2.1. Preparation of Carbamates
Carbamates
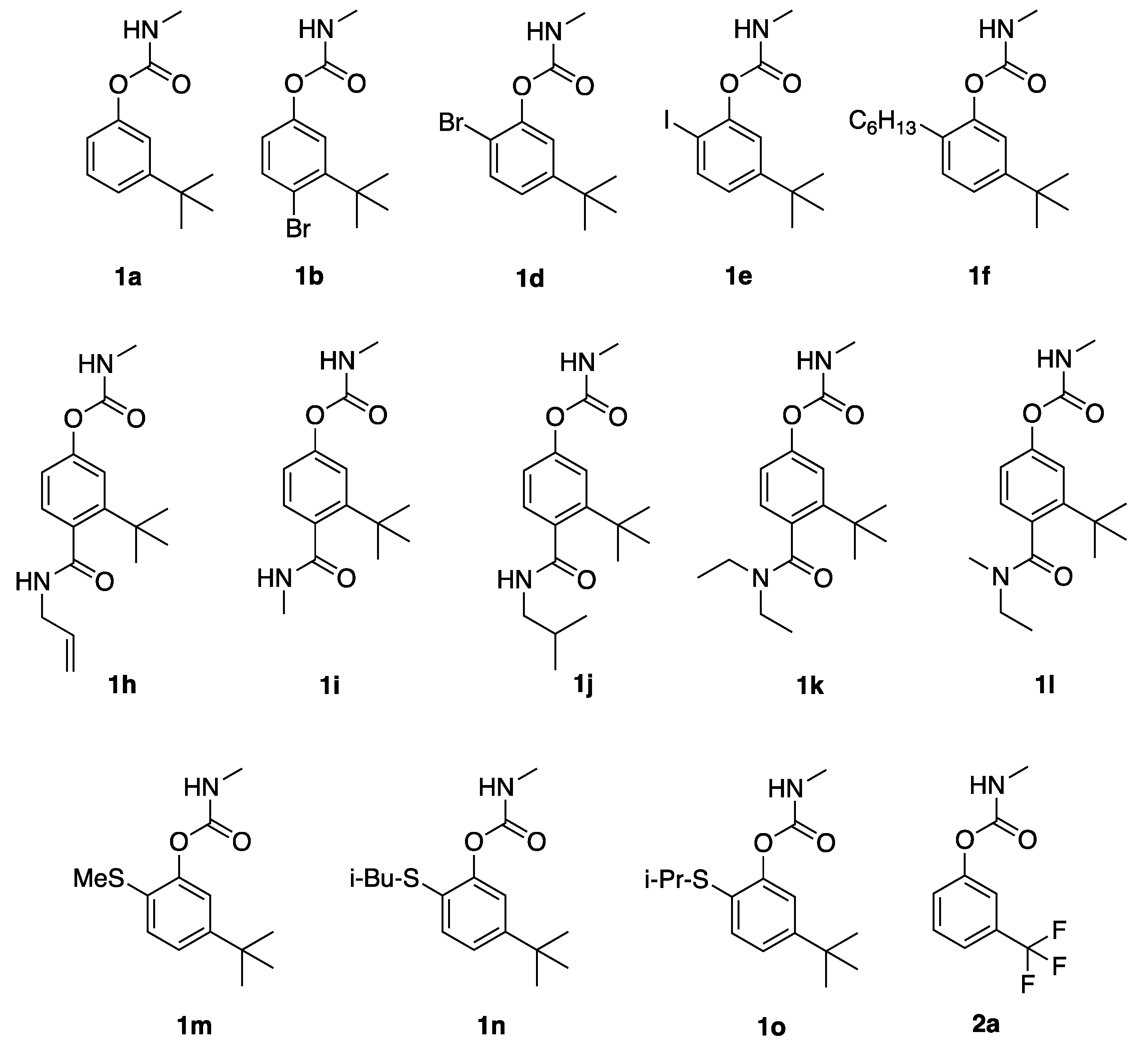
1a,
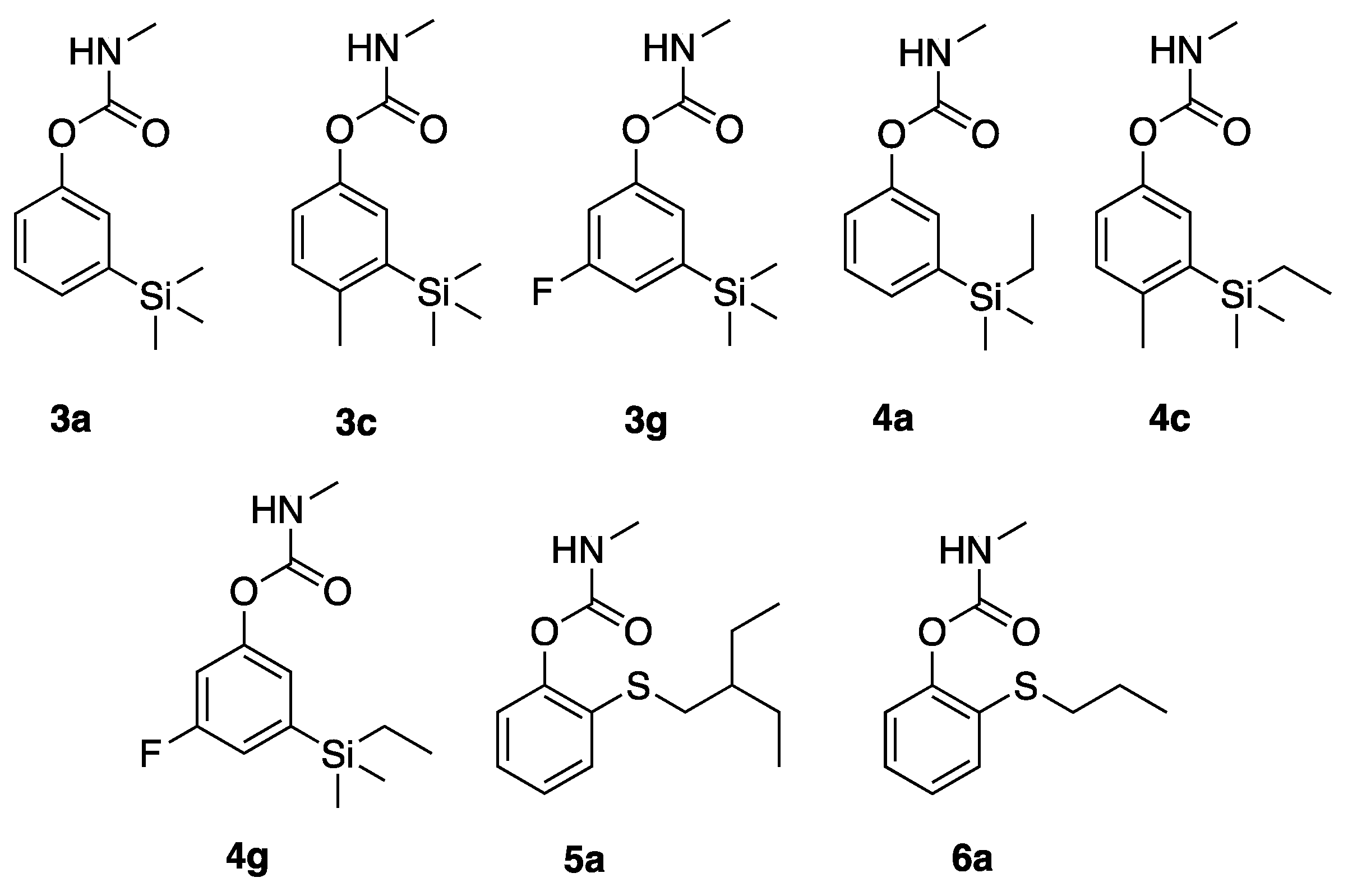
3a,
4a, and
5a were prepared as previously described by Hartsel et al. [
5] and the synthesis of other experimental compounds is described below.
4-bromo-3-(tert-butyl)phenyl methylcarbamate (1b): 3-t-butylphenol (3 g) was dissolved in 10% NaOH (10 mL), and iodine (5.6 g, 1.1 equivalent) and sodium iodide (5.6 g, 1.5 equivalent) were added to afford 5-(t-butyl)-2-iodophenol (4.96 g, 90% yield). Treatment with bromine in CH2Cl2 afforded 4-bromo-5-t-butyl-2-iodophenol; refluxing in N-methylmorpholine for 16 h, followed by column chromatography afforded 4-bromo-3-t-butylphenol in 50% yield over two steps. This compound (50 mg) was dissolved in dry tetrahydrofuran (THF) (2 mL), and potassium t-butoxide (0.29 mL, 1 M in THF) was added followed by methylcarbamoyl chloride (2 equiv). After 1 h, aqueous workup and column chromatography afforded 1b as a pale semi-solid (43 mg, 69%). 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 10.0 Hz, 1H), 7.19 (s, 1H), 6.85 (d, J = 10.0 Hz, 1H), 4.99 (br s, 1H), 2.95 (d, J = 3.0 Hz, 0.15H, minor carbamate rotamer), 2.88 (d, J = 3.0 Hz, 2.85H, major carbamate rotamer), 1.50 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.9, 150.2, 149.2, 136.2, 121.6, 120.7, 118.4, 36.7, 29.5, and 27.8.
