Evaluation of the Human Interference on the Microbial Diversity of Poyang Lake Using High-Throughput Sequencing Analyses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites
2.2. Sampling
2.3. Extraction of Genome DNA and High-Throughput Sequencing
2.4. Statistical Analysis
3. Results
3.1. Compositions and Relative Abundance of Bacterial Communities in Water Samples
3.2. Taxa Abundance of Bacterial Species and KEGG Analysis in Water Samples
3.3. Compositions and Relative Abundance of Bacterial Communities in Sludge Samples
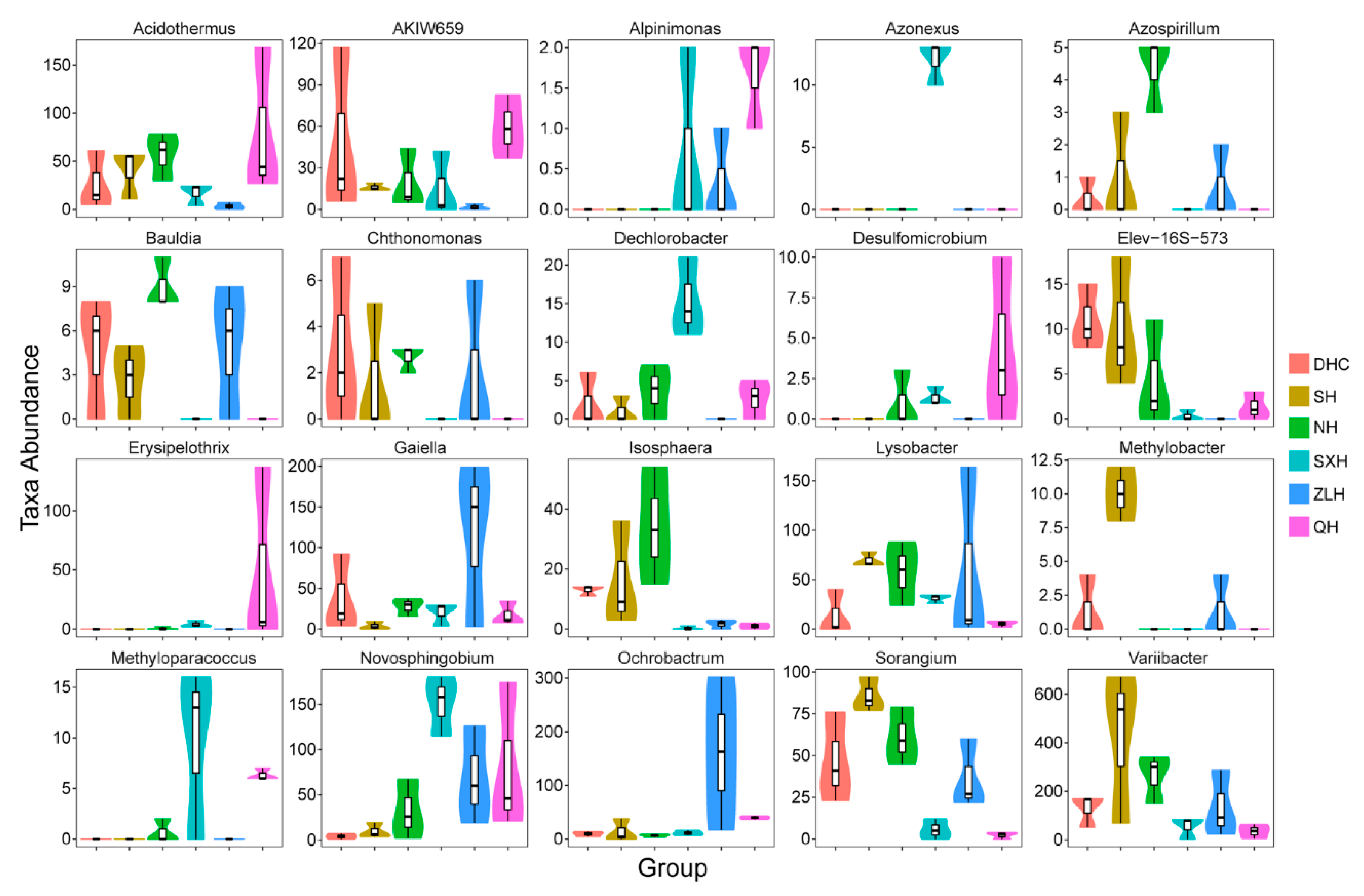
3.4. Taxa Abundance of Bacterial Species in Sludge Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Huang, X.; Hu, B.; Wang, P.; Chen, X.; Xu, B. Microbial diversity in lake–river ecotone of Poyang Lake, China. Environ. Earth Sci. 2016, 75. [Google Scholar] [CrossRef]
- Chandrajith, R.L.R.; Okumura, M.; Hashitani, H. Human influence on the Hg pollution in Lake Jinzai. Appl. Geochem. 1995, 10, 229–235. [Google Scholar] [CrossRef]
- Wu, L.; Ge, G.; Zhu, G.; Gong, S.; Li, S.; Wan, J. Diversity and composition of the bacterial community of Poyang Lake (China) as determined by 16S rRNA gene sequence analysis. World J. Microbiol. Biotechnol. 2012, 28, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.F.; Tao, C.Y.; Huang, Y. Study on Control Countermeasures of Agricultural Non-point Source Pollution in Lakeside Belt of Poyang Lake—Taking Duchang Section in the Lower Reaches of Poyang Lake as Example. Meteorol. Environ. Res. 2011, 2, 62–65. [Google Scholar]
- Quan, W.; Yan, L. Effects of Agricultural Non-point Source Pollution on Eutrophica tion of Water Body and Its Control Measure. Acta Ecol. Sin. 2002, 22, 291–299. [Google Scholar]
- Nilsson, C.; Reidy, C.A.; Dynesius, M.; Revenga, C. Fragmentation and flow regulation of the world’s large river systems. Science 2005, 308, 405–408. [Google Scholar] [CrossRef]
- Gutknecht, J.L.M.; Goodman, R.M.; Balser, T.C. Linking soil process and microbial ecology in freshwater wetland ecosystems. Plant Soil 2006, 289, 17–34. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.L.; Liu, B.G.; Qian, K.M.; Chen, Y.W.; Gao, J.F. Cyanobacteria in the complex river-connected Poyang Lake: Horizontal distribution and transport. Hydrobiologia 2016, 768, 95–110. [Google Scholar] [CrossRef]
- Lin, Y.R.; Du, Y.P.; Yang, H.M. Analysis of wetland ecosystem response of Poyang Lake with water level evolution base on TM images. Appl. Mech. Mater. 2013, 295–298, 1935–1940. [Google Scholar] [CrossRef]
- Mei, X.; Dai, Z.; Fagherazzi, S.; Chen, J. Dramatic variations in emergent wetland area in China’s largest freshwater lake, Poyang Lake. Adv. Water Resour. 2016, 96, 1–10. [Google Scholar] [CrossRef]
- Zhang, B.; Kang, X. The features of the natural resources and the renovation strategy of Poyang Lake. Chin. Geogr. Sci. 1994, 4, 352–370. [Google Scholar] [CrossRef]
- Shao, M.; Jiang, J.; Guo, H.; Zeng, B. Abundance, distribution and diversity variations of wintering water birds in Poyang Lake, Jiangxi Province, China. Pakistan J. Zool 2014, 46, 451–462. [Google Scholar]
