Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis

Abstract
1. Introduction
1.1. Early Life Stress and Biological Underpinnings of Epigenomic Modulation
1.2. Epigenetic Implications of Childhood Trauma
1.3. Structural Inequity in Childhood Trauma
1.4. Current Study Objective
2. Experimental Section
2.1. Design
2.2. Search Engine and Strategies
(gene expression OR DNA methylation OR histone acetylation OR microRNA OR gene transcription OR siRNA OR mRNA OR histone methylation OR epigenotype AND early childhood trauma) OR (gene expression OR DNA methylation OR histone acetylation OR microRNA OR gene transcription OR siRNA OR mRNA OR histone methylation OR epigenotype AND schizophrenia) OR (gene expression OR DNA methylation OR histone acetylation OR microRNA OR gene transcription OR siRNA OR mRNA OR histone methylation OR epigenotype AND neuroendocrine development (cortisol, pituitary hypothalamus, HPA axis, vasopressin, corticotropin hormone, CRF, ERA, glucocorticoid receptor gene, (NR3C1), brain derived neurotropic factor (BDNF), glucocorticoid receptor (GR)), GABA aminobutyric acid AND major depressive disorders AND human)
2.3. Eligibility Criteria
2.4. Data Extraction Reliability
2.5. Study Variables
2.6. Data Collection and Processing
2.7. Study Quality Assessment
2.8. Statistical Analysis: QES
3. Results and Discussion
3.1. Results
3.1.1. NR3C1 DNA Methylation in Early Life Stress and Major Depressive Disorder Correlation
3.1.2. NR3CI DNA Methylation in ELS
3.1.3. NR3CI DNA Methylation and MDD
3.2. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef]
- Vreugdenhil, E.; Verissimo, C.S.; Mariman, R.; Kamphorst, J.T.; Barbosa, J.S.; Zweers, T.; Champagne, D.L.; Schouten, T.; Meijer, O.C.; de Kloet, E.R.; et al. MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: Implications for glucocorticoid responsiveness in the brain. Endocrinology 2009, 150, 2220–2228. [Google Scholar] [CrossRef]
- Chen, E.; Miller, G.E.; Kobor, M.S.; Cole, S.W. Maternal warmth buffers the effects of low early-life socioeconomic status on pro-inflammatory signaling in adulthood. Mol. Psychiatry 2011, 16, 729–737. [Google Scholar] [CrossRef]
- Mulligan, C.J.; D’Errico, N.C.; Stees, J.; Hughes, D.A. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 2012, 7, 853–857. [Google Scholar] [CrossRef]
