Managing Cystic Fibrosis in Polish Healthcare





Abstract
1. Introduction
2. Materials and Methods
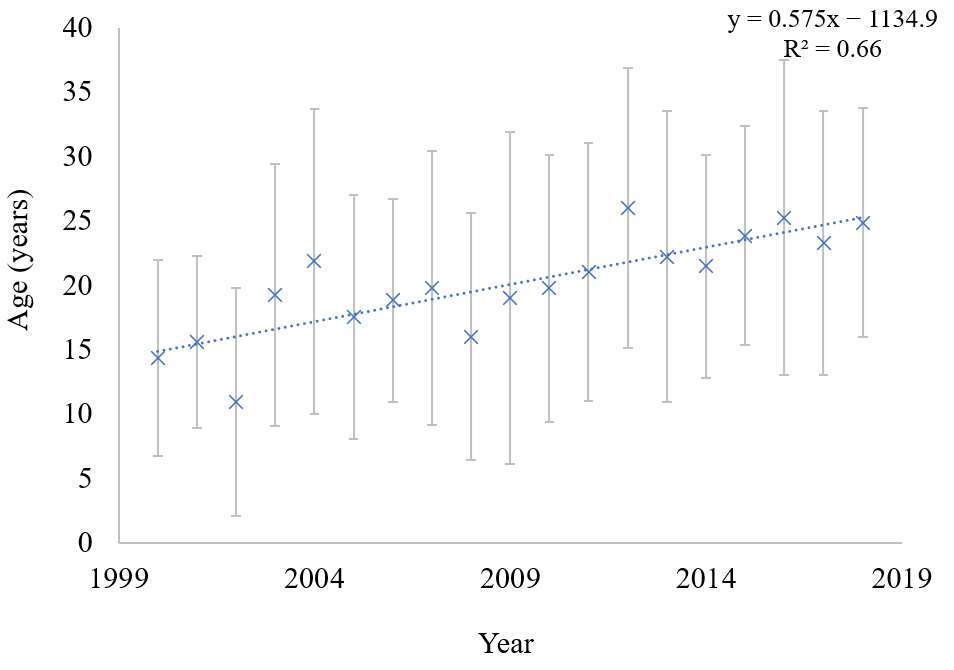
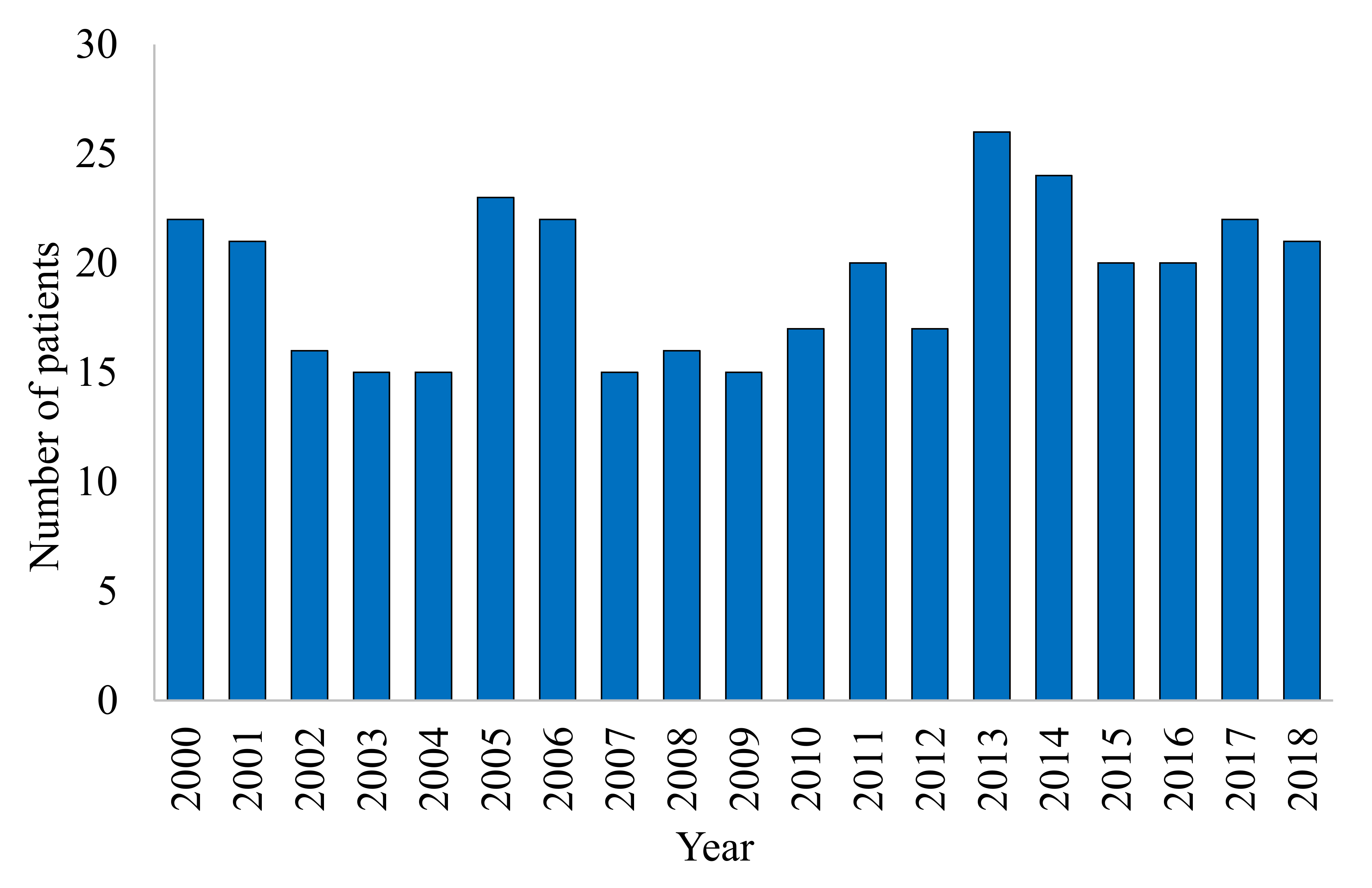
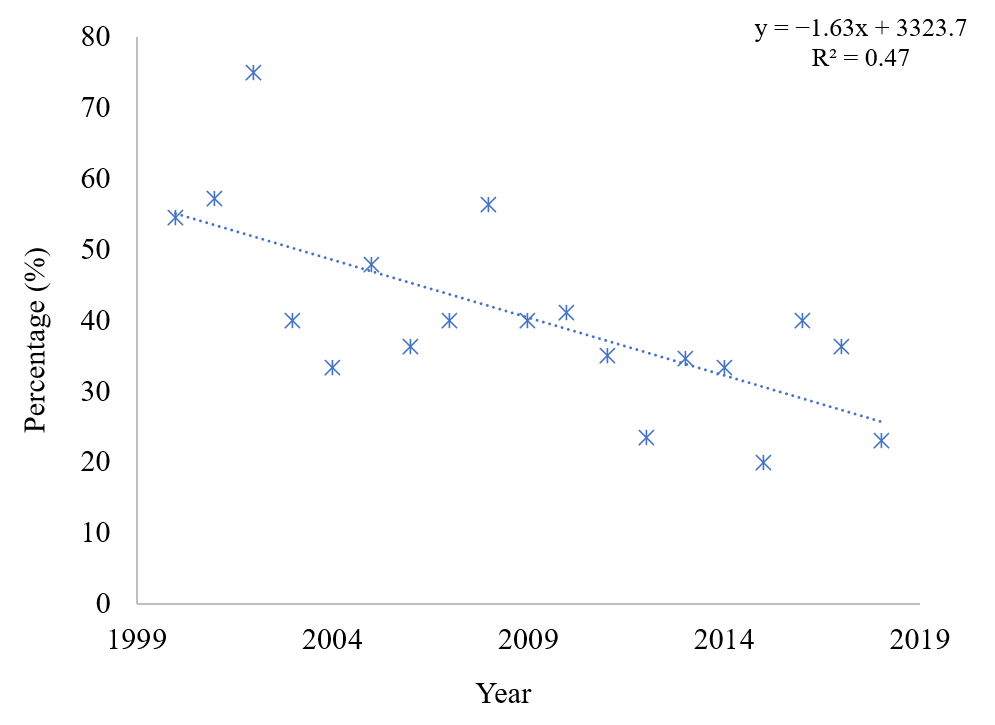
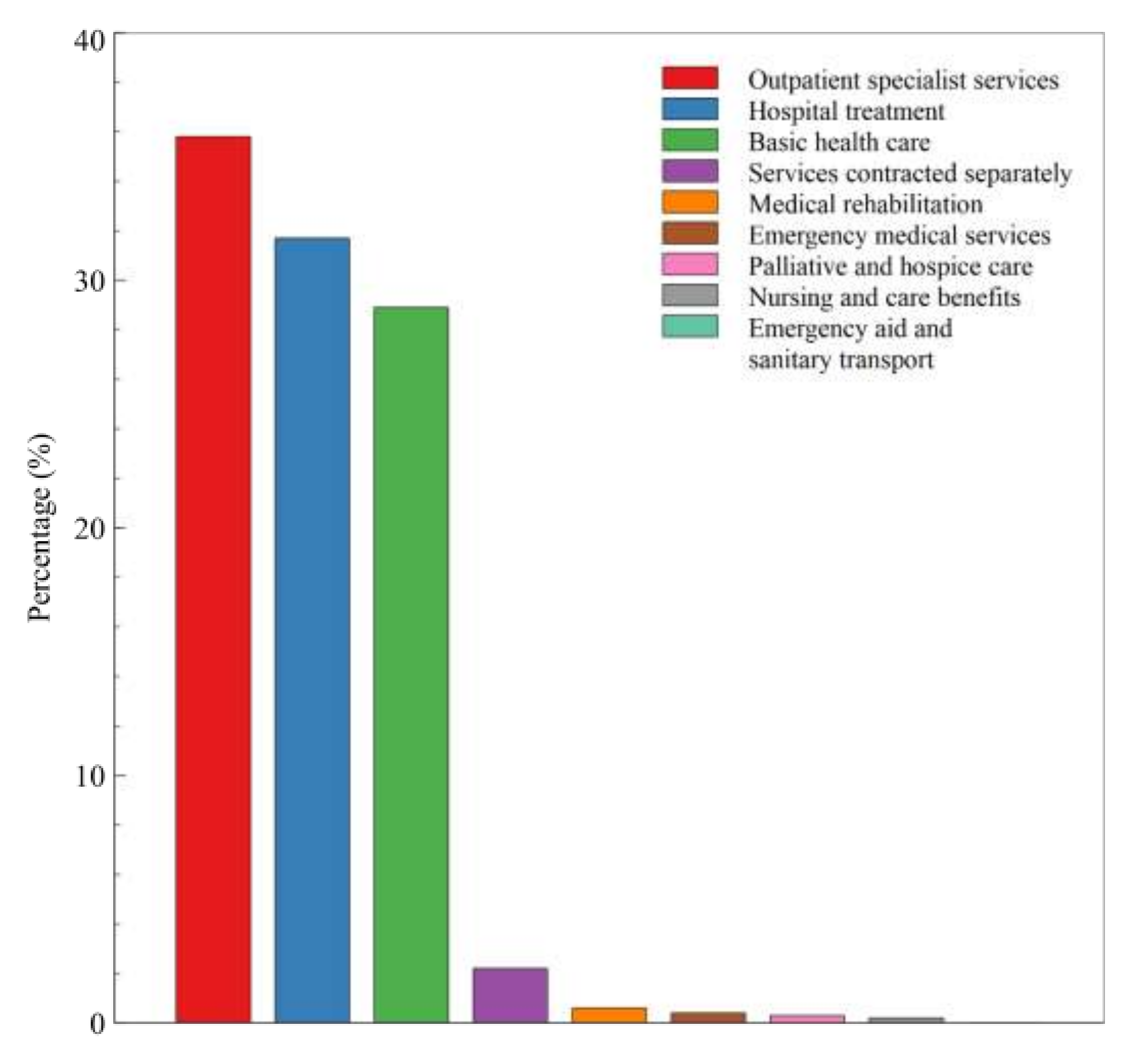
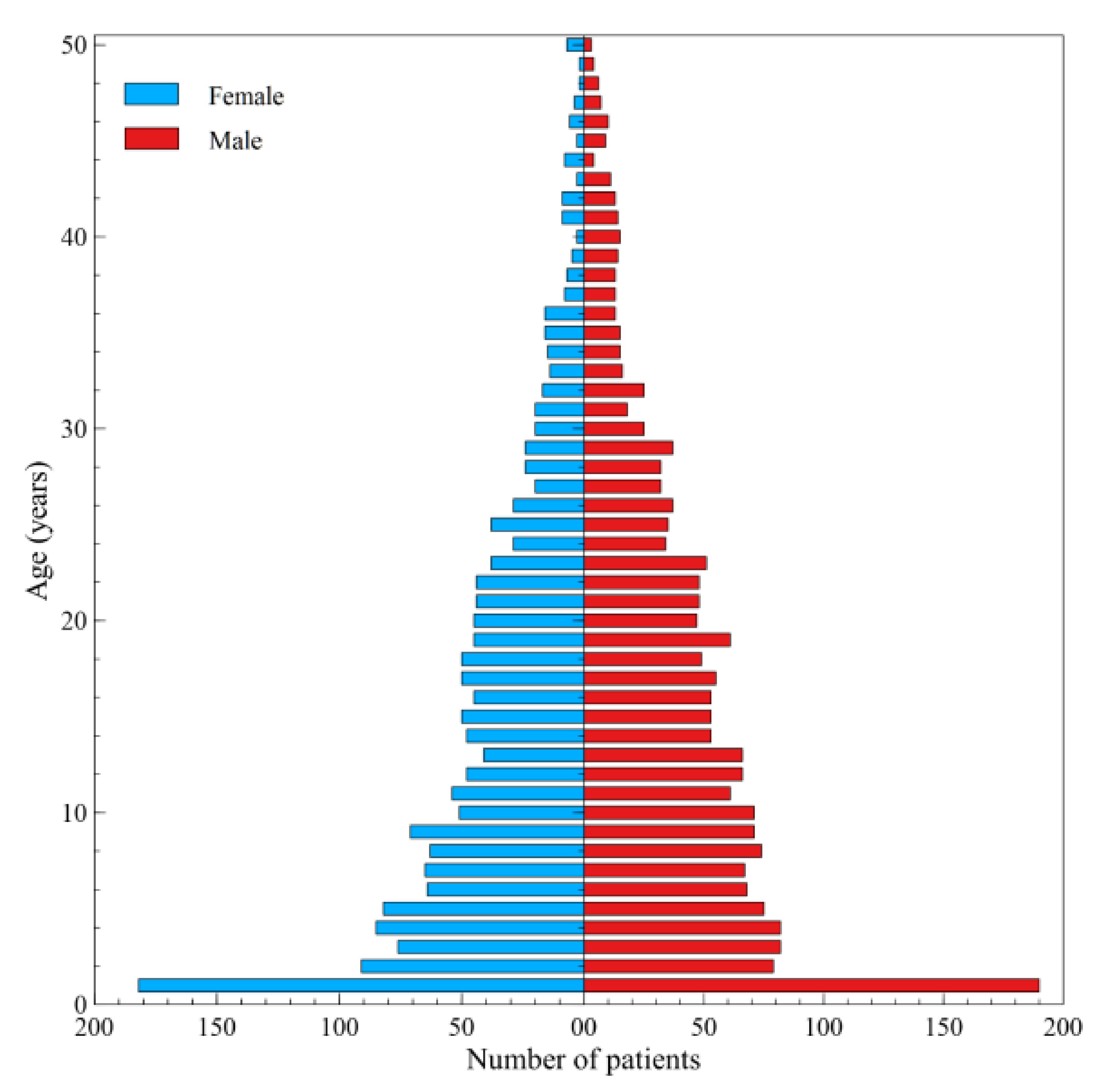
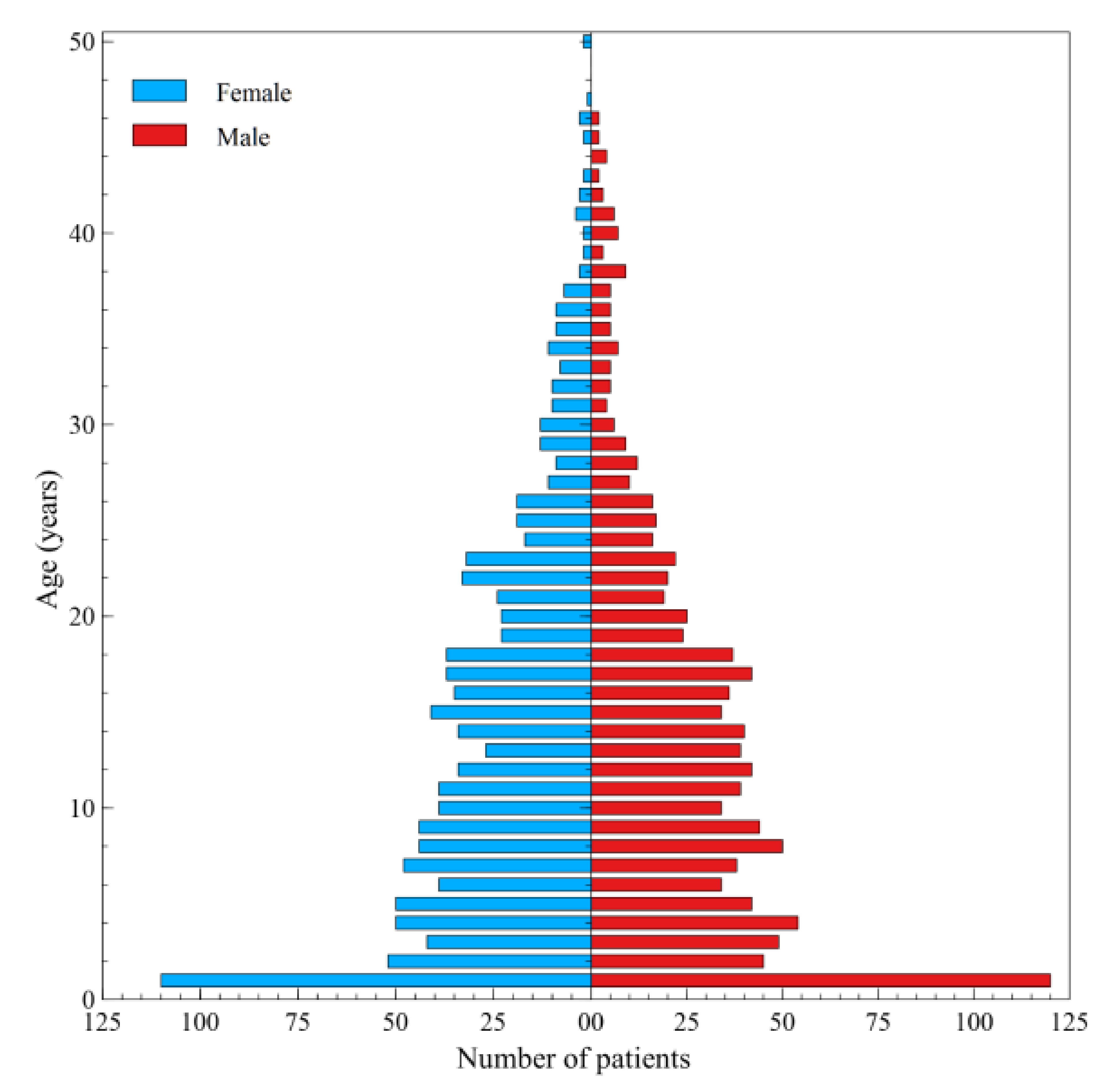
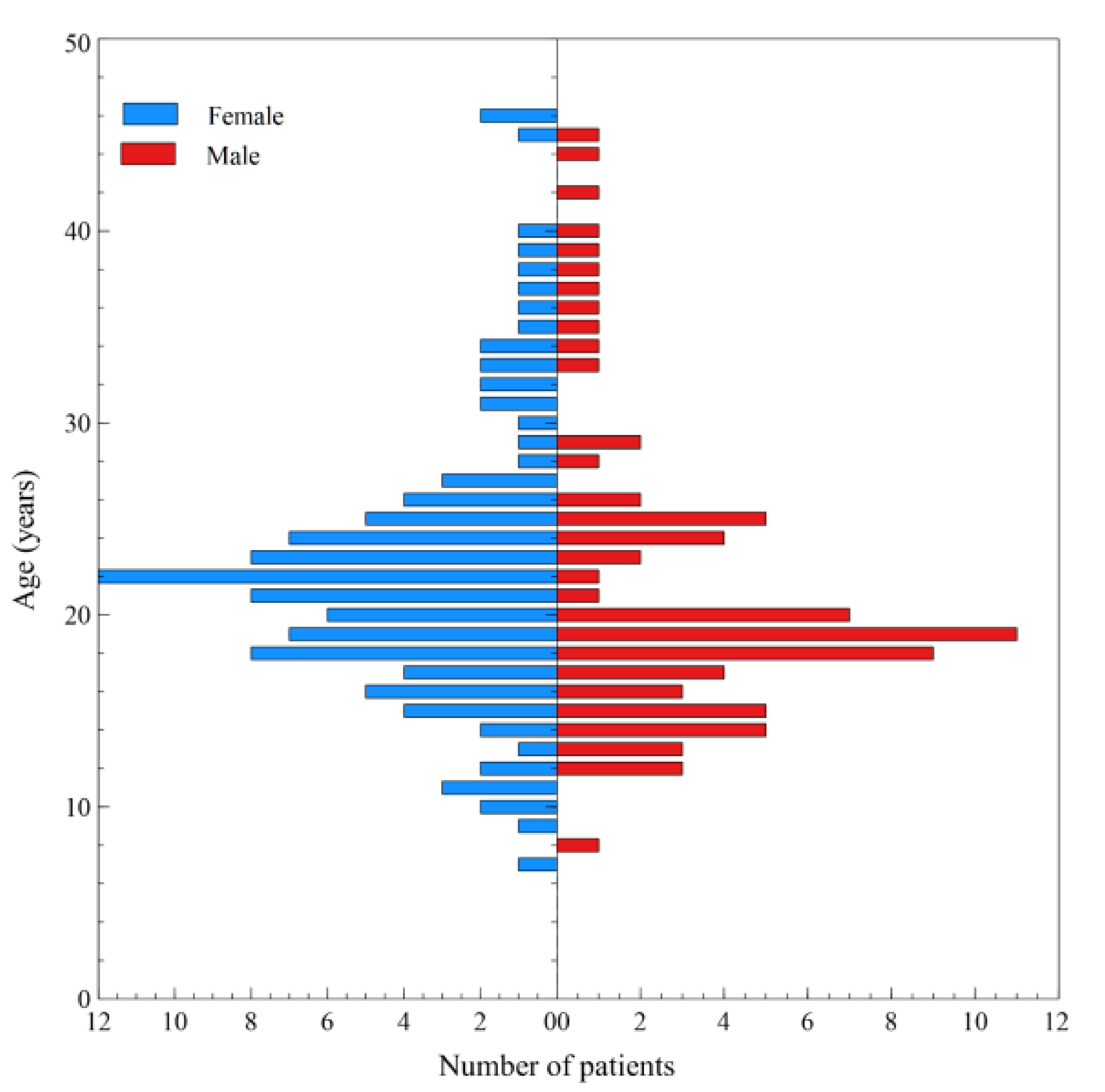
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sands, D.; Pogorzelski, A.; Skoczylas-Ligocka, A. Epidemiologia i Organizacja Opieki Medycznej Nad Chorymi na Mukowiscydozę w Polsce. In Mukowiscydoza Choroba Wieloukładowa; Sands, D., Ed.; Termedia: Poznań, Poland, 2018; pp. 15–24. [Google Scholar]
- Farrel, P.M. Case report: The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 2008, 7, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Rebordosa, C.; Bernholz, J.; Sharma, N. Epidemiology and genetics of cystic fibrosis in Asia: In preparation for the next-genaration treatments. Respirology 2015, 20, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Mirtajani, S.B.; Farnia, P.; Hassanzad, M.; Ghanavi, J.; Velayati, A.A. Geographical distribution of cystic fibrosis; The past 70 Years of data analysis. Biomed. Biotechnol. Res. J. 2017, 1, 105–112. [Google Scholar]
- Bodnar, R.; Kadar, L.; Holics, K.; Ujhelyi, R.; Kovacs, L.; Bolbas, K.; Szekely, G.; Gyurkovits, K.; Solyom, E.; Meszaros, A. Factors influencing quality of life and disease severity in Hungarian children and young adults with cystic fibrosis. Ital. J. Pediatr. 2014, 40, 50. [Google Scholar] [CrossRef]
- McColley, S.A.; Schechter, M.S.; Morgan, W.J.; Pasta, D.J.; Craib, M.L.; Konstan, M.W. Risk factors for mortality before age 18 years in cystic fibrosis. Pediatr. Pulmonol. 2017, 52, 909–915. [Google Scholar] [CrossRef]
- Barr, H.L.; Britton, J.; Smyth, A.R.; Fogarty, A.W. Association between socioeconomic status, sex, and age at death from cystic fibrosis in England and Wales (1959 to 2008): Cross sectional study. BMJ 2011, 343, d4662. [Google Scholar] [CrossRef]