- Venosa, A.D.; Lee, K.; Suidan, M.T.; Garcia-Blanco, S.; Cobanli, S.; Moteleb, M.; Haines, J.R.; Tremblay, G.; Hazelwood, M. Bioremediation and Biorestoration of a Crube Oil-Contaminated Freshwater Wetland on the St.Lawrence River. Biorem. J. 2002, 6, 261–281. [Google Scholar] [CrossRef]
- Lew, S.; Lew, M.; Mieszczyński, T.; Szarek, J. Selected fluorescent techniques for identification of the physiological state of individual water and soil bacterial cells—Review. Folia Microbiol. 2010, 55, 107–118. [Google Scholar] [CrossRef]
- Lew, S.; Glińska-Lewczuk, K.; Burandt, P.; Obolewski, K.; Goździejewska, A.; Lew, M.; Dunalska, J. Impact of environmental factors on bacterial communities in floodplain lakes differed by hydrological connectivity. Limnologica 2016, 58, 20–29. [Google Scholar] [CrossRef]
- Ren, Z.; Gao, H. Ecological networks reveal contrasting patterns of bacterial and fungal communities in glacier-fed streams in Central Asia. PeerJ 2019, 7, e7715. [Google Scholar] [CrossRef]
- Kou, W.; Zhang, J.; Lu, X.; Ma, Y.; Mou, X.; Wu, L. Identification of bacterial communities in sediments of Poyang Lake, the largest freshwater lake in China. SpringerPlus 2016, 5, 401. [Google Scholar] [CrossRef]
- Besemer, K.; Moeseneder, M.M.; Arrieta, J.M.; Herndl, G.J.; Peter, P. Complexity of bacterial communities in a river-floodplain system (Danube, Austria). Microb. Ecol. 2005, 71, 609–620. [Google Scholar] [CrossRef]
- Yu, X.; Wu, X.; Qiu, X.; Wang, D.; Gan, M.; Chen, X.; Wei, H.; Xu, F. Analysis of the intestinal microbial community structure of healthy and long-living elderly residents in Gaotian Village of Liuyang City. Appl. Microbiol. Biotechnol. 2015, 99, 9085–9095. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Meng, F.; Chen, T.; Wang, X.; Wang, X.; Wei, H.; Tian, P.; Wang, H.; Zhao, X.; Shen, L.; Xin, H. Evaluation of the accuracy and sensitivity of high-throughput sequencing technology using known microbiota. Mol. Med. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cao, X.; Peng, J. Analyzing the 2007 drought of Poyang Lake Watershed with MODIS-derived Normalized Difference Water Deviation Index. Remote Sens. Model. Ecosyst. Sustainability V 2008, 7083, 70831G. [Google Scholar]
- Yan, H.; Zhan, J.; Jiang, Q. Scenario simulation of changes of forest land in Poyang Lake watershed. Procedia Environ. Sci. 2010, 2, 1469–1478. [Google Scholar]
- Shao, M.; Zeng, B.; Tim, H.; Chen, L.; You, C.; Wang, H.; Dai, N. Winter ecology and conservation threats of scaly-sided Merganser Mergus squamatus in Poyang Lake watershed, China. Pakistan J. Zool. 2012, 44, 503–510. [Google Scholar]
- Sánchez, E.; Colmenarejo, M.F.; Vicente, J.; Rubio, A.; García, M.G.; Travieso, L.; Borja, R. Use of the water quality index and dissolved oxygen deficit as simple indicators of watersheds pollution. Ecol. Indic. 2007, 7, 315–328. [Google Scholar] [CrossRef]
- Carpenter, S.R.; Caraco, N.F.; Correll, D.L.; Howarth, R.W.; Sharpley, A.N.; Smith, V.H. Nonpoint pollution of surface waters with phosphorus and nitrogen. Ecol. Appl. 1998, 8, 559–568. [Google Scholar] [CrossRef]
- Brown, L.R.; Halweil, B.J. China’s water shortage could shake world food security. World Watch 1998, 11, 10. [Google Scholar]
