Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies





Abstract
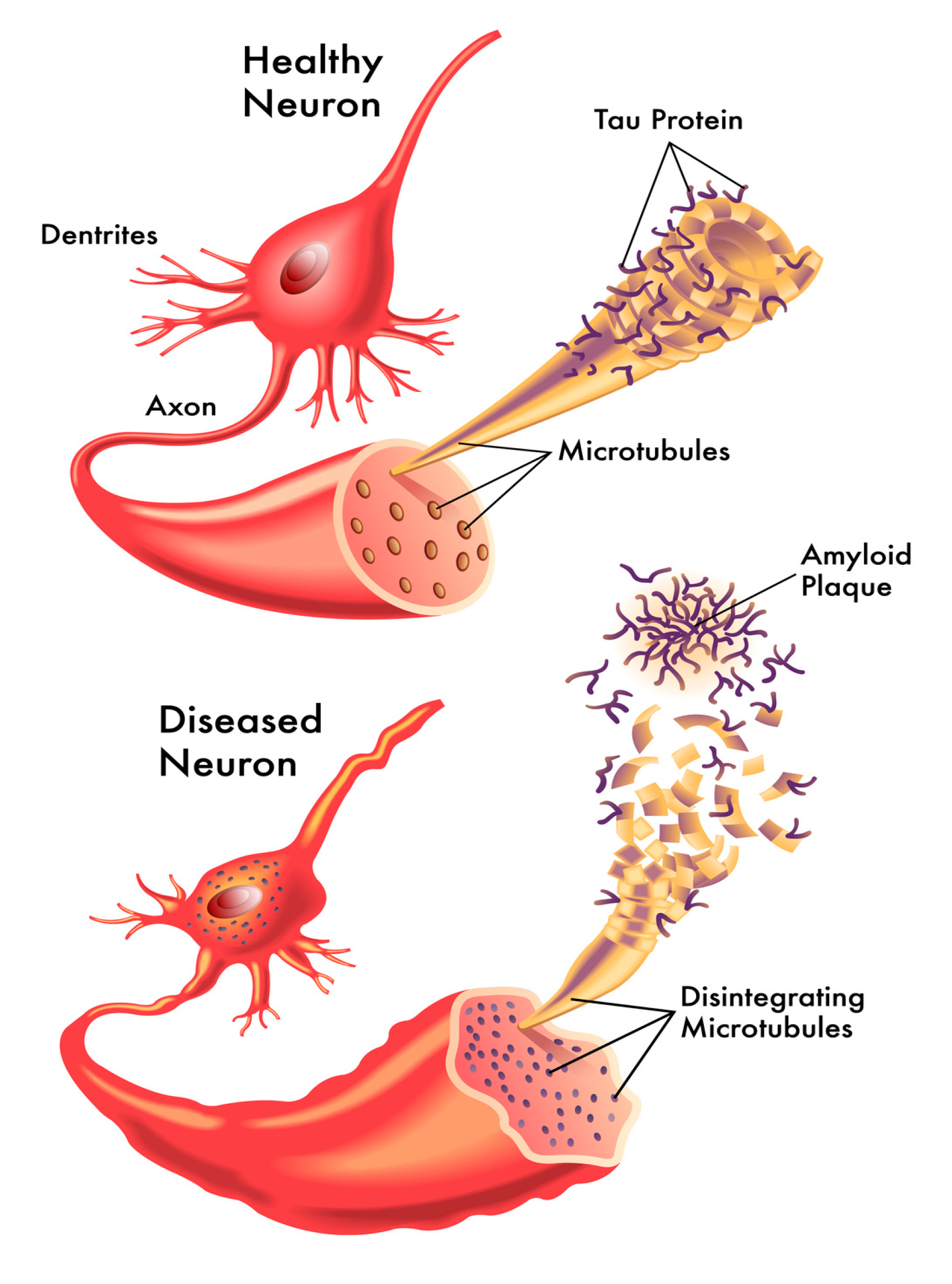
1. Introduction
2. Functional Roles of Tau Proteins
3. Clinical Types of Tauopathies
3.1. The Tau Hypothesis of Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Progressive Supranuclear Palsy
3.4. Examples of Geographic Clusters of Tauopathies
3.5. Frontotemporal Dementia and Pick’s Disease
4. Genetic Factors
5. Therapeutic and Preventive Possibilities
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reyes, J.F.; Reynolds, M.R.; Horowitz, P.M.; Fu, Y.; Guillozet-Bongaarts, A.L.; Berry, R.; Binder, L.I. A possible link between astrocyte activation and tau nitration in Alzheimer’s disease. Neurobiol. Dis. 2008, 31, 198–208. [Google Scholar] [CrossRef]
- Dehmelt, L.; Halpain, S. Protein family review The MAP2 / Tau family of microtubule-associated proteins. Genome Biol. 2004, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Shaw-Smith, C.; Pittman, A.M.; Willatt, L.; Martin, H.; Rickman, L.; Gribble, S.; Curley, R.; Cumming, S.; Dunn, C.; Kalaitzopoulos, D.; et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006, 38, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Kawamata, T.; Mukai, H.; Hasegawa, H.; Isagawa, T.; Yasuda, M.; Hashimoto, T.; Terashima, A.; Nakai, M.; Ono, Y.; et al. Phosphorylation of Tau Is Regulated by PKN. J. Biol. Chem. 2001, 276, 10025–10031. [Google Scholar] [CrossRef]
- Mawal-Dewan, M.; Henley, J.; Van De Voorde, A.; Trojanowski, J.Q.; Lee, V.M.Y. The phosphorylation state of tau in the developing rat brain is regulated by phosphoprotein phosphatases. J. Biol. Chem. 1994, 269, 30981–30987. [Google Scholar]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef]
- Roman, A.Y.; Devred, F.; Byrne, D.; La Rocca, R.; Ninkina, N.N.; Peyrot, V.; Tsvetkov, P.O. Zinc Induces Temperature-Dependent Reversible Self-Assembly of Tau. J. Mol. Biol. 2019, 431, 687–695. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation induces self-assembly of τ into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Kopeikina, K.J.; Koffie, R.M.; de Calignon, A.; Hyman, B.T. Are Tangles as Toxic as They Look? J. Mol. Neurosci. 2011, 45, 438–444. [Google Scholar] [CrossRef]
- Ittner, A.; Ke, Y.D.; Van Eersel, J.; Gladbach, A.; Götz, J.; Ittner, L.M. Brief update on different roles of tau in neurodegeneration. IUBMB Life 2011, 63, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hochgräfe, K.; Sydow, A.; Matenia, D.; Cadinu, D.; Könen, S.; Petrova, O.; Pickhardt, M.; Goll, P.; Morellini, F.; Mandelkow, E.; et al. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol. Commun. 2015, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 Activity Is Involved in Inducing Alzheimer’s Disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef]