2-bromo-5-(tert-butyl)phenyl methylcarbamate (1d): 3-t-butylphenol (3 g) was dissolved in dichloromethane (30 mL) and bromine (1.1 mL, 1.1 equivalent) was added dropwise. After stirring for 1 h, aqueous workup and concentration in vacuo afforded 2-bromo-5-t-butylphenol in quantitative yield. This phenol was treated as above for 1b to afford 1d as pale semi-solid (70% yield). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 2.0 Hz, 1H), 7.08 (dd, J = 8.4, 2.0 Hz, 1H), 5.05 (br s, 1H), 3.00 (d, J = 4.8 Hz, 0.15H, minor carbamate rotamer), 2.89 (d, J = 4.8 Hz, 2.85H, major carbamate rotamer), 1.26 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.3, 152.1, 147.9, 132.4, 124.3, 121.3, 113.0, 34.7, 31.1, and 27.8.
5-(tert-butyl)-2-iodophenyl methylcarbamate (1e): 5-(tert-butyl)-2-iodophenol (described above in synthesis of 1b) was then dissolved in dry THF (10 mL), treated with NaH (60% dispersion in mineral oil, 1.6 equivalent), and methylcarbamoyl chloride (2.5 equiv). After 1 h aqueous workup and column chromatography afforded 1e as a pale semi-solid (134 mg 50%). 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 8.3 Hz, 1H), 7.17(s, 1H), 6.97 (d, J = 8.3 Hz, 1H), 5.19 (br s, 1H), 3.01 (br s, 0.15H, minor carbamate rotamer), 2.89 (br s, 2.85H, major carbamate rotamer), 1.28 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.3, 153.3, 150.8, 138.4, 124.6, 120.6, 86.7, 34.6, 31.1, and 27.8.
5-(tert-butyl)-2-hexylphenyl methylcarbamate (1f): The requisite phenol was prepared from 5-(t-butyl)-2-iodophenol (see above) in a four-step sequence (62% overall yield) by (i) acetylation, (ii) Sonogashira coupling with 1-hexyne, catalytic hydrogenation, and deacetylation. This phenol was then reacted with NaH and methylcarbamoyl chloride according to the procedure for 1e, to afford 1f as a pale semi-solid (110 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 7.23–7.13 (m, 2H), 7.06 (s, 1H) 5.01 (br s, 1H), 2.94 (d, J = 4.8 Hz, 0.15 H, minor carbamate rotamer) 2.88 (d, J = 4.8 Hz, 2.85H, major carbamate rotamer), 2.50 (t, J = 7.7 Hz, 2H), 1.61–1.52 (m, 2H), 1.36–1.23 (m, 15H), 0.88 (t, J = 6.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 155.4, 150.0, 148.7, 131.8, 129.4, 122.5, 119.4, 34.4, 31.6, 31.2, 29.9, 29.6, 29.2, 27.7, 22.5, and 14.1.
4-(allylcarbamoyl)-3-(tert-butyl)phenyl methylcarbamate (1h): 4-bromo-3-t-butylphenol (above in 1b) was converted to the triiisopropylsilyl derivative (TIPS-Cl, imidazole, DMF), dissolved in diethyl ether, metalated with t-BuLi and trapped with ethylchloroformate, and then hydrolyzed (KOH, aq THF; HCl aq). Treatment with TBS-Cl and imidazole in dichloromethane afford 2-(t-butyl)-4-t-butyldimethylsilyloxybenzoic acid in 57% yield over four steps. Treatment with oxalyl chloride, followed by allyl amine afforded the allyl amide. Deprotection with HF/pyridine, deprotonation with potassium t-butoxide and acylation with methylcarbamoyl chloride afforded 1h as a pale oil (46% over three steps). 1H NMR (400 MHz, CDCl3): Note: chiral axis C4-C(O) renders the four CH2 protons diastereotopic δ 7.22 (d, J = 9.0 Hz, 1H), 7.15 (s, 1H), 6.88 (d, J = 9.0 Hz, 1H) 5.88 (ddt, J = 15.0, 10.0, 6.2 Hz, 1H), 5.81 (br s, 1H), 5.25 (dd, J = 15.0, 2.0 Hz, 1H), 5.22 (dd, J = 10.0, 2.0 Hz, 1H), 5.20 (br s, 1H), 4.00 (t, J = 6.2 Hz, 2H), 2.94 (d, J = 4.0 Hz, 0.15H, minor carbamate rotamer), 2.82 (d, J = 4.0 Hz, 2.85H, major carbamate rotamer), 1.45 (s, 9 H); 13C NMR (101 MHz, CDCl3) δ 172.3, 155.2, 151.6, 149.7, 133.7, 133.2, 129.4, 120.6, 118.8, 117.2, 42.5, 36.4, 31.4, and 27.8.