- Weaver, I.C.; Meaney, M.J.; Szyf, M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc. Natl. Acad. Sci. USA 2006, 103, 3480–3485. [Google Scholar] [CrossRef]
- McGowan, P.O.; Suderman, M.; Sasaki, A.; Huang, T.C.; Hallett, M.; Meaney, M.J.; Szyf, M. Broad epigenetic signature of maternal care in the brain of adult rats. PLoS ONE 2011, 6, e14739. [Google Scholar] [CrossRef]
- Dickerson, S.S.; Kemeny, M.E. Acute stressors and cortisol responses: A theoretical integration and synthesis of laboratory research. Psychol. Bull. 2004, 130, 355–391. [Google Scholar] [CrossRef]
- Gilbert, P.; McEwan, K.; Bellew, R.; Mills, A.; Gale, C. The dark side of competition: How competitive behaviour and striving to avoid inferiority are linked to depression, anxiety, stress and self-harm. Psychol. Psychother. 2009, 82 Pt 2, 123–136. [Google Scholar] [CrossRef]
- Layte, R. The association between income inequality and mental health: Testing status anxiety, social capital, and neo-materialist explanations. Eur. Soc. Rev. 2011, 28, 498–511. [Google Scholar] [CrossRef]
- Layte, R.; Whelan, C.T. Who feels inferior? A test of the status anxiety hypothesis of social inequalities in health. Eur. Soc. Rev. 2014, 30, 525–535. [Google Scholar] [CrossRef]
- Frankel, D.; Arbel, T. Group formation by two-year olds. Int. J. Behav. Dev. 1980, 3, 287–298. [Google Scholar] [CrossRef]
- McGowan, P.; Sasaki, A.; D’Alessio, A.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic Regulation of the Glucocorticoid Receptor in Human Brain Associates with Childhood Abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef]
- McGowan, P.O.; Szyf, M. The epigenetics of social adversity in early life: Implications for mental health outcomes. Neurobiol. Dis. 2010, 39, 66–72. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Razin, A. CpG methylation, chromatin structure and gene silencing-a three-way connection. EMBO J. 1998, 17, 4905–4908. [Google Scholar] [CrossRef]
- Rajewsky, N. MicroRNA Target Predictions in Animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef]
- Tuddenham, L.; Wheeler, G.; Hajihosseini, M.K.; Clark, I.; Dalmay, T. The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett. 2006, 580, 4214–4217. [Google Scholar] [CrossRef]
