Neonicotinoid Insecticides Alter the Gene Expression Profile of Neuron-Enriched Cultures from Neonatal Rat Cerebellum

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Ethics Statement
2.3. Neuron-Enriched Cultures of Neonatal Rat Cerebellum
2.4. RNA Preparation
2.5. Microarray
2.6. Data Normalization and Quality Control
2.7. Differential Expression Analysis and Statistics
2.8. Quantitative Real-Time PCR (qRT-PCR)
2.9. Gene Ontology and Bioinformatics Analysis
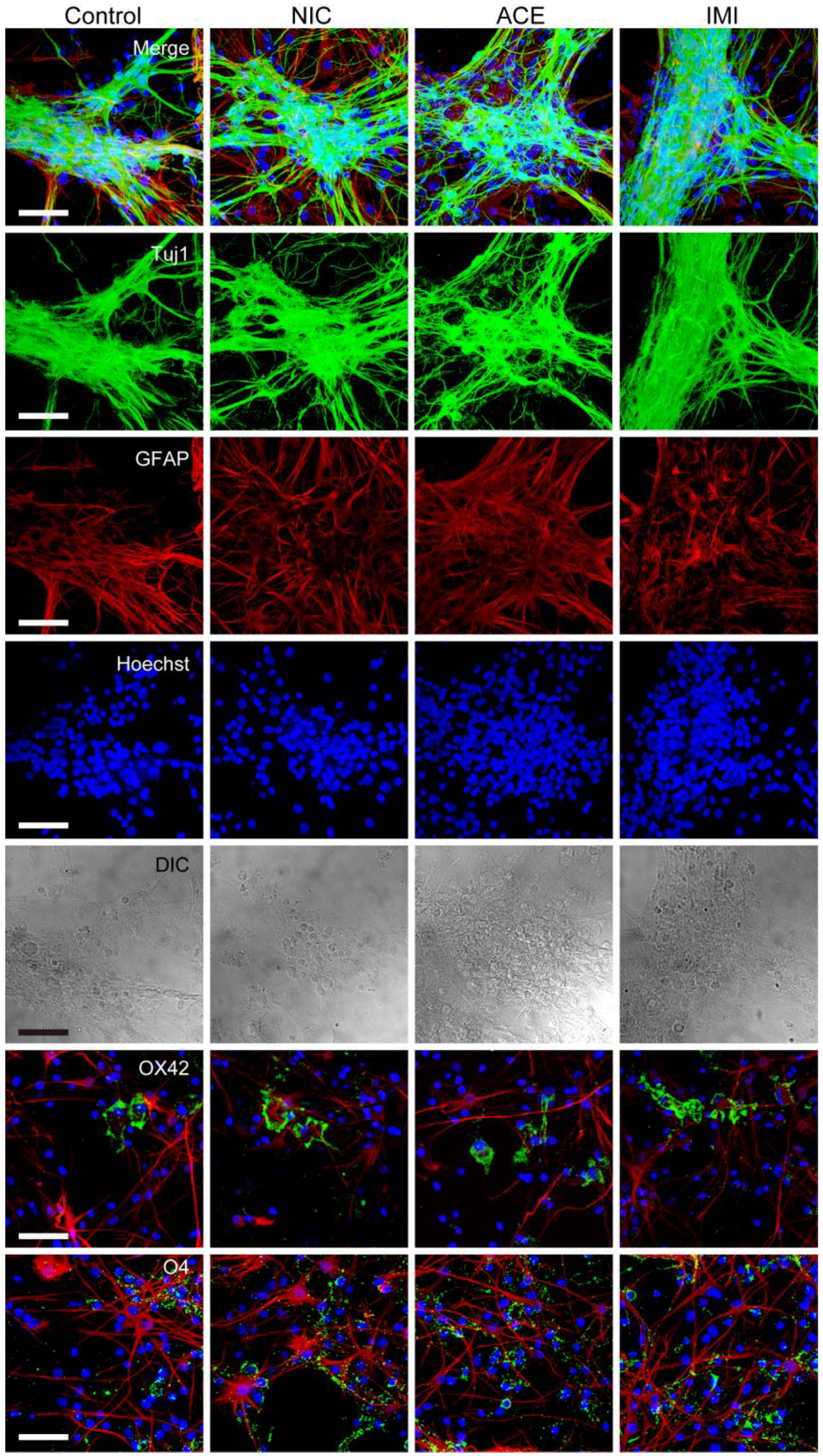
2.10. Immunocytochemistry
3. Results
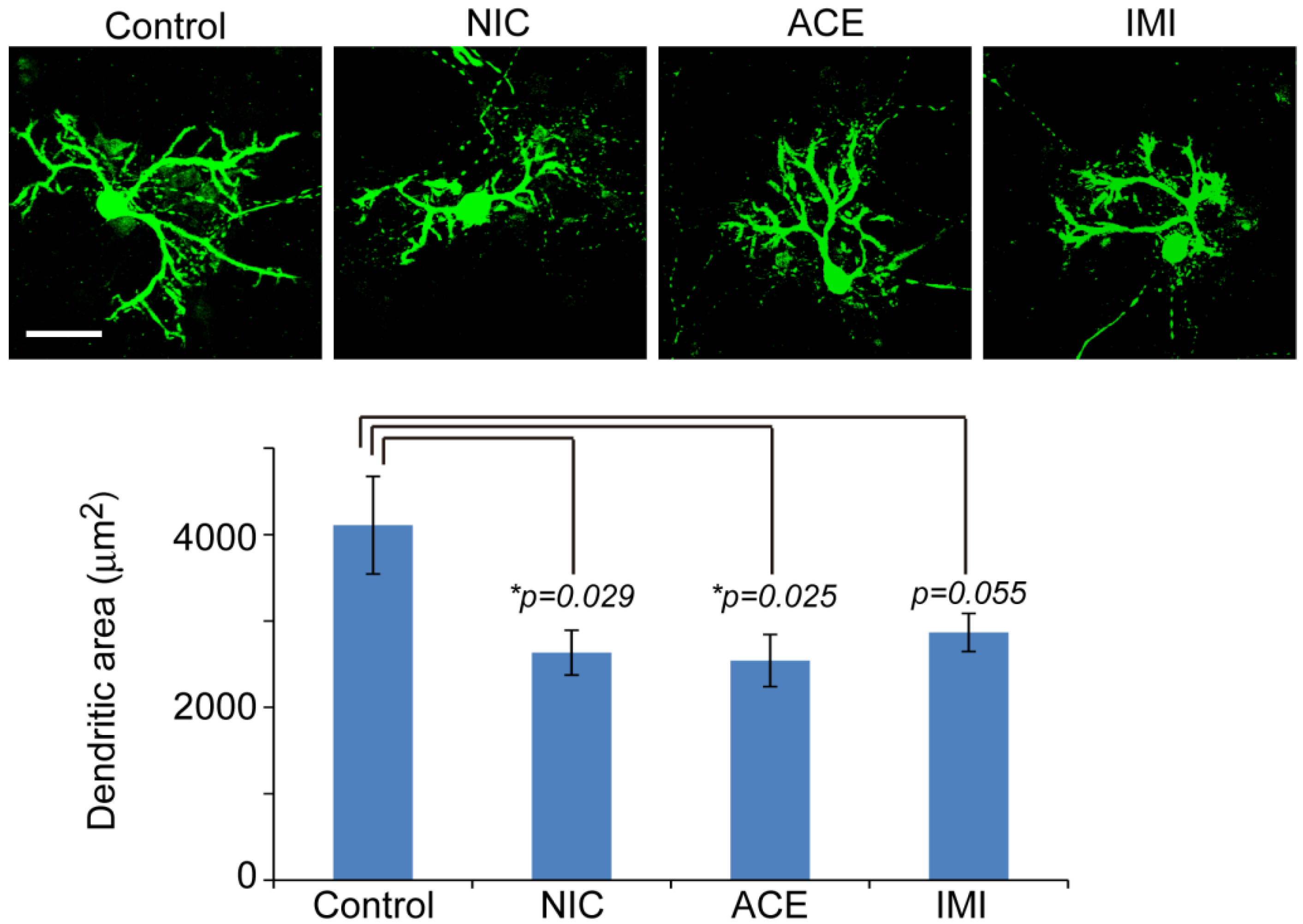
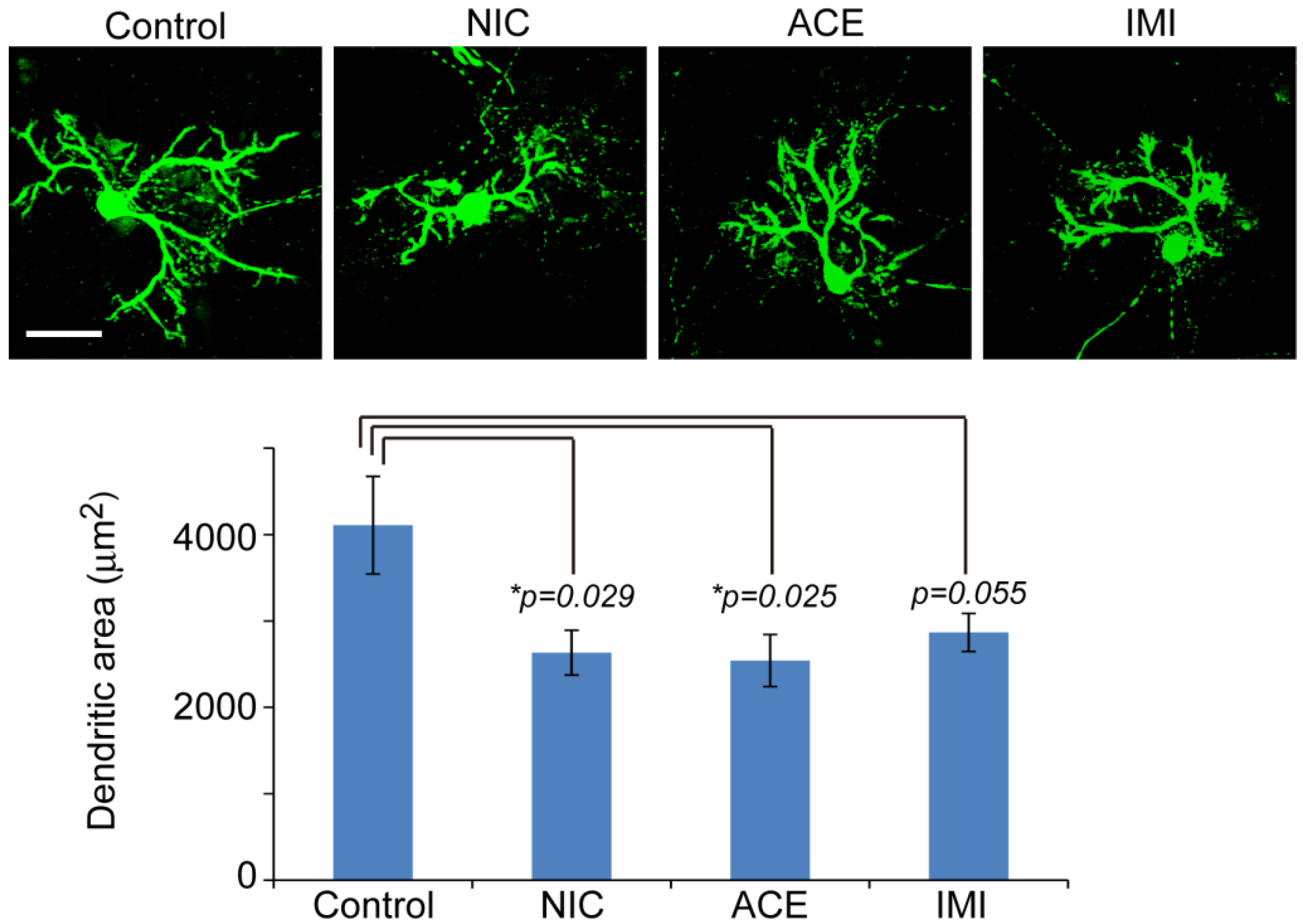
3.1. Morphological Characterics of 16 DIV Cerebellar Neuron-Enriched Cultures
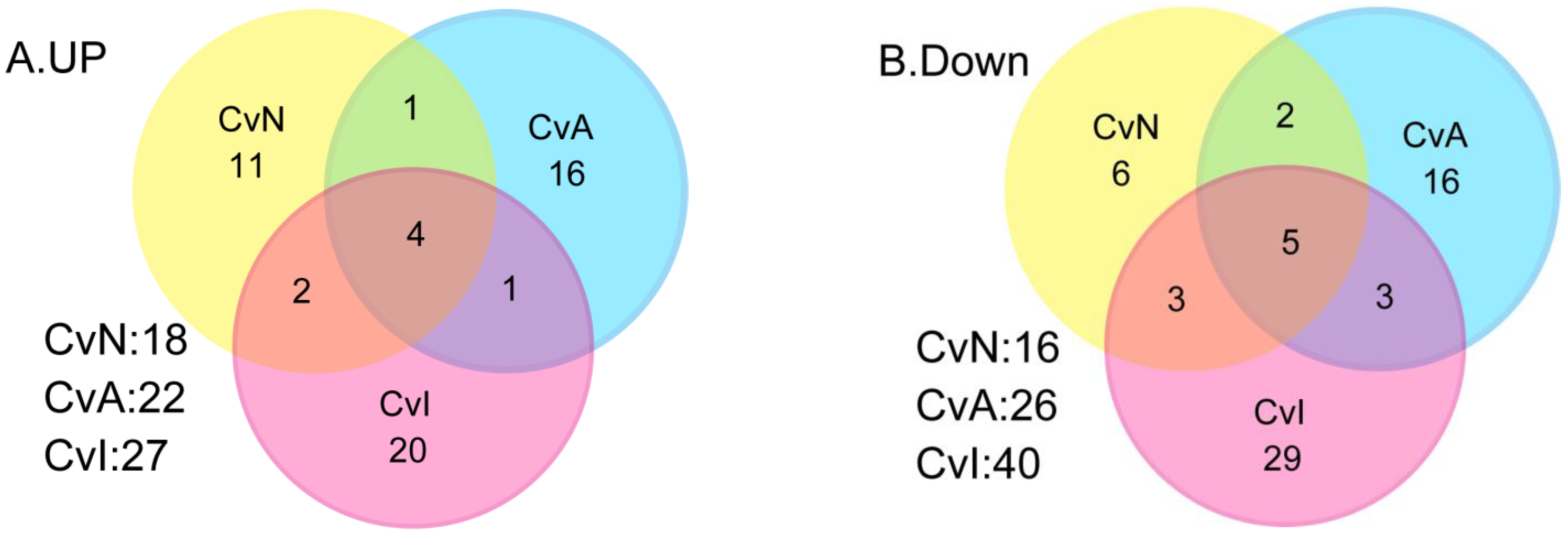
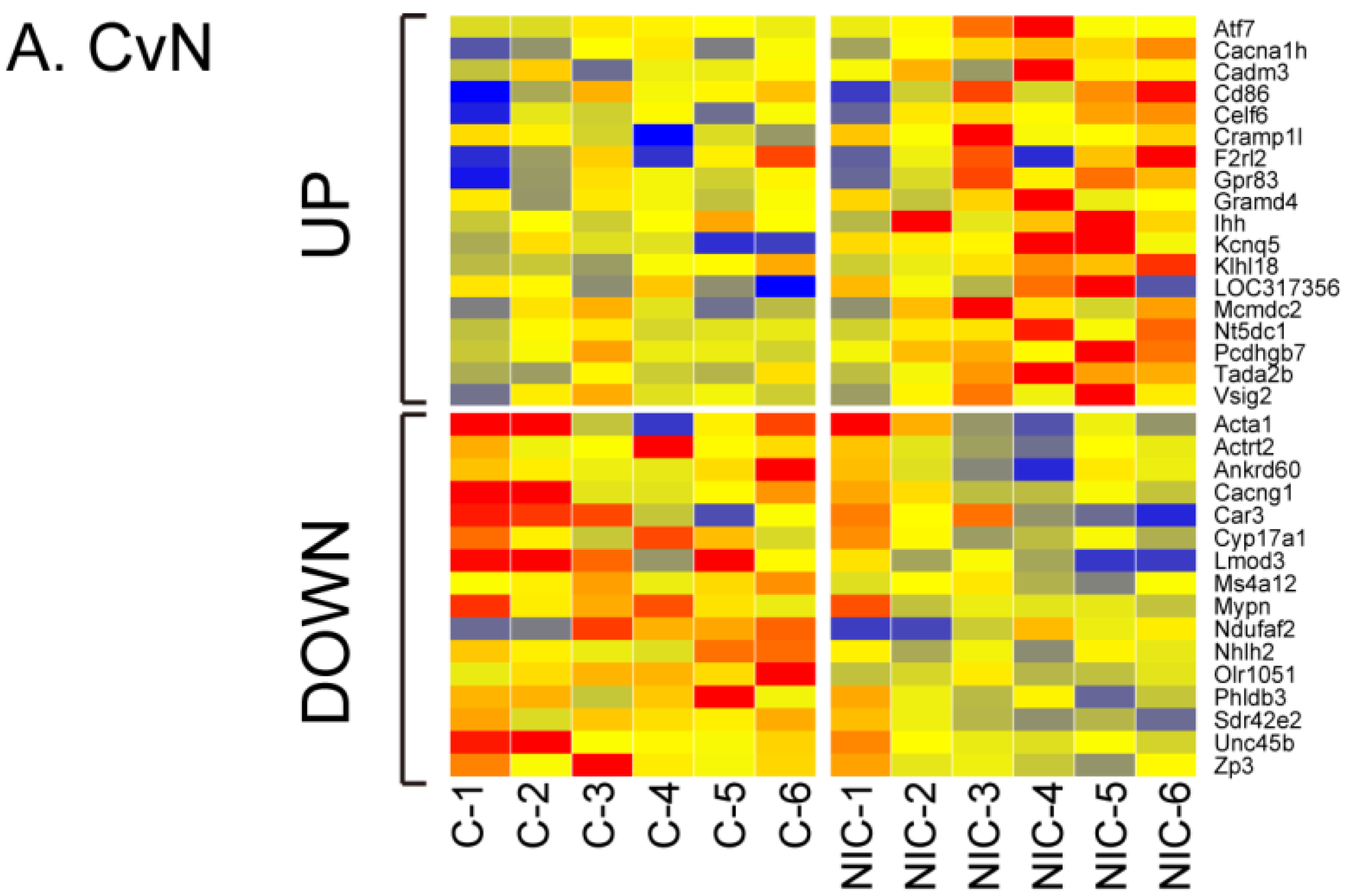
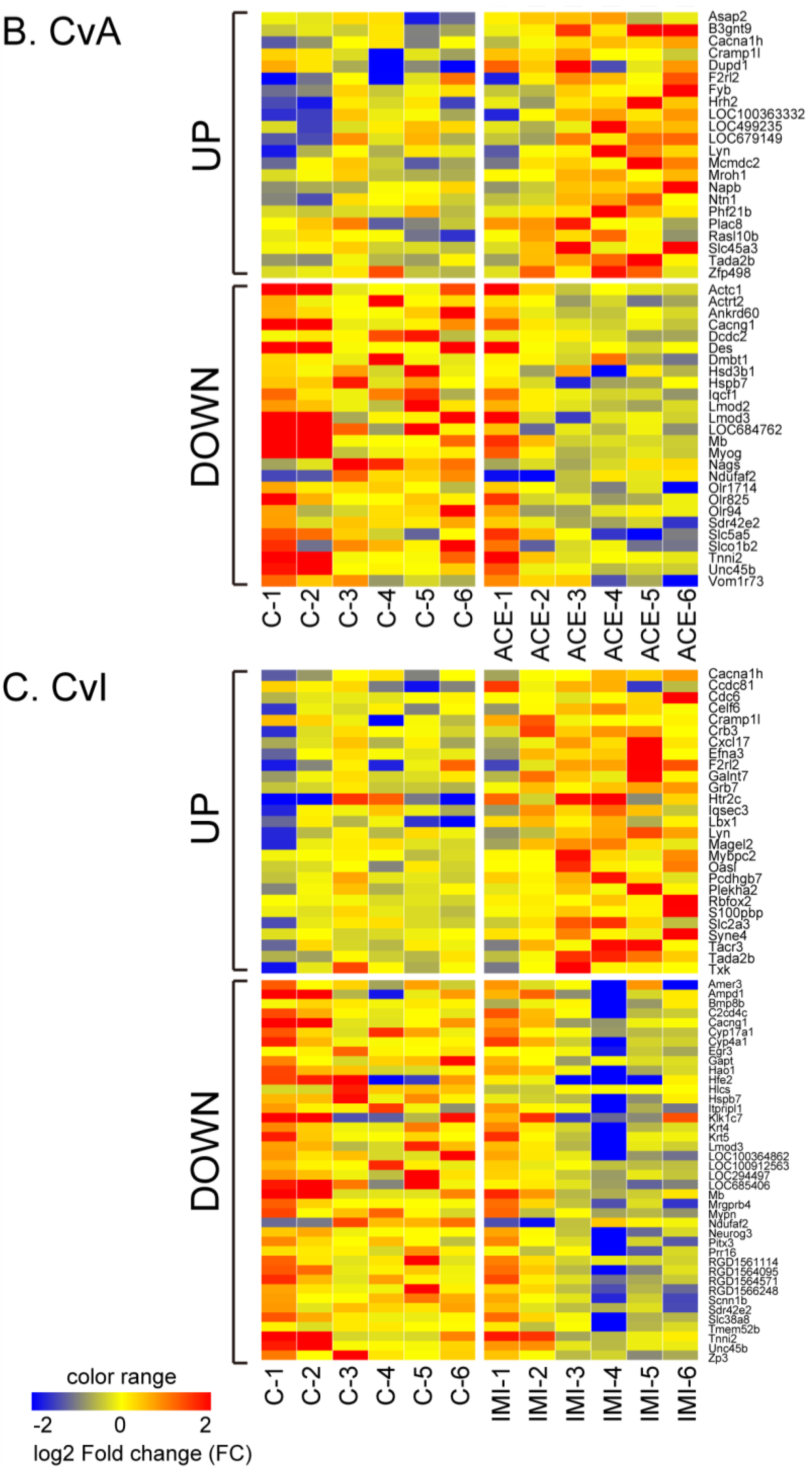
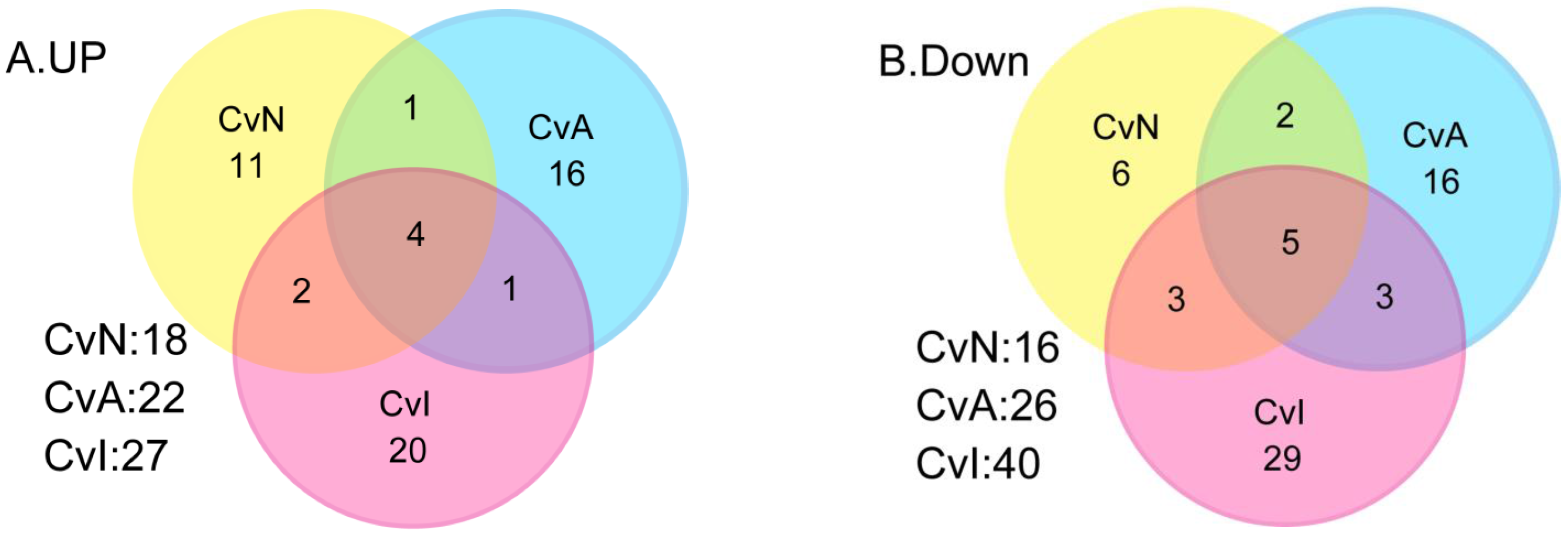
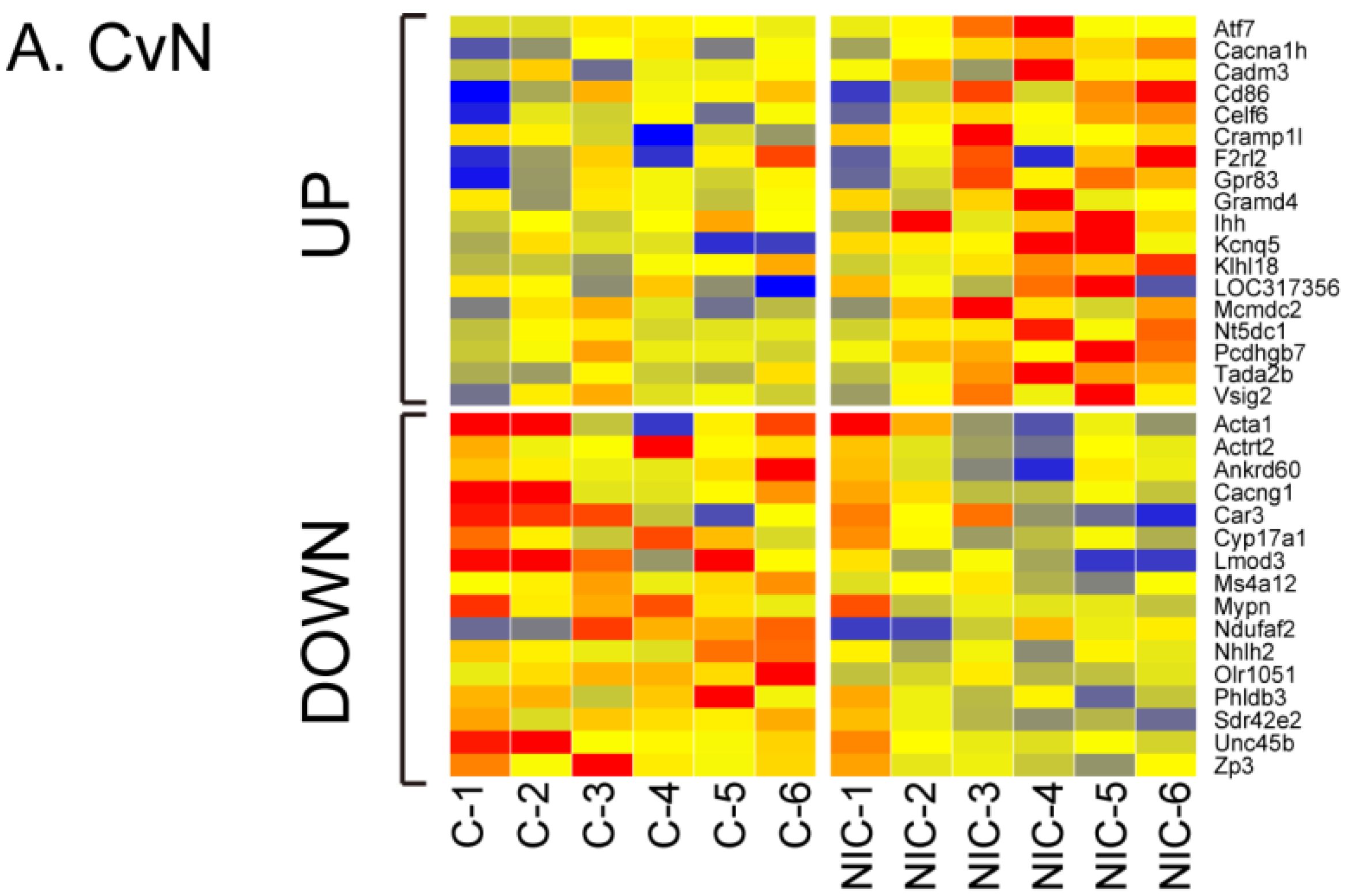
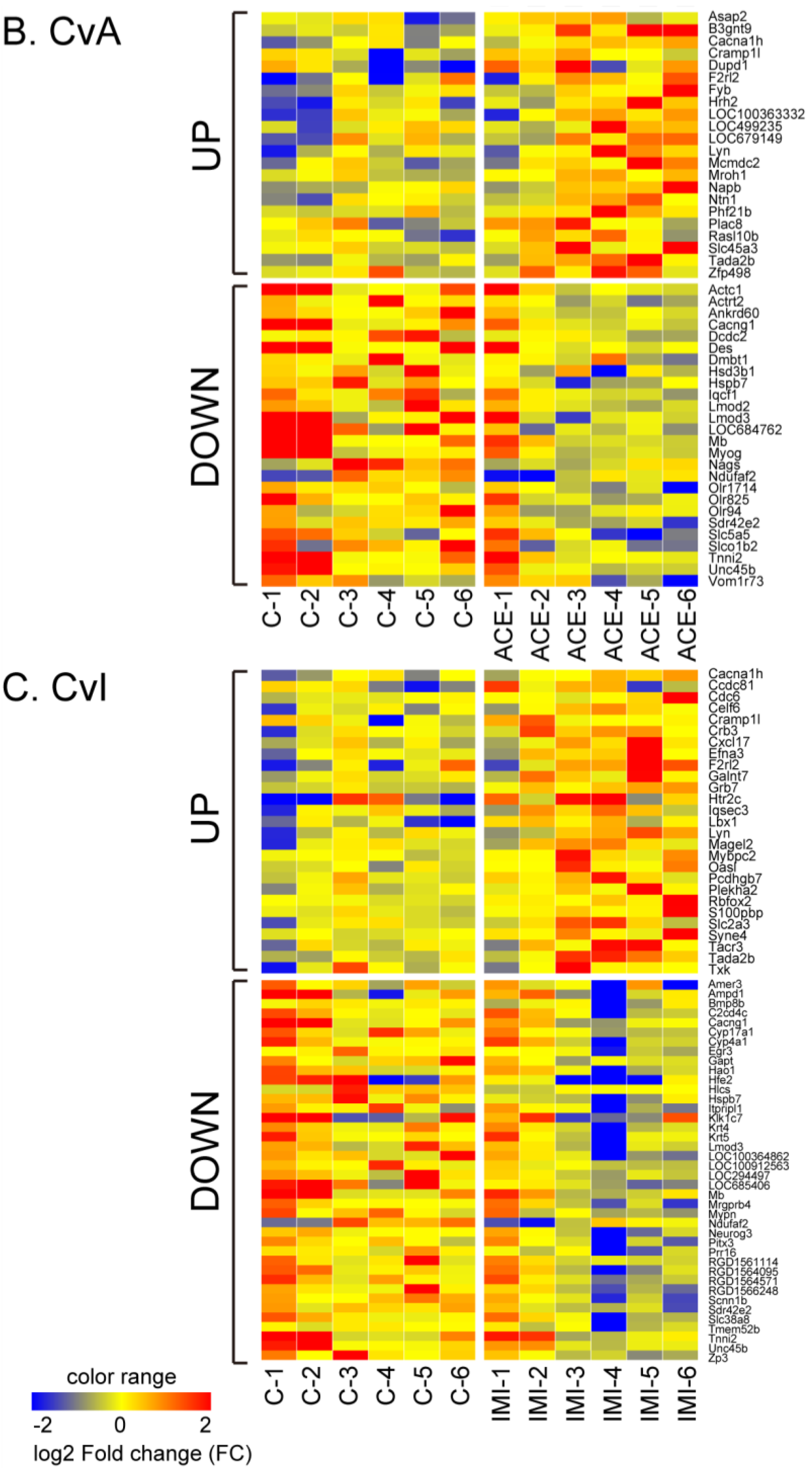
3.2. Differential Gene Expression after Exposure to NIC, ACE and IMI
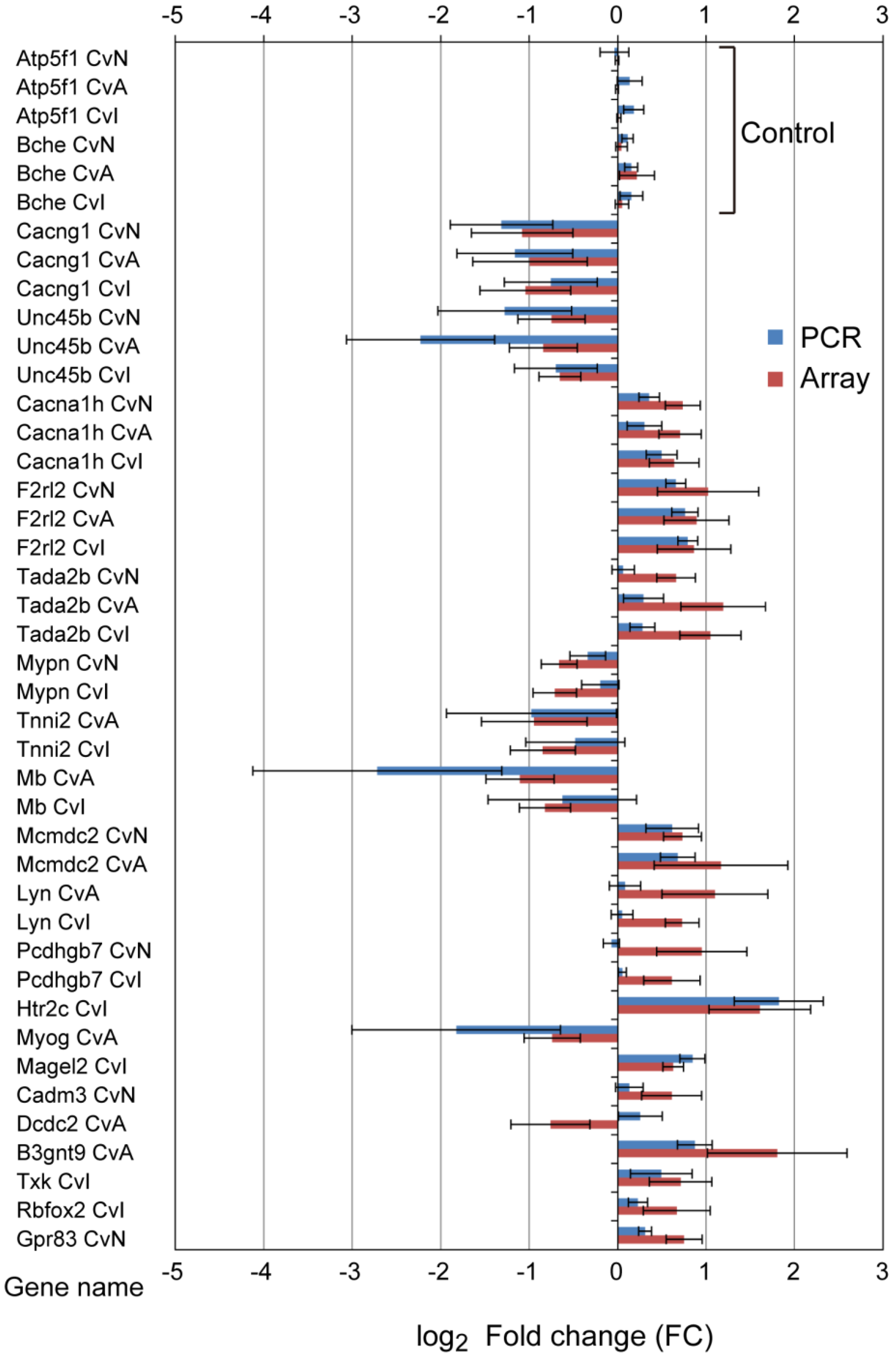
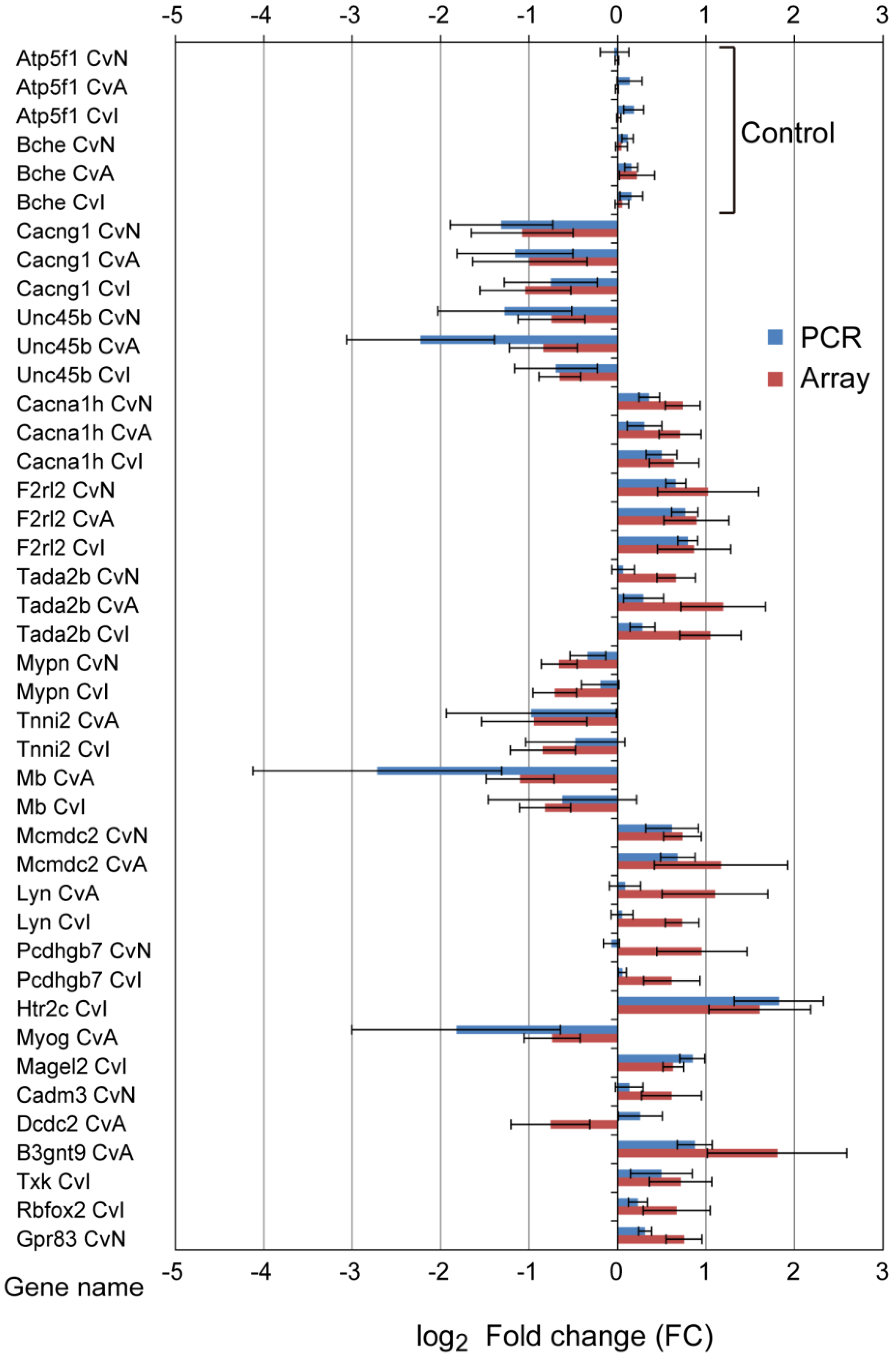
3.3. Quantitative qRT-PCR Validation
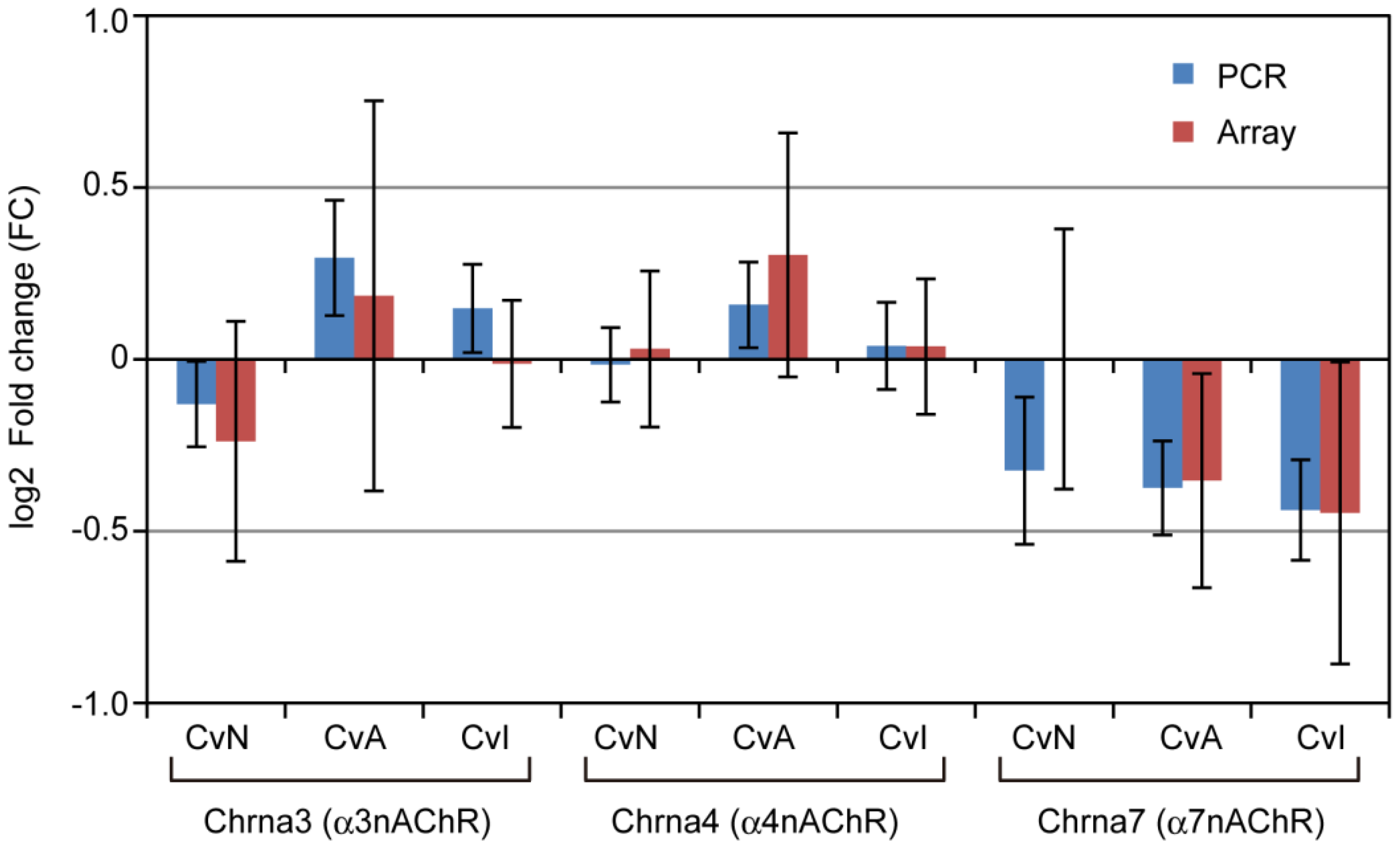
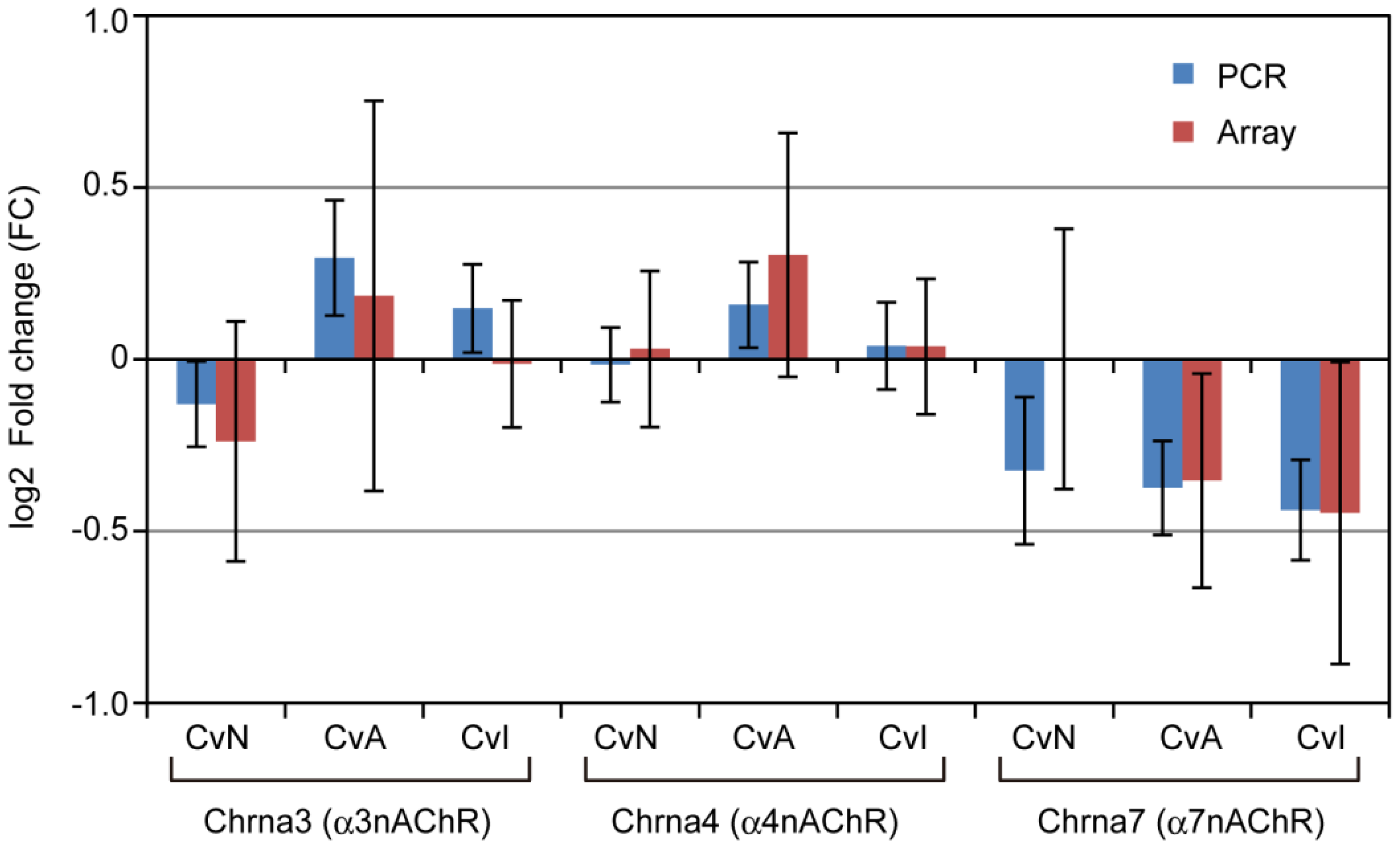
3.4. nAChRs Expressions
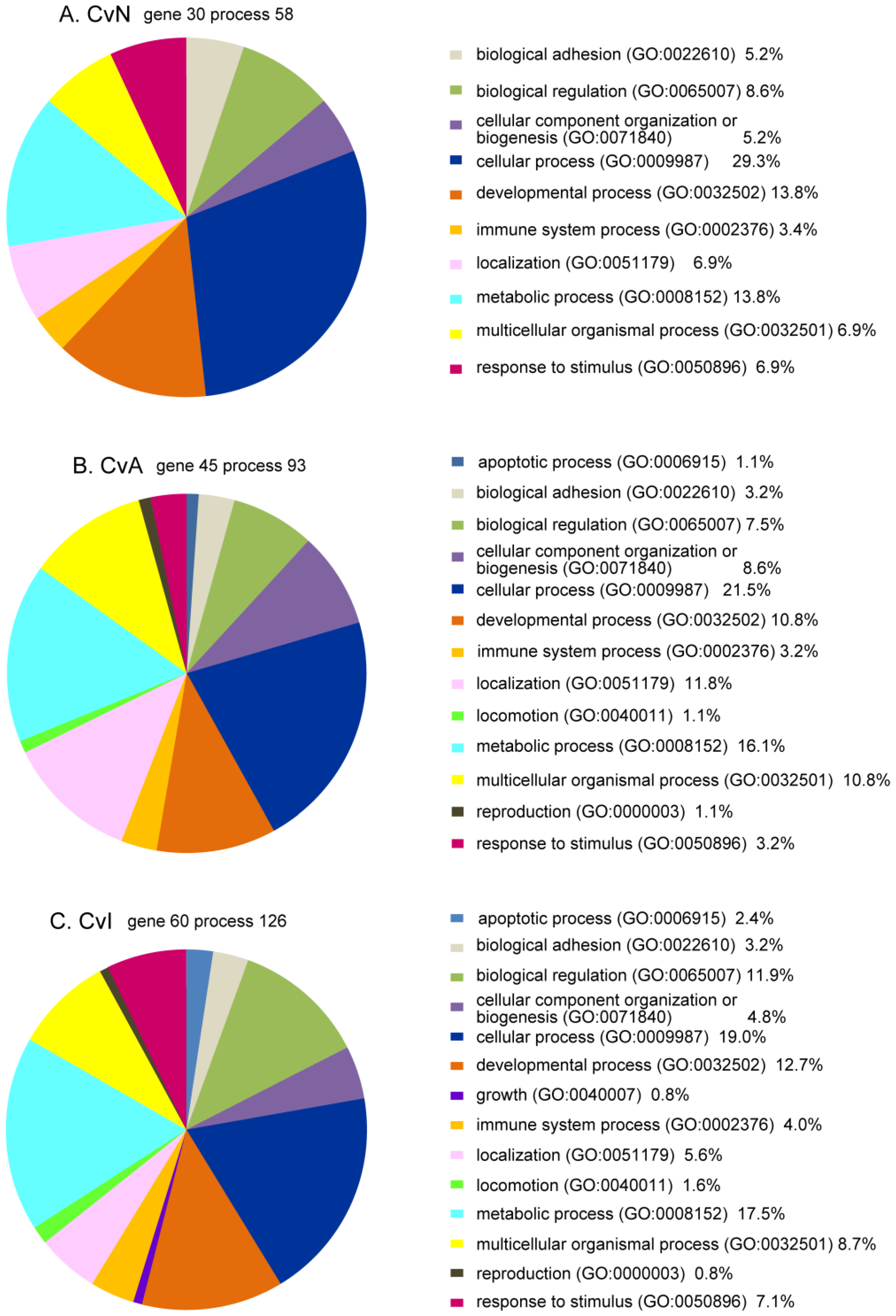
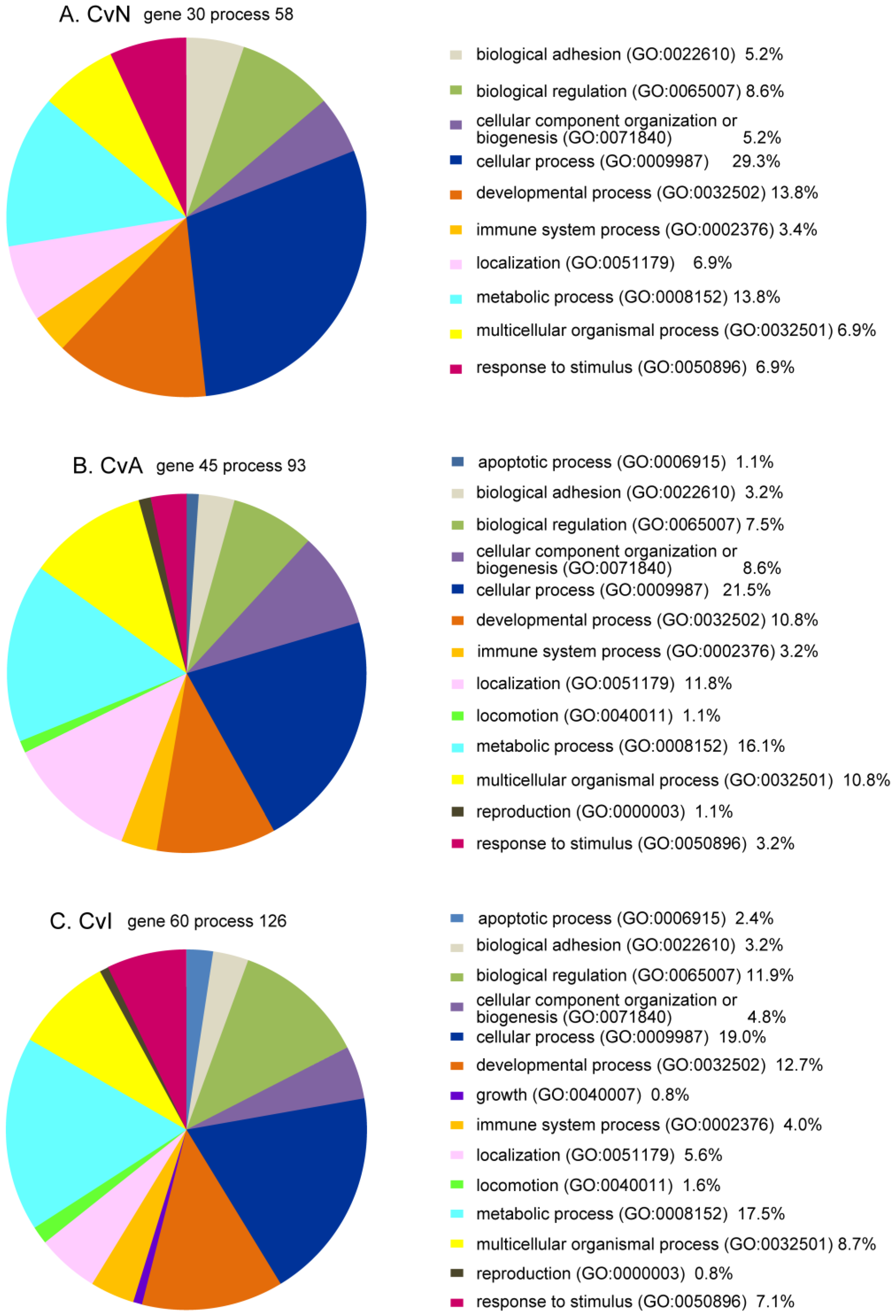
3.5. Bioinformatics Analysis of DE Genes for CvN, CvA, and CvI
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| NIC | nicotine |
| IMI | imidacloprid |
| ACE | acetamiprid |
| nAChR | nicotinic acetylcholine receptor |