- Villaverde-Hueso, A.; Sánchez-Díaz, G.; Molina-Cabrero, F.J.; Gallego, E.; Posada de la Paz, M.; Alonso-Ferreira, V. Mortality due to cystic fibrosis over a 36-year period in Spain: Time trends and geographic variations. Int. J. Environ. Res. Public Health 2019, 16, 119. [Google Scholar] [CrossRef]
- Quintana-Gallego, E.; Ruiz-Ramos, M.; Delgado-Pecellin, I.; Calero, C.; Soriano, J.B.; Lopez-Campos, J.L. Mortality from cystic fibrosis in Europe: 1994–2010. Pediatr. Pulmonol. 2016, 51, 133–142. [Google Scholar] [CrossRef]
- Elborn, J.S.; Bell, S.C.; Madge, S.L.; Burgel, P.R.; Castellani, C.; Conway, S.; De Rijcke, K.; Dembski, B.; Drevinek, P.; Heijerman, H.G.; et al. Report of the European Respiratory Society/European Cystic Fibrosis Society task force on the care of adults with cystic fibrosis. Eur. Respir. J. 2016, 47, 420–428. [Google Scholar] [CrossRef]
- Schechter, M.S.; Fink, A.K.; Homa, K.; Goss, C.H. The Cystic Fibrosis Foundation Patient Registry as a tool for use in quality improvement. BMJ Qual. Saf. 2014, 23, i9–i14. [Google Scholar] [CrossRef]