- Cole, S.W.; Conti, G.; Arevalo, J.M.; Ruggerio, A.M.; Heckman, J.J.; Suomi, S.J. Transcriptional modulation of the developing immune system by early life social adversity. Proc. Natl. Acad. Sci. USA 2012, 109, 20578–20583. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J. Antiinflammatory Action of Glucocorticoids—New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Binder Elisabeth, B. The Role of FKBP5, a Co-Chaperone of the Glucocorticoid Receptor in the Pathogenesis and Therapy of Affective and Anxiety Disorders. Psychoneuroendocrinology 2009, 34, S186–S195. [Google Scholar] [CrossRef]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef]
- Kadhim, M.; Holmes, L.; Gesheff, M.G.; Conway, J.D. Treatment Options for Nonunion with Segmental Bone Defects: Systematic Review and Quantitative Evidence Synthesis. J. Orthop. Trauma 2016, 31, 111–119. [Google Scholar] [CrossRef]
- Nawijn, F.; Ham, W.; Houwert, R.M.; Groenwold, R.; Hietbrink, F.; Smeeing, D. Quality of reporting of systematic reviews and meta-analyses in emergency medicine based on the PRISMA statement. BMC Emerg. Med. 2019, 19, 19. [Google Scholar] [CrossRef]
- Anacker, C.; O’Donnell, K.J.; Meaney, M.J. Early life adversity and the epigenetic programming of hypothalamic-pituitary-adrenal function. Dialogues Clin. Neurosci. 2014, 16, 321–333. [Google Scholar]
- Maccari, S.; Krugers, H.J.; Morley-Fletcher, S.; Szyf, M.; Brunton, P.J. The consequences of early-life adversity: Neurobiological, behavioural and epigenetic adaptations. J. Neuroendocrinol. 2014, 26, 707–723. [Google Scholar] [CrossRef]
- Kundakovic, M.; Champagne, F.A. Early-life experience, epigenetics, and the developing brain. Neuropsychopharmacology 2015, 40, 141–153. [Google Scholar] [CrossRef]
- Suderman, M.; McGowan, P.O.; Sasaki, A.; Huang, T.C.T.; Hallett, M.T.; Meaney, M.J.; Turecki, G.; Szyf, M. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. 2), 17266–17272. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Brockmϋhl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar] [CrossRef]
- Franklin, T.B.; Saab, B.J.; Mansuy, I.M. Neural mechanisms of stress resilience and vulnerability. Neuron 2012, 75, 747–761. [Google Scholar] [CrossRef]
