The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues

Abstract
1. Introduction
2. Materials and Methods
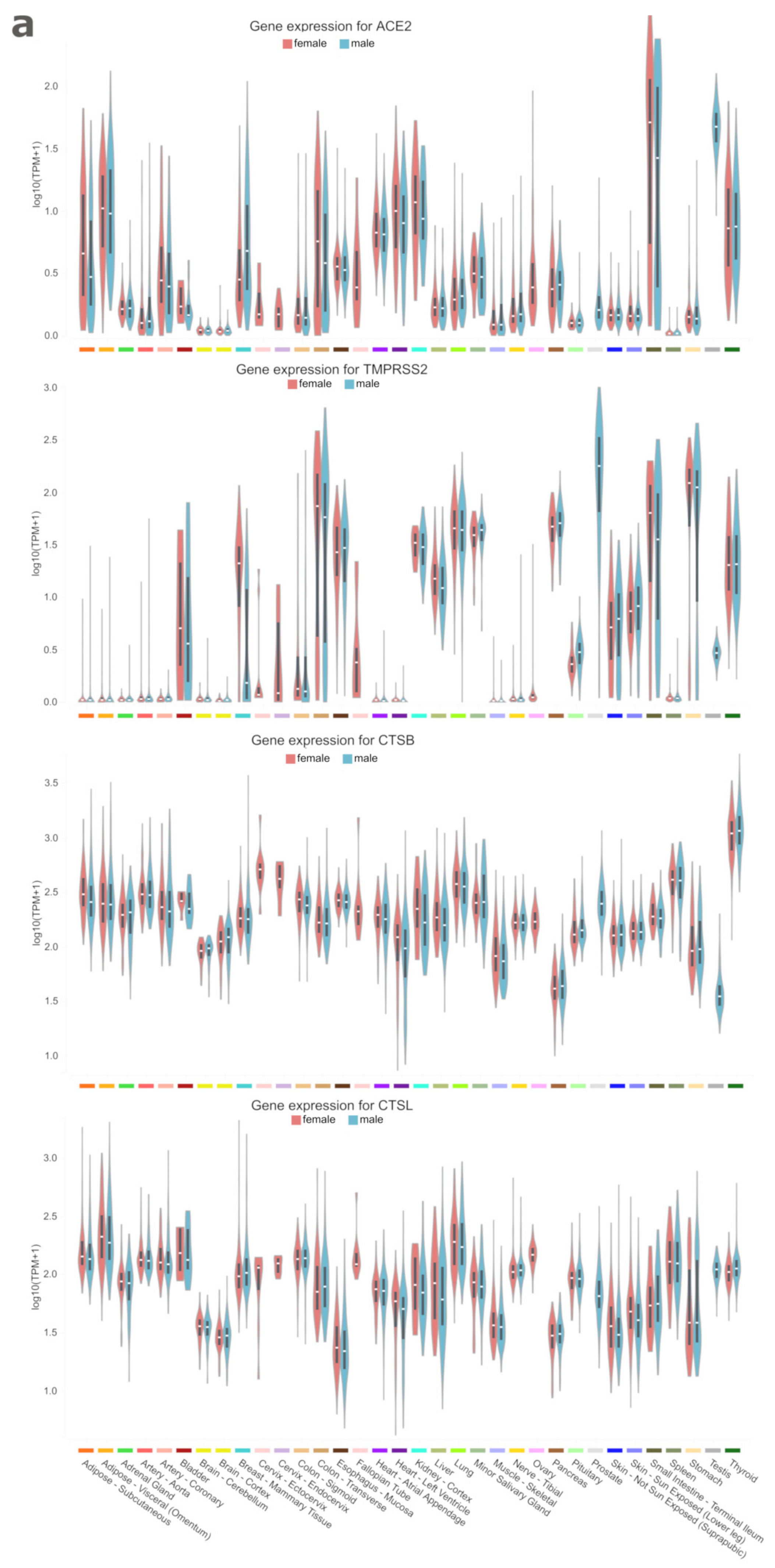
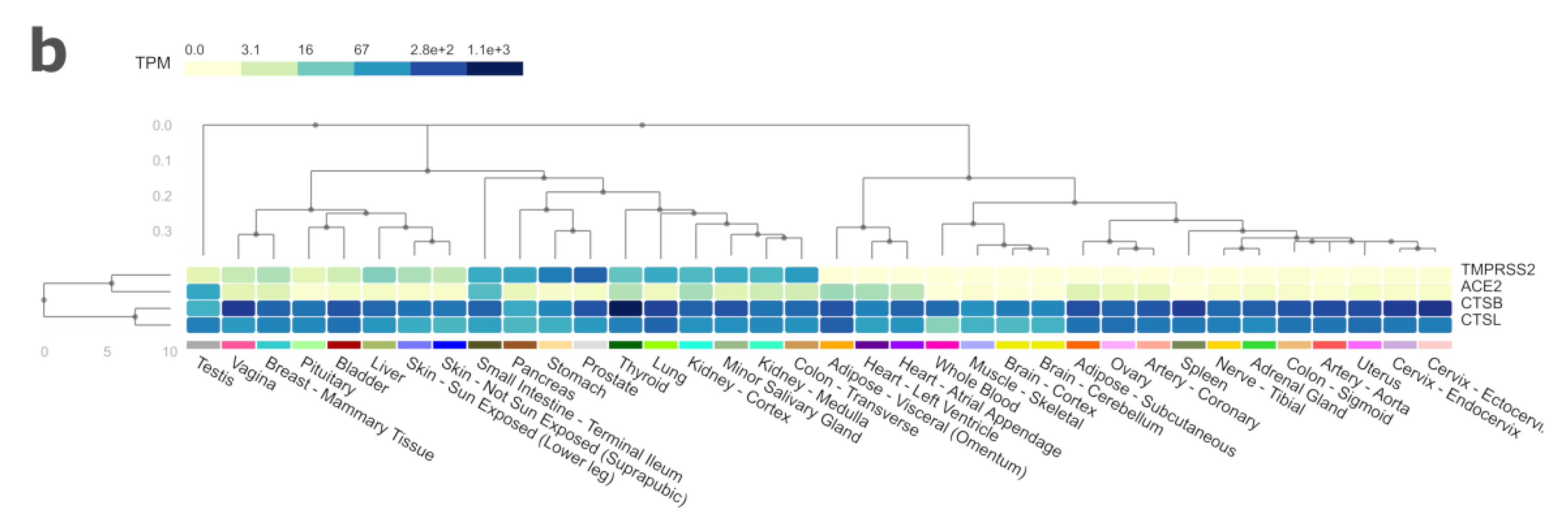
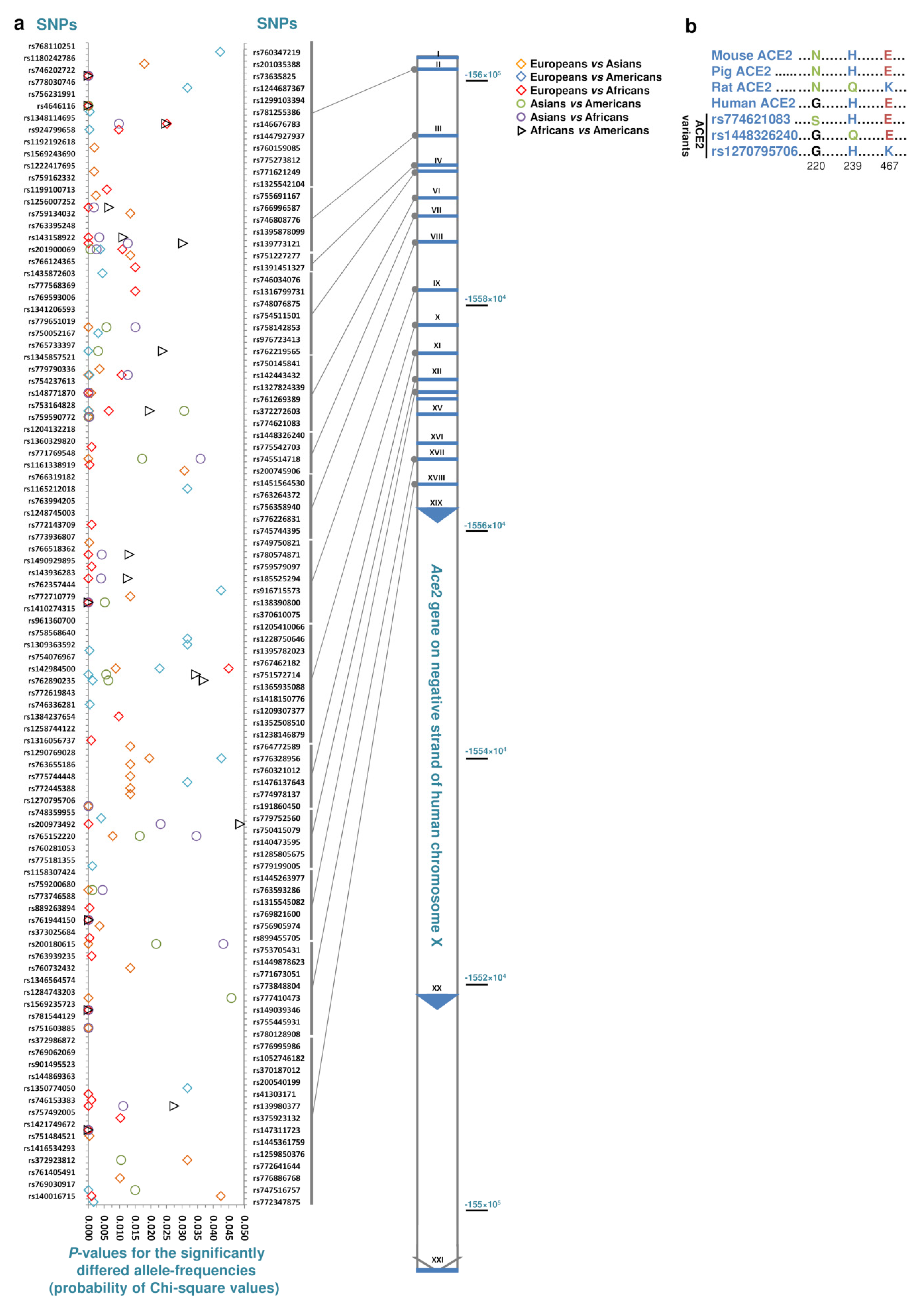
3. Results and Discussion
4. Conclusions
Funding
Conflicts of Interest
References
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, B.; Li, Q.; Wen, L.; Zhang, R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.-H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Novel Coronavirus Pneumonia Emergency Response Epidemiology Team. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China. Chin. J. Epidemiol. 2020, 41, 145–151. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease 2019 (COVID-19) Situation Report-92. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ (accessed on 21 April 2020).