- Aaseth, J.; Alexander, J.; Bjørklund, G.; Hestad, K.; Dusek, P.; Roos, P.M.; Alehagen, U. Treatment strategies in Alzheimer’s disease: A review with focus on selenium supplementation. BioMetals 2016, 29, 827–839. [Google Scholar] [CrossRef]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.K.T.S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-β (Aβ) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018, 140, 8138–8146. [Google Scholar] [CrossRef]
- Bjørklund, G.; Aaseth, J.; Dadar, M.; Chirumbolo, S. Molecular Targets in Alzheimer ’ s Disease. Mol. Neurobiol. 2019, 56, 7032–7044. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef]
- Dexter, D.T.; Jenner, P. Parkinson disease: From pathology to molecular disease mechanisms. Free Radic. Biol. Med. 2013, 62, 132–144. [Google Scholar] [CrossRef]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. BioMetals 2018, 31, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Kame, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Pearce, R.K.B.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Hofer, T.; Nurchi, V.M.; Aaseth, J. Iron and other metals in the pathogenesis of Parkinson’s disease: Toxic effects and possible detoxification. J. Inorg. Biochem. 2019, 199, 110717. [Google Scholar] [CrossRef]
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfó, E.; Carmona, M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm. 2011, 118, 821–839. [Google Scholar] [CrossRef]
- Wills, J.; Jones, J.; Haggerty, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia. Exp. Neurol. 2010, 225, 210–218. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein: A possible molecular link between parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated τ and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Litvan, I.; Agid, Y.; Calne, D.; Campbell, G.; Dubois, B.; Duvoisin, R.C.; Goetz, C.G.; Golbe, L.I.; Grafman, J.; Growdon, J.H.; et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): Report of the NINDS-SPSP international workshop. Neurology 1996, 47, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Kassubek, J.; Landwehrmeyer, B.G.; Mandelkow, E.; Mandelkow, E.M.; Burn, D.J.; Caparros-Lefebvre, D.; Frey, K.A.; De Yebenes, J.G.; Gasser, T.; et al. Tauopathies with parkinsonism: Clinical spectrum, neuropathologic basis, biological markers, and treatment options. Eur. J. Neurol. 2009, 16, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Caparros-Lefebvre, D.; Golbe, L.I.; Deramecourt, V.; Maurage, C.-A.; Huin, V.; Buée-Scherrer, V.; Obriot, H.; Sablonnière, B.; Caparros, F.; Buée, L.; et al. A geographical cluster of progressive supranuclear palsy in northern France. Neurology 2015, 85, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Aaseth, J.; Alexander, J.; Norseth, T. Uptake of 51Cr-chromate by human erythrocytes-a role of glutathione. Acta Pharmacol. Toxicol. 1982, 50, 310–315. [Google Scholar] [CrossRef]
- Alexander, J.; Aaseth, J.; Norseth, T. Uptake of chromium by rat liver mitochondria. Toxicology 1982, 24, 115–122. [Google Scholar] [CrossRef]
- Ryberg, D.; Alexander, J. Inhibitory action of hexavalent chromium (Cr(VI)) on the mitochondrial respiration and a possible coupling to the reduction of Cr(VI). Biochem. Pharmacol. 1984, 33, 2461–2466. [Google Scholar] [CrossRef]