- Ahern, S.; Sims, G.; Earnest, A.C.; Bell, S. Optimism, opportunities, outcomes: The Australian Cystic Fibrosis Data Registry. Intern. Med. J. 2018, 48, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E. Managing cystic fibrosis: Strategies that increase life expectancy and improve quality of life. Am. J. Respir. Crit. Care Med. 2011, 183, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Scotet, V.; L’Hostis, C.; Férec, C. The changing epidemiology of cystic fibrosis: Incidence: Survival and impact of the CFTR gene discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic fibrosis: Emergence of highly effective targeted therapeutics and potential clinical implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef]
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Schwarzenberg, S.J.; et al. European Cystic Fibrosis Society Standards of Care: Best practice guidelines. J. Cyst. Fibros. 2014, 13, S23–S42. [Google Scholar] [CrossRef]
- Stern, M.; Bertrand, D.P.; Bignamini, E.; Corey, M.; Dembski, B.; Goss, C.H.; Pressler, T.; Rault, G.; Viviani, L.; Elborn, J.S.; et al. European Cystic Fibrosis Society Standards of Care: Quality management in cystic fibrosis. J. Cyst. Fibros. 2014, 13, S43–S59. [Google Scholar] [CrossRef]
- Walicka-Serzysko, K.; Peckova, M.; Noordhoek, J.J.; Sands, D.; Drevinek, P. Insights into the cystic fibrosis care in Eastern Europe: Results of survey. J. Cyst. Fibros. 2018, 17, 475–477. [Google Scholar] [CrossRef]
- Madge, S.; Bell, S.C.; Burgel, P.R.; De Rijcke, K.; Blasi, F.; Elborn, J.S. ERS/ECFS task force: The provision of care for adults with cystic fibrosis in Europe. Limitations to providing adult cystic fibrosis care in Europe: Results of a care centre survey. J. Cyst. Fibros. 2017, 16, 85–88. [Google Scholar] [CrossRef]
- Sands, D. Opieka Nad Chorymi Na Mukowiscydozę W Polsce. Stan Obecny I Rekomendacje Poprawy. Raport. Warszawa-Kraków, Poland. 2019. Available online: http://www.mukowiscydoza.pl/images/aktual/Raport_Opieka_nad_chorymi_na_mukowiscydoz%C4%99_w_Polsce.pdf (accessed on 20 June 2020).