- Provencal, N.; Suderman, M.J.; Guillemin, C.; Massart, R.; Ruggiero, A.; Wang, D.; Bennett, A.J.; Pierre, P.J.; Friedman, D.P.; Côtè, S.M.; et al. The signature of maternal rearing in the methylome in rhesus macaque prefrontal cortex and T cells. J. Neurosci. 2012, 32, 15626–15642. [Google Scholar] [CrossRef] [PubMed]
- Elwenspoek, M.M.; Hengesch, X.; Leenen, F.A.; Schritz, A.; Sias, K.; Schaan, V.K.; Mériaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schächinger, H.; et al. Proinflammatory T Cell Status Associated with Early Life Adversity. J. Immunol. 2017, 199, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Suomi, S.J. Risk, resilience, and gene-environment interplay in primates. J. Can. Acad. Child. Adolesc. Psychiatry 2011, 20, 289–297. [Google Scholar] [PubMed]
- Dettmer, A.M.; Suomi, S.J. Nonhuman primate models of neuropsychiatric disorders: In uences of early rearing, genetics, and epigenetics. ILAR J. 2014, 55, 361–370. [Google Scholar] [CrossRef]
- Kinnally, E.L. Epigenetic plasticity following early stress predicts long-term health outcomes in rhesus macaques. Am. J. Phys. Anthropol. 2014, 155, 192–199. [Google Scholar] [CrossRef]
- Nieratschker, V.; Massart, R.; Gilles, M.; Luoni, A.; Suderman, M.J.; Krumm, B.; Meier, S.; Witt, S.H.; Nöthen, M.M.; Suomi, S.J.; et al. MORC1 exhibits cross-species differential methylation in association with early life stress as well as genome-wide association with MDD. Transl. Psychiatry 2014, 4, e429. [Google Scholar] [CrossRef]
- McEwen, B.S.; Nasca, C.; Gray, J.D. Stress effects on neuronal structure: Hippocampus, amygdala, and prefrontal cortex. Neuropsychopharmacology 2016, 41, 3–23. [Google Scholar] [CrossRef]
- Blaze, J.; Scheuing, L.; Roth, T.L. Differential methylation of genes in the medial prefrontal cortex of developing and adult rats following exposure to maltreatment or nurturing care during infancy. Dev. Neurosci. 2013, 35, 306–316. [Google Scholar] [CrossRef]
- Roth, T.L.; Matt, S.; Chen, K.; Blaze, J. Bdnf DNA methylation modi cations in the hippocampus and amygdala of male and female rats exposed to different caregiving environments outside the homecage. Dev. Psychobiol. 2014, 56, 1755–1763. [Google Scholar] [CrossRef]
- Doherty, T.S.; Forster, A.; Roth, T.L. Global and gene-speci c DNA methylation alterations in the adolescent amygdala and hippocampus in an animal model of caregiver maltreatment. Behav. Brain Res. 2016, 298 Pt A, 55–61. [Google Scholar] [CrossRef]