- Li, W.; Greenough, T.C.; Moore, M.J.; Vasilieva, N.; Somasundaran, M.; Sullivan, J.L.; Farzan, M.; Choe, H. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin-converting enzyme 2. J. Virol. 2004, 78, 11429–11433. [Google Scholar] [CrossRef]
- Li, K.K.B.; Yip, C.W.; Hon, C.C.; Lam, C.Y.; Zeng, F.; Leung, F.C.C. Characterisation of animal angiotensin-converting enzyme 2 receptors and use of pseudotyped virus to correlate receptor binding with susceptibility of SARS-CoV infection. Hong Kong Med. J. 2012, 18 (Suppl. 3), 35–38. [Google Scholar]
- Hou, Y.; Peng, C.; Yu, M.; Li, Y.; Han, Z.; Li, F.; Wang, L.-F.; Shi, Z. Angiotensin-converting enzyme 2 (ACE2) proteins of different bat species confer variable susceptibility to SARS-CoV entry. Arch. Virol. 2010, 155, 1563–1569. [Google Scholar] [CrossRef]
- Chaoxin, J.; Daili, S.; Yanxin, H.; Ruwei, G.; Chenlong, W.; Yaobin, T. The influence of angiotensin-converting enzyme 2 gene polymorphisms on type 2 diabetes mellitus and coronary heart disease. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2654–2659. [Google Scholar] [PubMed]
- Patel, S.K.; Wai, B.; Ord, M.; MacIsaac, R.J.; Grant, S.; Velkoska, E.; Panagiotopoulos, S.; Jerums, G.; Srivastava, P.M.; Burrell, L.M. Association of ACE2 genetic variants with blood pressure, left ventricular mass, and cardiac function in Caucasians with type 2 diabetes. Am. J. Hypertens. 2012, 25, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Huang, W.; Su, S.; Li, B.; Zhao, W.; Chen, S.; Gu, D. Association study of ACE2 (angiotensin I-converting enzyme 2) gene polymorphisms with coronary heart disease and myocardial infarction in a Chinese Han population. Clin. Sci. 2006, 111, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. PNAS 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS Coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Procko, E. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. bioRxiv 2020, 3, 994236. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Situation Update for the WHO European Region. Available online: http://www.euro.who.int/en/health-topics/health-emergencies/coronavirus-covid-19 (accessed on 29 April 2020).




{kind=link}
{kind=link}
{kind=link}
| Genes | Ace2 | Tmprss2 | CtsB | CtsL |
|---|---|---|---|---|
| Chromosome | X | 21 | 8 | 9 |
| Intronic-SNPs | 15,391 | 10,293 | 9470 | 1101 |
| Synonymous-SNPs | 104 | 173 | 149 | 50 |
| Missense SNPs, inframe insertion/deletion SNPs | 265 | 394 | 311 | 102 |
| Comparisons | Total | Specific for the Common Population of the Comparisons | ||
|---|---|---|---|---|
| Specific SNPs in Either of Comparisons | Specific SNPs in All of the Comparisons (Frequency) | Population Size (Avg./Median) | ||
| Europeans vs. Asians, Americans or Africans | 86 | 12 | 1 (0.000143) | 122,845/147,472 |
| Asians vs. Europeans, Americans or Africans | 55 | 43 | 4 (0.00013–0.00030) | 40,150/45,859 |
| Americans vs. Africans, Asians or Europeans | 52 | 29 | 2 (0.00016, 0.00018) | 29,909/35,302 |
| Africans vs. Americans, Asians or Europeans | 47 | 46 | 8 (0.00013–0.00336) | 20,482/21,424 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darbani, B. The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues. Int. J. Environ. Res. Public Health 2020, 17, 3433. https://doi.org/10.3390/ijerph17103433
Darbani B. The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues. International Journal of Environmental Research and Public Health. 2020; 17(10):3433. https://doi.org/10.3390/ijerph17103433
Chicago/Turabian StyleDarbani, Behrooz. 2020. "The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues" International Journal of Environmental Research and Public Health 17, no. 10: 3433. https://doi.org/10.3390/ijerph17103433
APA StyleDarbani, B. (2020). The Expression and Polymorphism of Entry Machinery for COVID-19 in Human: Juxtaposing Population Groups, Gender, and Different Tissues. International Journal of Environmental Research and Public Health, 17(10), 3433. https://doi.org/10.3390/ijerph17103433