef]
- Pla, A.; Pascual, M.; Renau-Piqueras, J.; Guerri, C. TLR4 mediates the impairment of ubiquitin-proteasome and autophagy-lysosome pathways induced by ethanol treatment in brain. Cell Death Dis. 2014, 5, e1066. [Google Scholar] [CrossRef]
- Brunois, C.; Ris, L. Multi-actions of microglia. In Neuroimmune Diseases. Form Cell to Living Brain; Mitoma, H., Manto, M., Eds.; Springer: Cham, Switzerland, 2019; pp. 303–328. [Google Scholar]
- Drew, P.D.; Kane, C.I. Fetal alcohol spectrum disorders and neuroimmune changes. Int. Rev. Neurobiol. 2014, 118, 41–80. [Google Scholar] [PubMed]
- Boyadjieva, N.I.; Sarkar, D.K. Role of microglia in ethanol’s apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol. Clin. Exp. Res. 2010, 34, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, C.L.; Alto, M.E. Interventions in fetal alcohol spectrum: An international perspective. Eur. J. Med. Genet. 2017, 60, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Risbud, R.D.; Mattson, S.N.; Chambers, C.D.; Thomas, J.D. Randomized, double-blind, placebo-controlled clinical trial of choline supplementation in school-aged children with fetal alcohol spectrum disorders. Am. J. Clin. Nutr. 2016, 104, 1683–1692. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, J.R.; Fuglestad, A.J.; Eckerle, J.K.; Fink, B.A.; Hoecker, H.L.; Boys, C.J.; Radke, J.P.; Kroupina, M.G.; Miller, N.C.; Brearley, A.M.; et al. Choline supplementation in children with fetal alcohol spectrum disorders: A randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2015, 102, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Granato, A.; Dering, B. Alcohol and the developing brain: Why neurons die and how survives change. Int. J. Mol. Sci. 2018, 19, 2992. [Google Scholar] [CrossRef] [PubMed]
- Nkpaa, K.W.; Owoeye, O.; Amadi, B.A.; Adedara, I.A.; Abolaji, A.O.; Wegwu, M.O.; Farombi, E.O. Ethanol exacerbates manganese-induced oxidative/nitrosative stress, pro-inflammatory cytokines, nuclear factor-kB activation, and apoptosis induction in rat cerebellar cortex. J. Biochem. Mol. Toxicol. 2021, 35, e22681. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.M.; Douglas, J.C.; Rafferty, T.; Johnson, J.W.; Niedzwiedz-Massey, V.M.; Phelan, K.D.; Majewska, A.K.; Drew, P.D. Ethanol modulation of cerebellar neuroinflammation in a postnatal mouse model of fetal alcohol spectrum disorders. J. Neurosci. Res. 2021, 99, 1986–2007. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Graus, F.; Honnorat, J.; Jarius, S.; Titulaer, M.; Manto, M.; Hoggard, N.; Sarrigiannis, P.; Mitoma, H. Diagnostic criteria for primary autoimmune cerebellar ataxia (PACA)-Guidelines from an International Task Force on Immune Mediated Cerebellar Ataxia. Cerebellum 2020, 19, 605–610. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| More Prevalent Neurological Deficits |
| Ataxic stance, titubation |
| Ataxic gait |
| Peripheral neuropathy |
| Wernicke’s encephalopathy |
| Acute confusional state |
| Hallucinations |
| Agitation |
| Korsakoff syndrome |
| Alcohol withdrawal syndrome |
| Seizures, myoclonus, asterixis |
| Less Prevalent Neurological Deficits |
| Gaze-evoked nystagmus |
| Ocular dysmetria |
| Ophthalmoparesis |
| Ataxic speech |
| 3 Hz postural leg tremor |
| Kinetic tremor |
| Hypotonia |
| Amyotrophy |
| Autonomic overactivity |
| Increased risk of stroke |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitoma, H.; Manto, M.; Shaikh, A.G. Mechanisms of Ethanol-Induced Cerebellar Ataxia: Underpinnings of Neuronal Death in the Cerebellum. Int. J. Environ. Res. Public Health 2021, 18, 8678. https://doi.org/10.3390/ijerph18168678
Mitoma H, Manto M, Shaikh AG. Mechanisms of Ethanol-Induced Cerebellar Ataxia: Underpinnings of Neuronal Death in the Cerebellum. International Journal of Environmental Research and Public Health. 2021; 18(16):8678. https://doi.org/10.3390/ijerph18168678
Chicago/Turabian StyleMitoma, Hiroshi, Mario Manto, and Aasef G. Shaikh. 2021. "Mechanisms of Ethanol-Induced Cerebellar Ataxia: Underpinnings of Neuronal Death in the Cerebellum" International Journal of Environmental Research and Public Health 18, no. 16: 8678. https://doi.org/10.3390/ijerph18168678
APA StyleMitoma, H., Manto, M., & Shaikh, A. G. (2021). Mechanisms of Ethanol-Induced Cerebellar Ataxia: Underpinnings of Neuronal Death in the Cerebellum. International Journal of Environmental Research and Public Health, 18(16), 8678. https://doi.org/10.3390/ijerph18168678