- Wu, Y.; Patchev, A.V.; Daniel, G.; Almeida, O.F.; Spengler, D. Early-life stress reduces DNA methylation of the Pomc gene in male mice. Endocrinology 2014, 155, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Bockmϋhl, Y.; Patchev, A.V.; Madejska, A.; Hoffmann, A.; Sousa, J.C.; Sousa, N.; Holsboer, F.; Almeida, O.F.X.; Spengler, D. Methylation at the CpG island shore region upregulates Nr3c1 promoter activity after early-life stress. Epigenetics 2015, 10, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.M.; Ruf, M.; Gunter, H.M.; Dohrmann, K.; Schauer, M.; Meyer, A.; Elbert, T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl. Psychiatry 2011, 1, e21. [Google Scholar] [CrossRef] [PubMed]
- Tyrka, A.R.; Parade, S.H.; Eslinger, N.M.; Marsit, C.J.; Lesseur, C.; Armstrong, D.A.; Philip, N.S.; Josefson, B.; Seifer, R. Methylation of exons 1D, 1F, and 1H of the glucocorticoid receptor gene promoter and exposure to adversity in preschool-aged children. Dev. Psychopathol. 2015, 27, 577–585. [Google Scholar] [CrossRef]
- Van der Knaap, L.J.; Riese, H.; Hudziak, J.J.; Verbiest, M.M.P.J.; Verhulst, F.C.; Oldehinkel, A.J.; van Oort, F.V.A. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study. Transl. Psychiatry 2014, 4, e381. [Google Scholar] [CrossRef]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 2015, 10, 408–417. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Quinn, J.P.; Sharp, H.M.; Pickles, A.; Hill, J. Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl. Psychiatry 2015, 5, e560. [Google Scholar] [CrossRef]
- Radtke, K.M.; Schauer, M.; Gunter, H.M.; Ruf-Leuschner, M.; Sill, J.; Meyer, A.; Elbert, T. Epigenetic modifications of the glucocorticoid receptor gene are associated with the vulnerability to psychopathology in childhood maltreatment. Transl. Psychiatry 2015, 5, e571. [Google Scholar] [CrossRef]
- Kertes, D.A.; Kamin, H.S.; Hughes, D.A.; Rodney, N.C.; Bhatt, S.; Mulligan, C.J. Prenatal maternal stress predicts methylation of genes regulating the hypothalamic–pituitary–adrenocortical system in mothers and newborns in the Democratic Republic of Congo. Child. Dev. 2016, 87, 61–72. [Google Scholar] [CrossRef]
- Kino, T. Stress, glucocorticoid hormones, and hippocampal neural progenitor cells: implications to mood disorders. Front Physiol. 2015, 6. [Google Scholar] [CrossRef]