- Aspli, K.T.; Flaten, T.P.; Roos, P.M.; Holmøy, T.; Skogholt, J.H.; Aaseth, J. Iron and copper in progressive demyelination—New lessons from Skogholt’s disease. J. Trace Elem. Med. Biol. 2015, 31, 183–187. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef]
- Caparros-Lefebvre, D.; Steele, J.; Kotake, Y.; Ohta, S. Geographic isolates of atypical Parkinsonism and tauopathy in the tropics: Possible synergy of neurotoxins. Mov. Disord. 2006, 21, 1769–1771. [Google Scholar] [CrossRef]
- Caparros-Lefebvre, D.; Elbaz, A. Possible relation of atypical parkinsonism in the French West Indies with consumption of tropical plants: A case-control study. Lancet 1999, 354, 281–286. [Google Scholar] [CrossRef]
- Escobar-Khondiker, M.; Höllerhage, M.; Muriel, M.P.; Champy, P.; Bach, A.; Depienne, C.; Respondek, G.; Yamada, E.S.; Lannuzel, A.; Yagi, T.; et al. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. 2007, 27, 7827–7837. [Google Scholar] [CrossRef]
- Caparros-Lefebvre, D.; Sergeant, N.; Lees, A.; Camuzat, A.; Daniel, S.; Lannuzel, A.; Brice, A.; Tolosa, E.; Delacourte, A.; Duyckaerts, C. Guadeloupean parkinsonism: A cluster of progressive supranuclear palsy-like tauopathy. Brain 2002, 125, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Van Swieten, J.; Spillantini, M.G. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007, 17, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tacik, P.; DeTure, M.; Hinkle, K.M.; Lin, W.L.; Sanchez-Contreras, M.; Carlomagno, Y.; Pedraza, O.; Rademakers, R.; Ross, O.A.; Wszolek, Z.K.; et al. A novel Tau Mutation in exon 12, P. Q336H, causes hereditary pick disease. J. Neuropathol. Exp. Neurol. 2015, 74, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Polito, L.; Greco, A.; Seripa, D. Genetic Profile, Environmental Exposure, and Their Interaction in Parkinson’s Disease. Parkinsons. Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.; Goldman, J.; Mander, K.S. Genetics aspects of AD. Neurologist 2009, 15, 80–86. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 2020, 4422, 1–10. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Buée, L.; Delacourte, A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999, 9, 681–693. [Google Scholar] [CrossRef]
- De Yébenes, J.G.; Sarasa, J.L.; Daniel, S.E.; Lees, A.J. Familial progressive supranuclear palsy: Description of a pedigree and review of the literature. Brain 1995, 118, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Litvan, I.; Houlden, H.; Adamson, J.; Dickson, D.; Perez-Tur, J.; Hardy, J.; Lynch, T.; Bigio, E.; Hutton, M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999, 8, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Melquist, S.; Craig, D.W.; Huentelman, M.J.; Crook, R.; Pearson, J.V.; Baker, M.; Zismann, V.L.; Gass, J.; Adamson, J.; Szelinger, S.; et al. Identification of a novel risk locus for progressive supranuclear palsy by a pooled genomewide scan of 500,288 single-nucleotide polymorphisms. Am. J. Hum. Genet. 