3-(tert-butyl)-4-(methylcarbamoyl)phenyl methylcarbamate (1i): 2-(t-butyl)-4-t-butyldimethylsilyloxybenzoic acid (from 1b above) was treated with oxalyl chloride, followed by methylamine. Deprotection with HF/pyridine, deprotonation with potassium t-butoxide and acylation with methylcarbamoyl chloride afforded 1i as a pale oil (42% over three steps). 1H NMR (400 MHz, CDCl3): Note: chiral axis C4-C(O) renders the four CH2 protons diastereotopic δ 7.22-7.13 (m, 2H), 6.89 (d, J = 9.0 Hz, 1H), 5.77 (br s, 1H), 5.18 (br s, 1H), 2.93 (d, J = 5.0 Hz, 3H), 2.85 (d, J = 4.4 Hz, 3H), 1.37 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 173.2, 155.2, 151.5, 149.6, 133.9, 129.3, 120.5, 118.8, 36.3, 31.3, 27.8, and 26.9.
3-(tert-butyl)-4-(isobutylcarbamoyl)phenyl methylcarbamate (1j): 2-(tert-butyl)-4-t-butyldimethylsilyloxybenzoic acid (from 1b above) was treated with oxalyl chloride, followed by isobutylamine. Deprotection with HF/pyridine, deprotonation with potassium t-butoxide and acylation with methylcarbamoyl chloride afforded 1j as a pale oil (28% over three steps). 1H NMR (400 MHz, CDCl3): Note: chiral axis C4-C(O) renders the four CH2 protons diastereotopic δ 7.22 (d, J = 8.3 Hz, 1H), 7.18 (s, 1H), 6.95 (d, J = 8.3 Hz, 1H), 5.80 (br s, 1H), 5.19 (br s, 1H), 3.23 (dd, J = 6.2, 5.8 Hz, 2H), 2.85 (d, J = 3.8 Hz, 0.15H, minor carbamate rotamer), 2.79 (d, J = 3.8 Hz, 2.85H, major carbamate rotamer), 1.90 (t sep, J = 6.8, 6.2 Hz, 1H), 1.44 (s, 9H), 0.92 (d, J = 6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 172.6, 155.2, 151.5, 149.6, 134.1, 129.4, 120.5, 118.8, 47.6, 36.3, 31.3, 28.3, 27.8, and 20.3.
3-(tert-butyl)-4-(diethylcarbamoyl)phenyl methylcarbamate (1k): 2-(t-butyl)-4-t-butyldimethylsilyloxybenzoic acid (from 1b above) was treated with oxalyl chloride, followed by diethylamine. Deprotection with HF/pyridine, deprotonation with potassium t-butoxide and acylation with methylcarbamoyl chloride afforded 1k as a pale oil (29% over three steps). 1H NMR (400 MHz, CDCl3): Note: chiral axis C4-C(O) renders the four CH2 protons diastereotopic δ 7.20 (s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 5.05 (br s, 1H), 3.72 (dq, J = 15.2, 6.2 Hz, 1H), 3.28 (dq, J = 15.6, 6.2 Hz, 1H), 3.20 (dq, J = 15.2, 6.2 Hz, 1H), 3.01 (dq, J = 15.6, 6.2 Hz, 1H), 2.95 (d, J = 4.0 Hz, 0.15 H, minor carbamate rotamer), 2.78 (d, J = 4.0 Hz, 2.85H, major carbamate rotamer), 1.29 (s, 9H), 1.23 (dd, J = 6.2, 6.2 Hz, 3H), 1.02 (dd, J = 6.2, 6.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.3, 154.6, 150.8, 147.0, 133.5, 127.7, 120.9, 118.1, 44.2, 38.6, 36.1, 32.0, 27.5, 13.8, and 12.7.