| ACh | acetylcholine |
| BBB | blood brain barrier |
| GFAP | glial fibrillary acidic protein |
| qRT-PCR | quantitative real-time PCR |
| DIV | days in vitro |
| DE | differentially expressed |
| FC | fold change |
| GO | gene ontology |
| FDR | false discovery rate |
| CvN | control versus NIC |
| CvA | control versus ACE |
| CvI | control versus IMI |
| GPCR | G-protein coupled receptor |
References
- Simon-Delso, N.; Amaral-Rogers, V.; Belzunces, L.P.; Bonmatin, J.M.; Chagnon, M.; Downs, C.; Furlan, L.; Gibbons, D.W.; Giorio, C.; Girolami, V.; et al. Systemic insecticides (neonicotinoids and fipronil): Trends, uses, mode of action and metabolites. Environ. Sci. Pollut. Res. Int. 2015, 22, 5–34. [Google Scholar] [CrossRef] [PubMed]
- Chagnon, M.; Kreutzweiser, D.; Mitchell, E.A.; Morrissey, C.A.; Noome, D.A.; Van der Sluijs, J.P. Risks of large-scale use of systemic insecticides to ecosystem functioning and services. Environ. Sci. Pollut. Res. Int. 2015, 22, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Casida, J.E. Minor structural changes in nicotinoid insecticides confer differential subtype selectivity for mammalian nicotinic acetylcholine receptors. Br. J. Pharmacol. 1999, 127, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Casida, J.E. Neonicotinoid insecticide toxicology: Mechanisms of selective action. Annu Rev. Pharmacol. Toxicol. 2005, 45, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.C.; Lin, H.J.; Liao, Y.Y.; Guo, H.R.; Chen, K.T. Acute Poisoning with Neonicotinoid Insecticides: A Case Report and Literature Review. Basic Clin. Pharmacol. Toxicol. 