- Pan-Vazquez, A.; Rye, N.; Ameri, M.; McSparron, B.; Smallwood, G.; Bickerdyke, J.; Rathbone, A.; Dajas-Bailador, F.; Toledo-Rodriguez, M. Impact of voluntary exercise and housing conditions on hippocampal glucocorticoid receptor, miR-124 and anxiety. Mol. Brain 2015, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child. Adolesc. Psychiatry 2014, 53, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Melas, P.A.; Wei, Y.; Wong, C.C.; Sjöholm, L.K.; Aberg, E.; Johnathan, M.; Schalling, M.; Forsell, Y.; Lavebratt, C. Genetic and epigenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int. J. Neuropsychopharmacol. 2013, 16, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef] [PubMed]
- Khulan, B.; Manning, J.R.; Dunbar, D.R.; Seckl, J.R.; Raikkonen, K.; Eriksson, J.G.; Drake, A.J. Epigenomic profiling of men exposed to early-life stress reveals DNA methylation differences in association with current mental state. Transl. Psychiatry 2014, 4, e448. [Google Scholar] [CrossRef] [PubMed]
- Non, A.L.; Hollister, B.M.; Humphreys, K.L.; Childebayeva, A.; Esteves, K.; Zeanah, C.H.; Fox, N.A.; Nelson, C.A.; Drury, S.S. DNA methylation at stress-related genes is associated with exposure to early life institutionalization. Am. J. Phys. Anthropol. 2016, 161, 84–93. [Google Scholar] [CrossRef]
- Houtepen, L.C.; Vinkers, C.H.; Carrillo-Roa, T.; Hiemstra, M.; van Lier, P.A.; Meeus, W.; Branje, S.; Heim, C.M.; Nemeroff, C.B.; Mill, J.; et al. Genome-wide DNA methylation levels and altered cortisol stress reactivity following childhood trauma in humans. Nat. Commun. 2016, 7, 10967. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
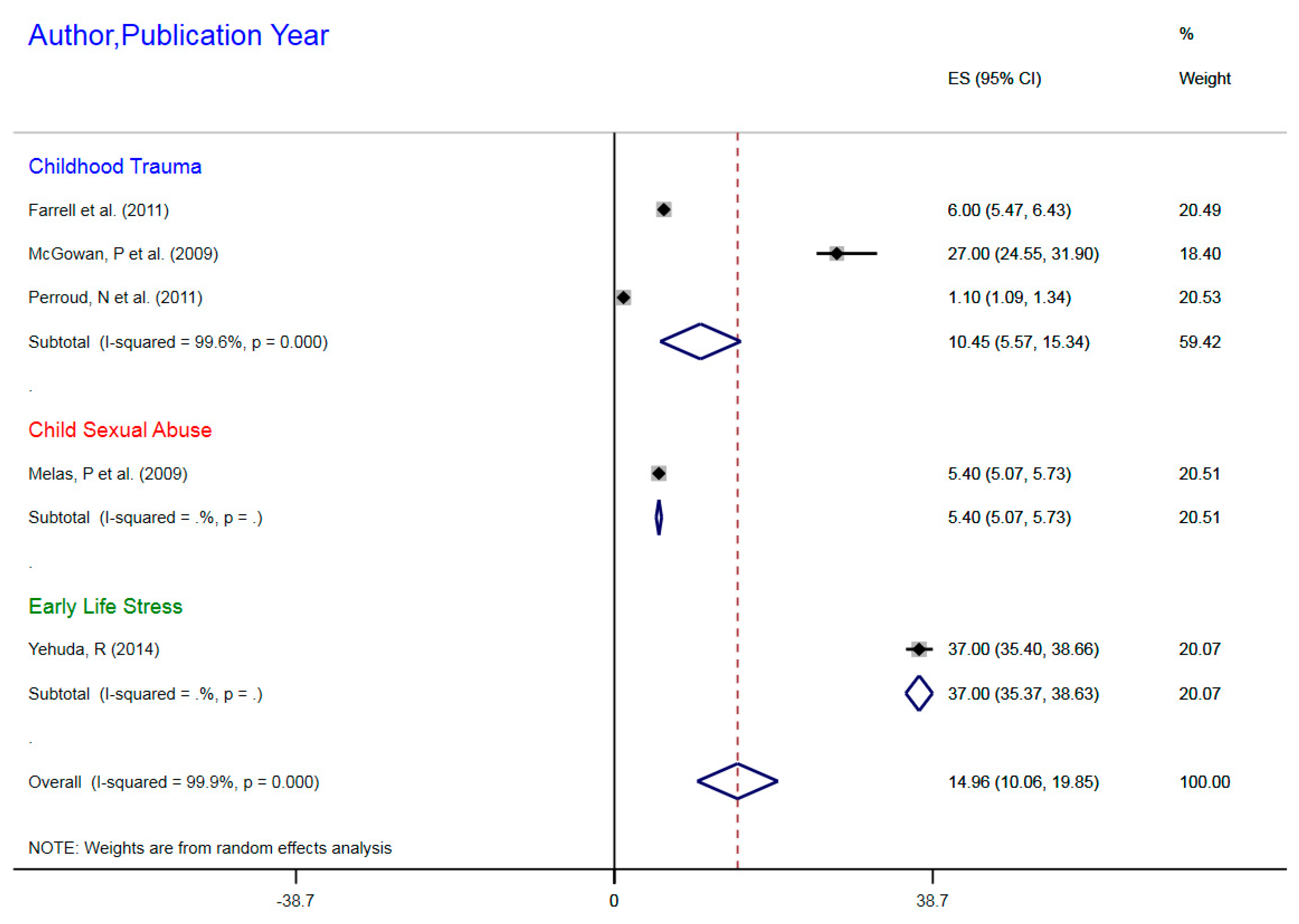
| Author, Year | Exposure | Outcomes | Sample Size | Epigenetic Mechanism | Gene | Gene Region | % Change (MI) | 95%CI |
|---|---|---|---|---|---|---|---|---|
| Farrell et al. (2011) | Childhood trauma | MDD | 77 | mDNA | NR3C1 | 1F Exon Promoter | 6% | 5.47–6.43 |
| McGowan, P et al. (2009) | Childhood trauma | MDD | 36 | mDNA | NR3CI | 1F Exon Promoter | 27% | 24.55–31.90 |
| Melas, P et al. (2009) | Childhood sexual abuse | MDD | 176 | mDNA | NR3C1 | 1F Exon Promoter | 5.4% | 5.07–5.73 |