3-(tert-butyl)-4-(ethyl(methyl)carbamoyl)phenyl methylcarbamate (1l): 2-(t-butyl)-4-t-butyldimethylsilyloxybenzoic acid (from 1b above) was treated with oxalyl chloride, followed by ethylmethylamine. Deprotection with HF/pyridine, deprotonation with potassium t-butoxide and acylation with methylcarbamoyl chloride afforded 1l as a pale oil (59% over three steps). 1H NMR (400 MHz, CDCl3): Note: chiral axis C4-C(O) renders the four CH2 protons diastereotopic δ 7.13 (d, J = 1.8 Hz, 1H), 7.02 (dd, J = 8.2, 1.8 Hz, 1H), 6.87 (d, J = 8.2 Hz, 1H), 5.32 (br s, 1H), 3.69 (dt, J = 15.2, 6.8 Hz, 0.5 H), 3.48 (dt, J = 15.2, 6.8 Hz, 0.5 H), 3.25 (dt, J = 15.2, 6.8 Hz, 0.5 H), 3.05 (s, 1.5 H), 2.98 (dt, J = 15.2, 6.8 Hz, 0.5 H), 2.97 (d, J = 3.8 Hz, 0.15 H, minor carbamate rotamer), 2.82 (d, J = 3.8 Hz, 2.85 H, major carbamate rotamer), 2.75 (s, 1.5 H), 1.32 (s, 9 H), 1.20 (dd, J = 6.8, 6.8 Hz, 1.5 H), 0.97 (dd, J = 6.8, 6.8 Hz, 1.5 H); 13C NMR (101 MHz, CDCl3) δ 176.0, 175.0, 155.3, 151.3, 151.2, 148.1, 148.0, 132.6, 131.9, 128.5, 127.8, 120.9, 119.1, 118.9, 46.0, 41.8, 36.9, 36.4, 31.7, 31.2, 27.7, 13.0, and 11.4 (two equally populated rotamers (ethymethylamide); 24 of a possible 28 resonances seen).
5-(tert-butyl)-2-(methylthio)phenyl methylcarbamate (1m): 3-t-butylphenol was treated with chlorosulfonic acid (10 equivalent) in dichloromethane for 4 h, and then poured into ice. Extractive workup, reduction with stannous chloride in acetic acid (18 h), and column chromatography afforded 6,6’-disulfanediylbis(3-(t-butyl)phenol). Reduction with sodium borohydride in THF gave 5-(t-butyl)-2-mercaptophenol in 64% yield over three steps. Treatment with methyl iodide (1.1 equiv) and sodium bicarbonate in DMF, followed by deprotonation with potassium t-butoxide in THF and acylation with methylcarbamoyl chloride gave 1m as a pale semi-solid in 37% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.23 (dd, J = 6.6, 1.6 Hz, 1H), 7.19 (d, J = 6.6 Hz, 1H), 7.12 (d, J = 1.6 Hz, 1H), 5.04 (br s, 1H), 3.07 (d, J = 4.0 Hz, 0.15 H, minor carbamate rotamer), 2.91 (d, J = 4.0 Hz, 2.85 H, major carbamate rotamer), 2.42 (s, 3H), 1.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 154.9, 150.1, 148.3, 128.1, 127.3, 123.5, 120.2, 34.6, 31.4, 28.0, 15.7.
5-(tert-butyl)-2-(isobutylthio)phenyl methylcarbamate (1n): 5-(t-butyl)-2-mercaptophenol (from 1m above) was treated with isobutyl iodide (2.9 equivalents) and sodium bicarbonate in DMF at 55 °C for 6 h, followed by deprotonation with potassium t-butoxide in THF and acylation with methylcarbamoyl chloride to gave 1n as a pale semi-solid in 63% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 6.6 Hz, 1H), 7.18 (d, J = 6.6, 1.6 Hz, 1H), 7.12 (d, J = 1.6 Hz, 1H), 5.04 (br s, 1H), 3.02, (d, J = 3.9 Hz, 0.15H, minor carbamate rotamer), 2.92 (d, J = 3.9 Hz, 2.85H, major carbamate rotamer), 2.73 (d, J = 5.5 Hz, 2H), 1.81 (nonet (9-let), J = 5.5 Hz, 1H), 1.29 (s, 9H), 1.02 (J = 5.3 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 155.0, 150.9, 149.5, 130.1, 127.0, 123.4, 120.3, 42.5, 34.7, 31.3, 28.4, 28.0, 22.2.
5-(tert-butyl)-2-(isopropylthio)phenyl methylcarbamate (1o): 5-(t-butyl)-2-mercaptophenol (from 1m above) was treated with isopropyl iodide (3.0 equiv) and sodium bicarbonate in DMF at 55 °C for 6 h, followed by deprotonation with potassium t-butoxide in THF and acylation with methylcarbamoyl chloride to give 1o as a pale semi-solid in 60% yield over two steps. 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 6.6 Hz, 1H), 7.19 (dd, J = 6.6, 1.6 Hz, 1H), 7.14 (d, J = 1.6 Hz, 1H), 5.03 (br s, 1H), 3.33 (sept, J = 5.3 Hz, 1H), 3.02 (d, J = 3.9 Hz, 0.15H, minor carbamate rotamer), 2.92 (d, J = 3.9 Hz, 2.85H, major carbamate rotamer), 1.30 (s, 9H), 1.27 (d, J = 5.3 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 155.1, 152.1, 150.7, 133.2, 125.3, 134.3, 120.4, 37.9, 34.8, 31.3, 28.0, and 23.4.