2007, 80, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Ros, R.; Garre, P.G.; Hirano, M.; Tai, Y.F.; Ampuero, I.; Vidal, L.; Rojo, A.; Fontan, A.; Vazquez, A.; Fanjul, S.; et al. Genetic linkage of autosomal dominant progressive supranuclear palsy to 1q31.1. Ann. Neurol. 2005, 57, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Lannuzel, A.; Michel, P.P.; Caparros-Lefebvre, D.; Abaul, J.; Hocquemiller, R.; Ruberg, M. Toxicity of annonaceae for dopaminergic neurons: Potential role in atypical parkinsonism in Guadeloupe. Mov. Disord. 2002, 17, 84–90. [Google Scholar] [CrossRef]
- Lannuzel, A.; Michel, P.P.; Höglinger, G.U.; Champy, P.; Jousset, A.; Medja, F.; Lombès, A.; Darios, F.; Gleye, C.; Laurens, A.; et al. The mitochondrial complex I inhibitor annonacin is toxic to mesencephalic dopaminergic neurons by impairment of energy metabolism. Neuroscience 2003, 121, 287–296. [Google Scholar] [CrossRef]
- Champy, P.; Höglinger, G.U.; Féger, J.; Gleye, C.; Hocquemiller, R.; Laurens, A.; Guérineau, V.; Laprévote, O.; Medja, F.; Lombès, A.; et al. Annonacin, a lipophilic inhibitor of mitochondrial complex I, induces nigral and striatal neurodegeneration in rats: Possible relevance for atypical parkinsonism in Guadeloupe. J. Neurochem. 2003, 88, 63–69. [Google Scholar] [CrossRef]
- Albers, D.S.; Swerdlow, R.H.; Manfredi, G.; Gajewski, C.; Yang, L.; Parker, W.D.; Beal, M.F. Further evidence for mitochondrial dysfunction in progressive supranuclear palsy. Exp. Neurol. 2001, 168, 196–198. [Google Scholar] [CrossRef]
- Karakaya, T.; Fußer, F.; Prvulovic, D.; Hampel, H. Treatment Options for Tauopathies. Curr. Treat. Options Neurol. 2012, 14, 126–136. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Drewes, G.; Biernat, J.; Gustke, N.; Van Lint, J.; Vandenheede, J.R.; Mandelkow, E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992, 314, 315–321. [Google Scholar] [CrossRef]
- Hernández, F.; Borrell, J.; Guaza, C.; Avila, J.; Lucas, J.J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3β in the brain but do not form tau filaments. J. Neurochem. 2002, 83, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Engel, T.; Goñi-Oliver, P.; Lucas, J.J.; Avila, J.; Hernández, F. Chronic lithium administration to FTDP-17 tau and GSK-3β overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006, 99, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. Neurodegenerative disease treatments by direct TNF reduction, SB623 cells, maraviroc and irisin and MCC950, from an inflammatory perspective–a Commentary. Expert Rev. Neurother. 2019, 19, 535–543. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczyłowska, B.; Kozłowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFα, and INFγ concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, S.M.; Hernan, M.A.; Schwarzschild, M.A.; Willett, W.C.; Colditz, G.A.; Speizer, F.E.; Ascherio, A. Nonsteroidal anti-inflammatory drugs and the risk of Parkinson disease. Arch. Neurol. 2003, 60, 1059–1064. [Google Scholar] [CrossRef]