- Polskie Towarzystwo Mukowiscydozy; Polskie Towarzystwo Walki z Mukowiscydozą. Polski Audyt Ośrodków Leczenia Mukowiscydozy—Opracowanie Własne; Polskie Towarzystwo Mukowiscydozy: Łomianki, Poland; Polskie Towarzystwo Walki z Mukowiscydozą: Rabka-Zdrój, Poland, 2016. [Google Scholar]
- Mehta, G.; Macek, M., Jr.; Mehta, A.; European Registry Working Group. Cystic fibrosis across Europe: EuroCareCF analysis of demographic data from 35 countries. J. Cyst. Fibros 2010, 9, S5–S21. [Google Scholar] [CrossRef]
- Zolin, A.; Orenti, A.; Naehrlich, L.; van Rens, J. ECFSPR Annual Report 2017; European Cystic Fibrosis Society: Karup, Demnark, 2017; Available online: https://www.ecfs.eu/sites/default/files/general-content-images/working-groups/ecfs-patient-registry/ECFSPR_Report2017_v1.3.pdf (accessed on 23 June 2020).
- Giordani, B.; Amato, A.; Majo, F.; Ferrari, G.; Quattrucci, S.; Minicucci, L.; Padoan, R.; Floridia, G.; Salvatore, D.; Carnovale, V.; et al. Registro italiano Fibrosi Cistica (RIFC). Rapporto 2015–2016 [Italian Cystic Fibrosis Registry (ICFR). Report 2015–2016]. Epidemiol. Prev. 2019, 43, 1–36. [Google Scholar]
- Stephenson, A.L.; Sykes, J.; Stanojevic, S.; Quon, B.S.; Marshall, B.C.; Petren, K.; Ostrenga, J.; Fink, A.K.; Elbert, A.; Goss, C.H. Survival comparison of patients with cystic fibrosis in Canada and the United States: A population-based cohort study. Ann. Intern. Med. 2017, 166, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Frischer, T.; Eber, E.; Ellemunter, H.; Zacharasiewicz, A.; Kaluza, I.; Riedler, J.; Renner, S. Cystic fibrosis in Austria. Wien. Klin. Wochenschr. 2017, 129, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Irish Cystic Fibrosis Registry. Annual Data Report. 2018. Available online: https://cfri.ie/annual-reports/ (accessed on 16 September 2020).
- UK Cystic Fibrosis Registry (Cystic Fibrosis Trust). Annual Data Report. 2018. Available online: https://www.cysticfibrosis.org.uk/~{}/media/documents/the-work-we-do/uk-cf-registry/2018-registry-annual-data-report.ashx?la=en (accessed on 16 September 2020).
- US Cystic Fibrosis Registry (Cystic Fibrosis Foundation). Annual Data Report. 2018. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2018-Patient-Registry-Annual-Data-Report.pdf (accessed on 16 September 2020).
- Canadian Cystic Fibrosis Registry. Annual Data Report. 2018. Available online: https://www.cysticfibrosis.ca/uploads/RegistryReport2018/2018RegistryAnnualDataReport.pdf (accessed on 16 September 2020).