3-(trifluoromethyl)phenyl methylcarbamate (2a): 3-trifluoromethylphenol (1.09 g) was deprotonated with potassium t-butoxide in THF and treated with methylcarbamoyl chloride to give 2a as a pale semi-solid (1.16 g, 81% yield). 1H NMR (400 MHz, CDCl3) δ 7.47-7.44 (m, 2H), 7.40 (br s, 1H), 7.33-7.31 (m, 1H), 5.17 (br s, 1H), 2.95 (d, J = 4.0 Hz, 0.15H, minor carbamate rotamer), 2.88 (d, J = 4.0 Hz, 2.85H, major carbamate rotamer); 13C NMR (101 MHz, CDCl3) δ 154.8, 151.3, 132.3, 129.9, 125.3, 123.3 (q, 1JCF = 238 Hz), 122.1, 118.9, and 27.8.
4-methyl-3-(trimethylsilyl)phenyl methylcarbamate (3c): 3-bromo-4-methylphenol (0.70 g) was dissolved in dry THF (5 mL), cooled to -78 °C, treated with BuLi (3.4 mL, 2.5 M in hexanes, 2.2 equivalent) and trimethylsilyl chloride (1.2 mL, 1.1 g, 2.6 equivalent). After stirring for 30 min the reaction was allowed to warm to 25 °C and the reaction was quenched with aqueous HCl. Extractive workup and column chromatograph afforded 4-methyl-3-trimethylsilylphenol as a colorless oil (630 mg, 93%). Applying the procedure above, the phenol (630 mg) was converted to the methyl carbamate, affording 3c as a yellow oil (591 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ 7.12 (s, 1H), 7.11 (d, J = 9.6 Hz, 1H), 6.98 (d, J = 9.6 Hz, 1H), 4.97 (br s, 1H), 2.93 (d, J = 4.0 Hz, 0.15H, minor carbamate rotamer), 2.85 (d, J = 4.0 Hz, 2.85H, major carbamate rotamer), 2.40 (s, 3H), 0.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 155.8, 148.7, 140.6, 140.1, 130.8, 127.1, 122.4, 27.9, 22.5, and −0.13.
3-fluoro-5-(trimethylsilyl)phenyl methylcarbamate (3g): 3-bromo-5-fluorophenol (0.19 g) was dissolved in dry THF (9 mL), cooled to −78 °C, treated with BuLi (1.4 mL, 2.5 M in hexanes, 3.3 equivalent) and trimethylsilyl chloride (0.45 mL, 0.39 g, 3.6 equivalent). After stirring for 30 min the reaction was allowed to warm to 25 °C and the reaction was quenched with aqueous HCl. Extractive workup and column chromatograph afforded 5-fluoro-3-trimethylsilylphenol as a colorless oil (66 mg, 66%). Applying the procedure above, the phenol (66 mg) was converted to the methyl carbamate, affording 3g as a yellow oil (78 mg, 92% yield). 1H NMR (400 MHz, CDCl3) δ 7.08-6.99 (m, 2H), 6.87 (ddd, J = 9.6, 2.3, 2.1 Hz, 1H), 5.12 (br s, 1H), 2.92 (d, J = 4.7 Hz, 0.15H, minor carbamate rotamer), 2.87 (d, J = 4.7 Hz, 2.85H, major carbamate rotamer), 0.25 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 162.5 (d, 1JCF = 248.4 Hz) 154.9, 151.5, 144.2 (d, 3JCF = 4.7 Hz), 121.6 (d, 3JCF = 2.9 Hz), 116.5 (d, 2JCF = 18.2 Hz), 110.0 (d, 2JCF = 24.1 Hz), 27.7, and −1.4.
3-(ethyldimethylsilyl)-4-methylphenyl methylcarbamate (4c): 3-bromo-4-methylphenol (0.70 g) was dissolved in dry THF (5 mL), cooled to −78 °C, treated with BuLi (3.4 mL, 2.5 M in hexanes, 2.2 equivalent) and ethyldimethylsilyl chloride (1.36 mL, 1.19 g, 2.6 equivalent). After stirring for 30 min, the reaction was allowed to warm to 25 °C and the reaction was quenched with aqueous HCl. Extractive workup and column chromatograph afforded 4-methyl-3-trimethylsilylphenol as a colorless oil (665 mg, 91%). Applying the procedure above, the phenol (419 mg) was converted to the methyl carbamate, affording 4c as a yellow oil (419 mg, 77% yield). 1H NMR (400 MHz, CDCl3) δ 7.13 (s, 1H), 7.11 (d, J = 9.0 Hz, 1H), 6.99 (d, J = 9.0 Hz, 1H), 5.05 (br s), 2.90 (d, J = 4.9 Hz, 0.15H, minor carbamate rotamer), 2.83 (d, J = 4.9 Hz, 2.85H, major carbamate rotamer), 2.40 (s, 3H), 0.93 (t, J = 8.0 Hz, 3H), 0.79 (q, J = 8.0 Hz, 2H), 0.28 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 155.9, 148.7, 140.7, 139.2, 130.8, 127.5, 122.3, 27.9, 22.5, 7.75, 7.74, and −2.35.