| Perroud, N et al. (2011) | Childhood trauma | MDD | 200 | mDNA | NR3C1 | 1F Exon Promoter | 1.1% | 1.09–1.34 |
| Yehuda, R (2014) | ELS due to parental PTSD | MDD | 42 | mDNA | NR3C1 | 1F Exon Promoter | 37% | 35.4–38.66 |
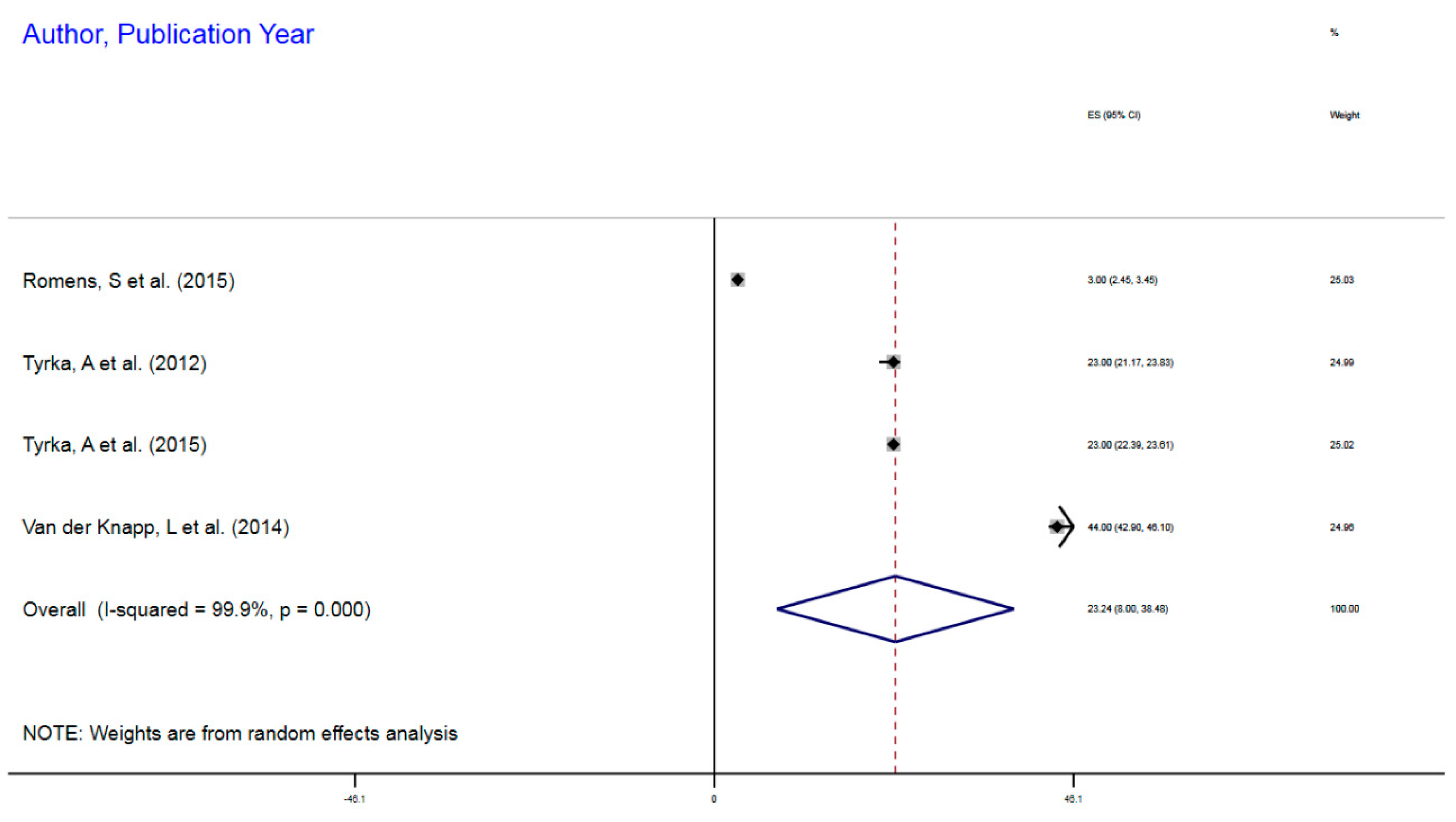
| Author, Year | Exposure | Sample Size | Epigenetic Mechanism | Gene | Gene Region | % Change (MI) | 95%CI |
|---|---|---|---|---|---|---|---|
| Romens, S et al. (2015) | Childhood trauma | 56 | mDNA | NR3C1 | 1F Exon Promoter | 3% | 2.45–3.45 |
| Tyrka, A et al. (2012) | Childhood trauma | 99 | mDNA | NR3C1 | 1F Exon Promoter | 23% | 21.17–23.83 |
| Tyrka, A et al. (2015) | Childhood sexual abuse | 184 | mDNA | NR3C1 | 1F Exon Promoter | 23% | 22.39–23.61 |
| van der Knapp, L et al. (2014) | Childhood trauma | 22 | mDNA | NR3C1 | 1F Exon Promoter | 44% | 42.9–46.1 |
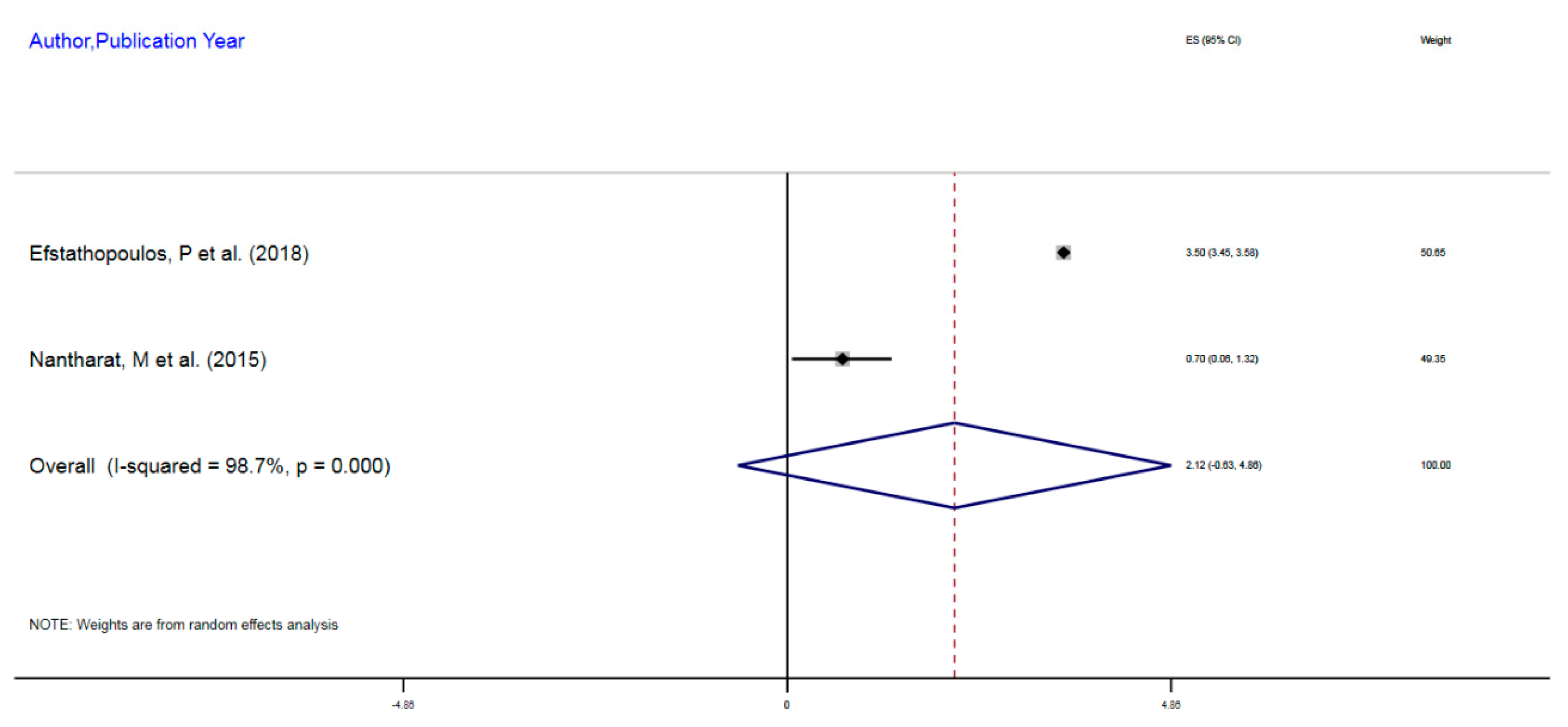
| Author, Year | Exposure | Sample Size | Epigenetic Mechanism | Gene | Region on Gene | % Change (MI) | 95%CI |
|---|---|---|---|---|---|---|---|
| Efstathopoulos, P et al. (2018) | Adult MDD | 580 | mDNA | NR3C1 | 1F Exon Promoter | 3.5% | 3.45–3.55 |
| Nantharat, M et al. (2015) | Adult MDD | 62 | mDNA | NR3CI | 1F Exon Promoter | 0.7% | 0.06–1.32 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmes, L., Jr.; Shutman, E.; Chinaka, C.; Deepika, K.; Pelaez, L.; Dabney, K.W. Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis. Int. J. Environ. Res. Public Health 2019, 16, 4280. https://doi.org/10.3390/ijerph16214280
Holmes L Jr., Shutman E, Chinaka C, Deepika K, Pelaez L, Dabney KW. Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis. International Journal of Environmental Research and Public Health. 2019; 16(21):4280. https://doi.org/10.3390/ijerph16214280
Chicago/Turabian StyleHolmes, Laurens, Jr., Emily Shutman, Chinacherem Chinaka, Kerti Deepika, Lavisha Pelaez, and Kirk W. Dabney. 2019. "Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis" International Journal of Environmental Research and Public Health 16, no. 21: 4280. https://doi.org/10.3390/ijerph16214280
APA StyleHolmes, L., Jr., Shutman, E., Chinaka, C., Deepika, K., Pelaez, L., & Dabney, K. W. (2019). Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis. International Journal of Environmental Research and Public Health, 16(21), 4280. https://doi.org/10.3390/ijerph16214280