- Keogh, R.H.; Szczesniak, R.; Taylor-Robinson, D.; Bilton, D. Up-to-date and projected estimates of survival for people with cystic fibrosis using baseline characteristics: A longitudinal study using UK patient registry data. J. Cyst. Fibros. 2018, 17, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Hollmeyer, H.; Schreyögg, J.; Wahn, U.; Staab, D. Staff costs of hospital-based outpatient care of patients with cystic fibrosis. Health Econ. Rev. 2011, 1, 10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Horvais, V.; Touzet, S.; François, S.; Bourdy, S.; Bellon, G.; Colin, C.; Durieu, I. Cost of home and hospital care for patients with cystic fibrosis followed up in two reference medical centers in France. Int. J. Technol. Assess. Health Care 2006, 22, 525–531. [Google Scholar] [CrossRef]
- Grosse, S.D.; Do, T.Q.N.; Vu, M.; Feng, L.B.; Berry, J.G.; Sawicki, G.S. Healthcare expenditures for privately insured US patients with cystic fibrosis, 2010–2016. Pediatr. Pulmonol. 2018, 53, 1611–1618. [Google Scholar] [CrossRef]
- van Gool, K.; Norman, R.; Delatycki, M.B.; Hall, J.; Massie, J. Understanding the costs of care for cystic fibrosis: An analysis by age and health state. Value Health 2013, 16, 345–355. [Google Scholar] [CrossRef]
- Zolin, A.; Bossi, A.; Cirilli, N.; Kashirskaya, N.; Padoan, R. Cystic fibrosis mortality in childhood. Data from European Cystic Fibrosis Society Patient Registry. Int. J. Environ. Res. Public Health 2018, 15, 2020. [Google Scholar] [CrossRef]
- Kerem, E.; Conway, S.; Elborn, S.; Heijerman, H.; Consensus Committee. Standards of care for patients with cystic fibrosis: A European consensus. J. Cyst. Fibros. 2005, 4, 7–26. [Google Scholar] [CrossRef]
- Colombo, C.; Littlewood, J. The implementation of standards of care in Europe: State of the art. J. Cyst. Fibros. 2011, 10, S7–S15. [Google Scholar] [CrossRef]
- Post, P.N.; Wittenberg, J.; Burgers, J.S. Do specialized centers and specialists produce better outcomes for patients with chronic diseases than primary care generalists? A systematic review. Int. J. Qual. Health Care 2009, 21, 387–396. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burnett, D.M.; Barry, A.N.; Mermis, J.D. Physical activity level and perception of exercise in cystic fibrosis. Respir. Care 2020, 65, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.E.; Button, B.M.; International Physiotherapy Group for Cystic Fibrosis, Australian Chapter. Physiotherapy for cystic fibrosis in Australia: Knowledge and acceptance of the Consensus Statement recommendations. Respirology 2013, 18, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Donadio, M.V.F.; Campos, N.E.; Vendrusculo, F.M.; Stofella, A.M.; Almeida, A.C.D.S.; Ziegler, B.; Schivinski, C.I.S.; Santuzzi, C.H.; Sarges, E.D.S.N.F.; Gonçalves, F.M.; et al. Respiratory physical therapy techniques recommended for patients with cystic fibrosis treated in specialized centers. Braz. J. Phys. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Rowbotham, N.J.; Regan, K.H. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 3, CD001021. [Google Scholar] [CrossRef]
- McIlwaine, M.; Button, B.; Nevitt, S.J. Positive expiratory pressure physiotherapy for airway clearance in people with cystic fibrosis. Cochrane Database Syst. Rev. 2019, 2019, CD003147. [Google Scholar] [CrossRef]
- Shteinberg, M.; Elborn, J.S. Use of inhaled tobramycin in cystic fibrosis. Adv. Ther. 2015, 32, 1–9. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthcare Services | |
|---|---|
| 01 | Basic health care |
| 02 | Ambulatory specialist benefits |
| 03 | Hospital treatment |
| 04 | Psychiatric care and addictive treatment |
| 05 | Medical rehabilitation |
| 06 | Long-term care |
| 07 | Dental treatment |
| 09 | Advisory assistance and sanitary transport |
| 10 | Preventive health programs |
| 11 | Independent contracting benefits |
| 12 | Care and care |
| 13 | Palliative and hospitable care |
| 14 | Medical rescue |
| 15 | Advisory assistance and sanitary transport |
| Voivodeship | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dolnośląskie | 180 | 173 | 176 | 170 | 172 | 190 | 183 | 201 | 208 | 227 | 190 |
| Kujawsko-Pomorskie | 131 | 153 | 123 | 129 | 126 | 118 | 133 | 137 | 157 | 151 | 117 |
| Lubelskie | 82 | 92 | 100 | 91 | 93 | 96 | 97 | 94 | 80 | 90 | 47 |
| Lubuskie | 60 | 85 | 70 | 58 | 53 | 53 | 54 | 46 | 51 | 55 | 42 |
| Łódzkie | 164 | 177 | 220 | 268 | 303 | 343 | 303 | 221 | 197 | 173 | 163 |