3-(ethyldimethylsilyl)-5-fluorophenyl methylcarbamate (4g): 3-bromo-5-fluorophenol (0.27 g) was dissolved in dry THF (9 mL), cooled to −78 Ç, treated with BuLi (2.1 mL, 2.5 M in hexanes, 3.5 equivalent) and ethyldimethylsilyl chloride (0.80 mL, 0.70 g, 4.0 equivalent). After stirring for 30 min the reaction was allowed to warm to 25 °C and the reaction was quenched with aqueous HCl. Extractive workup and column chromatograph afforded 3-ethyldimethylsilyl-5-fluorophenol as a colorless oil (178 mg, 62%). Applying the procedure above, the phenol (58 mg) was converted to the methylcarbamate, affording 4g as a yellow oil (55 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.08–6.98 (m, 2H), 6.87 (ddd, J = 9.6, 2.2, 2.2 Hz, 1H), 5.09 (br s, 1H), 2.93 (d, J = 4.0 Hz, 0.15H, minor carbamate rotamer), 2.87 (d, J = 4.0 Hz, 2.85Hz, major carbamate rotamer), 0.94 (t, J = 7.8 Hz, 3H), 0.70 (q, J = 7.8 Hz, 2H), 0.23 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 162.5 (d, 1JCF = 248.4 Hz), 154.8, 151.5 (d, 2JCF = 9.7 Hz), 143.2 (d, 3JCF = 4.6Hz), 121.8 (d, 3JCF = 2.9 Hz), 116.6, 109.9 (d, 2JCF = 24.2), 27.7, 7.23, 7.13, and −3.73.
2-(propylthio)phenyl methylcarbamate (6a) 2-mercaptophenol (300 mg, 2.5 mmol) was dissolved in dry DMF (5 mL) and NaHCO3 (630 mg, 3 equivalent) and propyl bromide (0.45 g, 5.0 equivalent) were added. After heating to 55 °C for 16 h, aqueous workup, and column chromatography, 2-propylthiophenol was isolated as a pale oil (391 mg, 98%). 2-propylthiophenol (354 mg, 2.11 mmol) was dissolved in dry THF (20 mL), treated with NaH (60% in mineral oil, 110 mg, 2.75 equivalent), and methylcarbamoyl chloride (394 mg, 4.2 equivalent) was added. Aqueous workup and column chromatography afforded 6a as a pale oil (360 mg, 76%). 1H NMR (400 MHz, CDCl3) δ 7.33 (dd, J = 5.6, 1.8 Hz, 1H), 7.21–7.15 (m, 2H), 7.11 (dd, J = 5.3, 1.9 Hz, 1H), 5.08 (br s, 1H), 3.02 (d, J = 4.0 Hz, 0.15H, minor carbamate rotamer), 2.91 (d, J = 4.0 Hz, 2.85Hz, major carbamate rotamer), 2.85 (t, J = 5.8 Hz, 2H), 1.66 (hextet, J = 5.9 Hz, 2H), 1.02 (t, J = 5.9 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 154.8, 149.3, 130.5, 129.5, 126.7, 126.2, 123.1, 34.9, 28.0, 22.5, and 13.6.