- Ansola, G.; Arroyo, P.; de Miera, L.E.S. Characterisation of the soil bacterial community structure and composition of natural and constructed wetlands. Sci. Total Environ. 2014, 473–474, 63–71. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495. [Google Scholar] [CrossRef]
- Bullerjahn, G.S.; Post, A.F. Physiology and molecular biology of aquatic cyanobacteria. Front. Microbiol. 2014, 5, 359. [Google Scholar] [CrossRef]
- Achenbach, L.A.; Michaelidou, U.; Bruce, R.A.; Fryman, J.; Coates, J.D. Dechloromonas agitata gen. nov., sp. nov. and Dechlorosoma suillum gen. nov., sp. nov., two novel environmentally dominant (per)chlorate-reducing bacteria and their phylogenetic position. Int. J. Syst. Evol. Microbiol. 2001, 51, 527–533. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. The genus Aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hameed, A.; Arun, A.B.; Liu, Y.-C.; Hsu, Y.-H.; Lai, W.-A.; Rekha, P.D.; Young, C.-C. Description of Noviherbaspirillum malthae gen. nov. sp. nov. isolated from an oil contaminated soil, and proposal to reclassify Herbaspirillum soli, Herbaspirillum aurantiacum, Herbaspirillum canariense and Herbaspirillum psychrotolerans into the genus Nov. Int. J. Syst. Evol. Microbiol. 2013, 63, 4100–4107. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, Y.; Wang, Y.; Wu, Z.; Xie, S.; Liu, Y. Distribution of bacterial communities across plateau freshwater lake and upslope soils. J. Environ. Sci. 2016, 43, 61–69. [Google Scholar] [CrossRef]
- Miura, Y.; Watanabe, Y.; Okabe, S. Significance of Chloroflexi in performance of submerged membrane bioreactors (MBR) treating municipal wastewater. Environ. Sci. Technol. 2007, 41, 7787–7794. [Google Scholar] [CrossRef]
- Bryant, D.A.; Liu, Z.; Li, T.; Zhao, F.; Costas, A.M.G.; Klatt, C.G.; Ward, D.M.; Frigaard, N.-U.; Overmann, J. Comparative and Functional Genomics of Anoxygenic Green Bacteria from the Taxa Chlorobi, Chloroflexi, and Acidobacteria. Funct. Genomics Evol. Photosynth. 2012, 33, 47–102. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, H.; Cui, L.; Cao, X.; Lv, Q.; Chen, T. Evaluation of the Human Interference on the Microbial Diversity of Poyang Lake Using High-Throughput Sequencing Analyses. Int. J. Environ. Res. Public Health 2019, 16, 4218. https://doi.org/10.3390/ijerph16214218
Qin H, Cui L, Cao X, Lv Q, Chen T. Evaluation of the Human Interference on the Microbial Diversity of Poyang Lake Using High-Throughput Sequencing Analyses. International Journal of Environmental Research and Public Health. 2019; 16(21):4218. https://doi.org/10.3390/ijerph16214218
Chicago/Turabian StyleQin, Haiming, Lanyue Cui, Xinyi Cao, Qian Lv, and Tingtao Chen. 2019. "Evaluation of the Human Interference on the Microbial Diversity of Poyang Lake Using High-Throughput Sequencing Analyses" International Journal of Environmental Research and Public Health 16, no. 21: 4218. https://doi.org/10.3390/ijerph16214218
APA StyleQin, H., Cui, L., Cao, X., Lv, Q., & Chen, T. (2019). Evaluation of the Human Interference on the Microbial Diversity of Poyang Lake Using High-Throughput Sequencing Analyses. International Journal of Environmental Research and Public Health, 16(21), 4218. https://doi.org/10.3390/ijerph16214218