2013, 112, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Shadnia, S.; Moghaddam, H.H. Fatal intoxication with imidacloprid insecticide. Am. J. Emerg. Med. 2008, 26, 634.e1–634.e4. [Google Scholar] [CrossRef] [PubMed]
- Vinod, K.V.; Srikant, S.; Thiruvikramaprakash, G.; Dutta, T.K. A fatal case of thiacloprid poisoning. Am. J. Emerg. Med. 2015, 33, 310.e5–310.e6. [Google Scholar] [CrossRef] [PubMed]
- Marfo, J.T.; Fujioka, K.; Ikenaka, Y.; Nakayama, S.M.; Mizukawa, H.; Aoyama, Y.; Ishizuka, M.; Taira, K. Relationship between Urinary N-Desmethyl-Acetamiprid and Typical Symptoms including Neurological Findings: A Prevalence Case-Control Study. PLoS ONE 2015, 10, e0142172. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ann, J.; Akk, G. Activation and modulation of human α4β2 nicotinic acetylcholine receptors by the neonicotinoids clothianidin and imidacloprid. J. Neurosci. Res. 2011, 89, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Cimino, A.M.; Boyles, A.L.; Thayer, K.A.; Perry, M.J. Effects of Neonicotinoid Pesticide Exposure on Human Health: A Systematic Review. Environ. Health Perspect. 2016. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Goldstein, L.B.; Bullman, S.; Tu, T.; Khan, W.A.; Dechkovskaia, A.M.; Abdel-Rahman, A.A. Imidacloprid induces neurobehavioral deficits and increases expression of glial fibrillary acidic protein in the motor cortex and hippocampus in offspring rats following in utero exposure. J. Toxicol. Environ. Health A 2008, 71, 119–130. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, I.M.; Nunes, B.V.; Barbosa, D.R.; Pallares, A.M.; Faro, L.R. Effects of the neonicotinoids thiametoxam and clothianidin on in vivo dopamine release in rat striatum. Toxicol. Lett. 2010, 192, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.J.; Santana, M.B.; Do Nascimento, J.L.; Picanco-Diniz, D.L.; Maues, L.A.; Santos, S.N.; Ferreira, V.M.; Alfonso, M.; Duran, R.; Faro, L.R. Behavioral and biochemical effects of neonicotinoid thiamethoxam on the cholinergic system in rats. Ecotoxicol. Environ. Saf. 2010, 73, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Effects of maternal clothianidin exposure on behavioral development in F(1) generation mice. Toxicol. Ind. Health 2012, 28, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yanai, S.; Omotehara, T.; Hashimoto, R.; Umemura, Y.; Kubota, N.; Minami, K.; Nagahara, D.; Matsuo, E.; Aihara, Y.; et al. The combined effect of clothianidin and environmental stress on the behavioral and reproductive function in male mice. J. Vet. Med. Sci. 2015, 77, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Kara, M.; Yumrutas, O.; Demir, C.F.; Ozdemir, H.H.; Bozgeyik, I.; Coskun, S.; Eraslan, E.; Bal, R. Insecticide imidacloprid influences cognitive functions and alters learning performance and related gene expression in a rat model. Int. J. Exp. Pathol. 2015, 96, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Isobe, T.; Yang, J.; Win-Shwe, T.