- Gagne, J.J.; Power, M.C. Anti-inflammatory drugs and risk of Parkinson disease. Neurology 2010, 74, 995–1002. [Google Scholar] [CrossRef]
- Pickhardt, M.; Gazova, Z.; Von Bergen, M.; Khlistunova, I.; Wang, Y.; Hascher, A.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer’s paired helical filaments in vitro and in cells. J. Biol. Chem. 2005, 280, 3628–3635. [Google Scholar] [CrossRef]
- Freyssin, A.; Page, G.; Fauconneau, B.; Rioux Bilan, A. Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen. Res. 2018, 13, 955–961. [Google Scholar] [CrossRef]
- Baell, J.B.; Nissink, J.W.M. Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017—Utility and Limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling Nature’s PAINS: Natural Products, Natural Product Drugs, and Pan Assay Interference Compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Kebede, M.T.; Kemeh, M.M.; Islam, S.; Lee, B.; Bleck, S.D.; Wurfl, L.A.; Lazo, N.D. Inhibition of the self-assembly of Aβ and of tau by polyphenols: Mechanistic studies. Molecules 2019, 24, 2316. [Google Scholar] [CrossRef] [PubMed]
- Götz, J.; Deters, N.; Doldissen, A.; Bokhari, L.; Ke, Y.; Wiesner, A.; Schonrock, N.; Ittner, L.M. A decade of tau transgenic animal models and beyond. Brain Pathol. 2007, 17, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A walk through tau therapeutic strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef]
- Medina, M. An overview on the clinical development of tau-based therapeutics. Int. J. Mol. Sci. 2018, 19, 1160. [Google Scholar] [CrossRef]


{kind=link}
| Disease | Location | Management |
|---|---|---|
| Alzheimer’s disease | Hippocampus and entorhinal cortex | Cholinesterase inhibitors, memantine |
| Parkinson’s disease | Substantia nigra in the basal ganglia | Levodopa |
| Progressive supranuclear palsy | Basal ganglia and brain stem/spinal cord | Levodopa (in some cases only) |
| Frontotemporal dementia | Frontal and temporal lobes | Antipsychotics and antidepressants |
| Chronic traumatic encephalopathy | Sulcus depths | No approved therapy |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aaseth, J.; Buha, A.; Wallace, D.R.; Bjørklund, G. Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies. Int. J. Environ. Res. Public Health 2020, 17, 1269. https://doi.org/10.3390/ijerph17041269
Aaseth J, Buha A, Wallace DR, Bjørklund G. Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies. International Journal of Environmental Research and Public Health. 2020; 17(4):1269. https://doi.org/10.3390/ijerph17041269
Chicago/Turabian StyleAaseth, Jan, Aleksandra Buha, David R. Wallace, and Geir Bjørklund. 2020. "Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies" International Journal of Environmental Research and Public Health 17, no. 4: 1269. https://doi.org/10.3390/ijerph17041269
APA StyleAaseth, J., Buha, A., Wallace, D. R., & Bjørklund, G. (2020). Xenobiotics, Trace Metals and Genetics in the Pathogenesis of Tauopathies. International Journal of Environmental Research and Public Health, 17(4), 1269. https://doi.org/10.3390/ijerph17041269