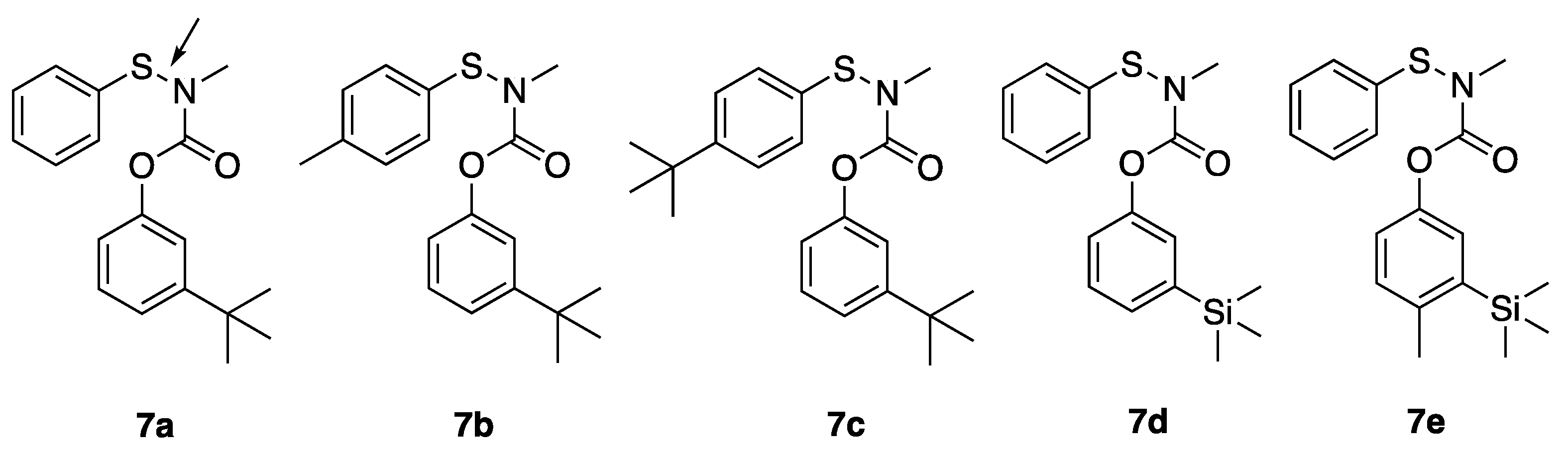
3-(tert-butyl)phenyl methyl(phenylthio)carbamate (7a): Compound 1a (150 mg) was treated with triethylamine (4 equiv) and benzenesulfenyl chloride (1.5 equvalent) in carbon tetrachloride (4 mL) at 45 °C for 18 h. Following aqueous workup the residue was chromatographed in 30:1 hexanes:ethyl acetate to afford 7a as a yellow oil (186 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 7.40–7.36 (m, 2H), 7.34 (d, J = 9.5 Hz, 2H), 7.30–7.22 (m, 3H), 7.08 (s, 1H), 6.92 (d, J = 8.2 Hz, 1H), 3.43 (s, 3H), 1.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.8, 153.0, 151.3, 137.5, 129.3, 128.8, 127.3, 126.3, 125.3, 122.8, 118.5, 42.0, 34.8, and 32.2.
3-(tert-butyl)phenyl methyl(p-tolylthio)carbamate (7b): Following the procedure for 7a, 1a, and p-toluylsulfenyl chloride were reacted to afford 7b as a yellow oil (213 mg, 89%). 1H NMR (400 MHz, CDCl3) δ 7.36–7.22 (m, 6H), 7.11 (s, 1H), 6.96 (br s, 1H), 3.42 (s, 3H), 2.38 (s, 3H), 1.32 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.8, 154.9, 151.4, 138.2, 134.1, 130.0, 128.8, 127.2, 122.7, 118.6, 41.9, 34.8, 31.3, and 21.2.
3-(tert-butyl)phenyl ((4-(tert-butyl)phenyl)thio)(methyl)carbamate (7c): following the procedure for 7a, 1a, and 4-t-butylphenylsulfenyl chloride were reacted to afford 7c as a yellow oil (152 mg (73%). 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 9.7 Hz, 2H), 7.37 (d, J = 9.7 Hz, 2H), 7.30 (t, J = 8.0 Hz, 1H), 7.25 (d, 8.0 Hz, 1H), 7.10 (s, 1H), 6.95 (d, J = 8.0 Hz, 1H), 3.42 (s, 3H), 1.34 (s, 9H), 1.31 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.8, 152.9, 151.4, 151.2, 134.3, 128.8, 126.9, 126.3, 122.7, 118.6, 41.9, 34.8, 34.7, 31.6, 31.3, 31.2, 22.7, and 14.1.
3-(trimethylsilyl)phenyl methyl(phenylthio)carbamate (7d): Following the procedure for 7a, 3a and benzenesulfenyl chloride were reacted to afford 7d as a yellow oil (68 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 7.44–7.30 (m, 6H), 7.28–7.23 (m, 1H), 7.19 (s, 1H), 7.09 (s, 1H), 3.47 (s, 3H), 0.27 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.9, 151.2, 142.7, 133.7, 130.7, 129.4, 129.0, 127.5, 125.9, 122.0, 42.1, and −1.11.
4-methyl-3-(trimethylsilyl)phenyl methyl(phenylthio)carbamate (7e): Following the procedure for 7a, 4c and benzenesulfenyl chloride were reacted to afford 7e as a yellow oil (166 mg, 58%). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.7 Hz, 2H), 7.39–7.35 (m, 2H), 7.29 (t, J = 8.7, Hz, 1H), 7.19–7.10 (m, 2H), 7.00 (d, J = 8.0 Hz, 1H), 3.44 (s, 3H), 2.44 (s, 3H), 0.27 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 157.0, 149.0, 140.9, 140.1, 130.7, 129.3, 129.0, 127.3, 126.7, 125.6, 122.0, 42.0, 22.3, and −0.4.

 ,
, 



{kind=link}
{kind=link}
{kind=link}