-T.; Yoshikane, M.; Nakayama, S.F.; Kawashima, T.; Suzuki, G.; Hashimoto, S.; Nohara, K.; et al. In utero and Lactational Exposure to Acetamiprid Induces Abnormalities in Socio-Sexual and Anxiety-Related Behaviors of Male Mice. Front. Neurosci 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.A.; Casida, J.E. Chloropyridinyl Neonicotinoid Insecticides: Diverse Molecular Substituents Contribute to Facile Metabolism in Mice. Chem. Res. Toxicol. 2006, 19, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Kimura-Kuroda, J.; Komuta, Y.; Kuroda, Y.; Hayashi, M.; Kawano, H. Nicotine-like effects of the neonicotinoid insecticides acetamiprid and imidacloprid on cerebellar neurons from neonatal rats. PLoS ONE 2012, 7, e32432. [Google Scholar] [CrossRef] [PubMed]
- Hellström-Lindahl, E.; Gorbounova, O.; Seiger, A.; Mousavi, M.; Nordberg, A. Regional distribution of nicotinic receptors during prenatal development of human brain and spinal cord. Dev. Brain Res. 1998, 108, 147–160. [Google Scholar] [CrossRef]
- Miwa, J.M.; Freedman, R.; Lester, H.A. Neural systems governed by nicotinic acetylcholine receptors: Emerging hypotheses. Neuron 2011, 70, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Lozada, A.F.; Wang, X.; Gounko, N.V.; Massey, K.A.; Duan, J.; Liu, Z.; Berg, D.K. Glutamatergic synapse formation is promoted by α7-containing nicotinic acetylcholine receptors. J. Neurosci. 2012, 32, 7651–7661. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, T.J.; Xu, H.P.; Ackman, J.B.; Ge, X.; Zhang, Y.; Ye, M.J.; Zhou, Z.J.; Xu, J.; Contractor, A.; Crair, M.C. Visual circuit development requires patterned activity mediated by retinal acetylcholine receptors. Neuron 2014, 84, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.R.; Garland, M.; Stark, R.I.; Myers, M.M.; Fifer, W.P.; Mokler, D.J.; Kinney, H.C. Prenatal nicotine exposure selectively affects nicotinic receptor expression in primary and associative visual cortices of the fetal baboon. Brain Pathol. 2015, 25, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, X. Desensitized nicotinic receptors in brain. Brain Res. Rev. 2005, 48, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Tate, C.A.; Cousins, M.M.; Seidler, F.J. Prenatal nicotine exposure alters the responses to subsequent nicotine administration and withdrawal in adolescence: Serotonin receptors and cell signaling. Neuropsychopharmacology 2006, 31, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Plant Protection Products and their Residues (PPR). Scientific Opinion on the developmental neurotoxicity potential of acetamiprid and imidacloprid. EFSA J. 2013, 11, 3471. [Google Scholar]
- Bal-Price, A.K.; Hogberg, H.T.; Buzanska, L.; Lenas, P.; van Vlietd, E.; Hartung, T. ln vitro developmental neurotoxicity (DNT) testing: Relevant models and endpoints. Neurotoxicology 2010, 31, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.; Barone, S.J. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. S3), 511–533. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Storey, J.D.; Noble, W.S. QVALITY: Non-parametric estimation of q-values and posterior error probabilities. Bioinformatics 2009, 25, 964–966. [Google Scholar] [CrossRef] [PubMed]