| Małopolskie | 359 | 433 | 483 | 466 | 486 | 533 | 523 | 526 | 572 | 577 | 590 |
| Mazowieckie | 617 | 675 | 673 | 666 | 660 | 667 | 633 | 624 | 590 | 740 | 710 |
| Opolskie | 74 | 68 | 68 | 58 | 63 | 57 | 61 | 60 | 60 | 52 | 50 |
| Podkarpackie | 85 | 103 | 105 | 129 | 138 | 126 | 107 | 109 | 116 | 113 | 92 |
| Podlaskie | 79 | 92 | 83 | 97 | 98 | 109 | 92 | 78 | 86 | 70 | 57 |
| Pomorskie | 617 | 738 | 675 | 628 | 523 | 553 | 251 | 234 | 234 | 226 | 211 |
| Śląskie | 263 | 373 | 383 | 384 | 355 | 397 | 377 | 366 | 363 | 345 | 325 |
| Świętokrzyskie | 49 | 69 | 65 | 60 | 62 | 71 | 73 | 65 | 69 | 66 | 50 |
| Warmińsko-Mazurskie | 46 | 50 | 42 | 44 | 58 | 51 | 48 | 56 | 56 | 53 | 41 |
| Wielkopolskie | 239 | 298 | 271 | 287 | 307 | 323 | 335 | 331 | 356 | 363 | 320 |
| Zachodniopomorskie | 70 | 71 | 84 | 102 | 102 | 102 | 100 | 108 | 102 | 121 | 76 |
| TOTAL | 3115 | 3650 | 3621 | 3637 | 3599 | 3789 | 3370 | 3256 | 3297 | 3422 | 3081 |
| Voivodeship | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dolnośląskie | 55 | 61 | 55 | 77 | 98 | 97 | 108 | 116 | 139 | 139 | 123 |
| Kujawsko-Pomorskie | 58 | 38 | 39 | 28 | 38 | 33 | 35 | 35 | 44 | 48 | 37 |
| Lubelskie | 24 | 30 | 22 | 25 | 25 | 23 | 22 | 28 | 22 | 20 | 21 |
| Lubuskie | 4 | 5 | 5 | 8 | 4 | 5 | 5 | 7 | 6 | 8 | 6 |
| Łódzkie | 40 | 36 | 43 | 34 | 47 | 43 | 60 | 52 | 37 | 47 | 33 |
| Małopolskie | 313 | 330 | 393 | 369 | 413 | 468 | 454 | 456 | 487 | 494 | 510 |
| Mazowieckie | 306 | 295 | 415 | 406 | 357 | 400 | 384 | 389 | 311 | 503 | 690 |
| Opolskie | 12 | 6 | 2 | 7 | 3 | 5 | 4 | 6 | 5 | 8 | 4 |
| Podkarpackie | 26 | 27 | 18 | 23 | 32 | 27 | 17 | 34 | 32 | 40 | 51 |
| Podlaskie | 23 | 22 | 19 | 22 | 16 | 19 | 18 | 22 | 26 | 31 | 20 |
| Pomorskie | 108 | 124 | 121 | 114 | 108 | 120 | 125 | 116 | 87 | 95 | 82 |
| Śląskie | 56 | 56 | 53 | 50 | 62 | 46 | 46 | 47 | 66 | 83 | 74 |
| Świętokrzyskie | 7 | 18 | 15 | 15 | 18 | 21 | 21 | 18 | 21 | 22 | 19 |
| Warmińsko-Mazurskie | 6 | 9 | 4 | 4 | 12 | 8 | 8 | 7 | 6 | 4 | 4 |
| Wielkopolskie | 145 | 173 | 197 | 204 | 215 | 205 | 223 | 221 | 238 | 234 | 210 |
| Zachodniopomorskie | 28 | 31 | 25 | 37 | 31 | 35 | 26 | 24 | 25 | 37 | 24 |
| TOTAL | 1211 | 1261 | 1426 | 1423 | 1479 | 1555 | 1556 | 1578 | 1552 | 1813 | 1908 |
| Voivodeship | 2008 | 2009 | 2010 | 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | 2018 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dolnośląskie | n/a | 7 | 12 | 10 | 13 | 17 | 16 | 16 | 18 | 17 | n/a |
| Kujawsko-Pomorskie | n/a | n/a | 4 | 4 | 4 | 4 | 4 | 4 | 5 | 5 | n/a |
| Lubelskie | n/a | n/a | 1 | 1 | 3 | 1 | 1 | 7 | 9 | 6 | n/a |
| Lubuskie | n/a | 4 | 5 | 9 | 9 | 7 | 6 | 7 | 8 | 0 | n/a |
| Łódzkie | n/a | 14 | 17 | 10 | 7 | 7 | 9 | 10 | 12 | 8 | n/a |
| Małopolskie | n/a | 5 | 7 | 6 | 8 | 5 | 9 | 19 | 22 | 15 | n/a |
| Mazowieckie | n/a | 3 | 3 | 4 | 3 | 6 | 3 | 2 | 1 | 22 | n/a |
| Opolskie | n/a | 3 | 2 | 1 | 4 | 4 | 4 | 6 | 7 | 0 | n/a |
| Podkarpackie | n/a | 5 | 5 | 4 | 5 | 7 | 7 | 5 | 5 | 0 | n/a |
| Podlaskie | n/a | 4 | 5 | 5 | 5 | 7 | 8 | 8 | 8 | 9 | n/a |
| Pomorskie | n/a | n/a | 4 | 5 | 6 | 7 | 7 | 8 | 7 | 7 | n/a |
| Śląskie | n/a | n/a | 1 | 2 | 6 | 9 | 12 | 10 | 9 | 8 | n/a |
| Świętokrzyskie | n/a | n/a | n/a | n/a | n/a | n/a | 1 | 1 | 1 | 5 | n/a |
| Warmińsko-Mazurskie | n/a | 7 | 12 | 10 | 13 | 17 | 16 | 16 | 18 | n/a | n/a |
| Wielkopolskie | n/a | n/a | 4 | 4 | 4 | 4 | 4 | 4 | 5 | 12 | n/a |
| Zachodniopomorskie | n/a | n/a | 1 | 1 | 3 | 1 | 1 | 7 | 9 | 1 | n/a |
| TOTAL | n/a | 52 | 83 | 76 | 93 | 103 | 108 | 130 | 144 | 115 | n/a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachel, M.; Topolewicz, S.; Śliwczyński, A.; Galiniak, S. Managing Cystic Fibrosis in Polish Healthcare. Int. J. Environ. Res. Public Health 2020, 17, 7630. https://doi.org/10.3390/ijerph17207630
Rachel M, Topolewicz S, Śliwczyński A, Galiniak S. Managing Cystic Fibrosis in Polish Healthcare. International Journal of Environmental Research and Public Health. 2020; 17(20):7630. https://doi.org/10.3390/ijerph17207630
Chicago/Turabian StyleRachel, Marta, Stanisław Topolewicz, Andrzej Śliwczyński, and Sabina Galiniak. 2020. "Managing Cystic Fibrosis in Polish Healthcare" International Journal of Environmental Research and Public Health 17, no. 20: 7630. https://doi.org/10.3390/ijerph17207630
APA StyleRachel, M., Topolewicz, S., Śliwczyński, A., & Galiniak, S. (2020). Managing Cystic Fibrosis in Polish Healthcare. International Journal of Environmental Research and Public Health, 17(20), 7630. https://doi.org/10.3390/ijerph17207630