- Primer3Plus: Pick Primers from a DNA Sequence. Available online: http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi (accessed on 20 July 2016).
- NCBI/Primer-BLAST: Finding Primers Specific to Your PCR Template. Available online: Http://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 20 July 2016).
- Zhang, Y.; Xu, G.; Ling, Q.; Da, C. Expression of Aquaporin 4 and Kir4.1 in Diabetic Rat Retina: Treatment with Minocycline. J. Int. Med. Res. 2011, 39, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, Y.; Muraski, J.; Shiraishi, I.; Kato, T.; Kajstura, J.; Anversa, P.; Sussman, M.A. Nuclear targeting of Akt antagonizes aspects of cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11946–11951. [Google Scholar] [CrossRef] [PubMed]
- The PANTHER (Protein ANalysis THrough Evolutionary Relationships) Classification System. Available online: http://pantherdb.org/about.jsp (accessed on 20 July 2016).
- Kimura-Kuroda, J.; Nagata, I.; Negishi-Kato, M.; Kuroda, Y. Thyroid hormone-dependent development of mouse cerebellar Purkinje cells in vitro. Dev. Brain Res. 2002, 137, 55–65. [Google Scholar] [CrossRef]
- Levine, J.M.; Stincone, F.; Lee, Y.S. Development and differentiation of glial precursor cells in the rat cerebellum. Glia 1993, 7, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Kessler, J.D.; Read, T.A.; Kaiser, C.; Corbeil, D.; Huttner, W.B.; Johnson, J.E.; Wechsler-Reya, R.J. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 2005, 8, 723–729. [Google Scholar] [CrossRef] [PubMed]
- SFARI Gene: A Modular Database for Autism Research. Available online: https://gene.sfari.org/autdb/Welcome.do (accessed on 20 July 2016).
- Vitko, I.; Chen, Y.; Arias, J.M.; Shen, Y.; Wu, X.R.; Perez-Reyes, E. Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J. Neurosci. 2005, 25, 4844–4855. [Google Scholar] [CrossRef] [PubMed]
- Eckle, V.S.; Shcheglovitov, A.; Vitko, I.; Dey, D.; Yap, C.C.; Winckler, B.; Perez-Reyes, E. Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J. Physiol. 2014, 592, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Engbers, J.D.; Anderson, D.; Asmara, H.; Rehak, R.; Mehaffey, W.H.; Hameed, S.; McKay, B.E.; Kruskic, M.; Zamponi, G.W.; Turner, R.W. Intermediate conductance calcium-activated potassium channels modulate summation of parallel fiber input in cerebellar Purkinje cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.L.; Gefrides, L.A.; Foreman, P.J.; Noebels, J.L. A cluster of three novel Ca2+ channel gamma subunit genes on chromosome 19q13.4: Evolution and expression profile of the gamma subunit gene family. Genomics 2001, 71, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, T.; Shiraishi-Yamaguchi, Y.; Sato, A.; Sadakata, T.; Huang, J.; Shinoda, Y.; Hayashi, K.; Mishima, Y.; Tomomura, M.; Nishibe, H.; et al. Systematizing and cloning of genes involved in the cerebellar cortex circuit development. Neurochem. Res. 2011, 36, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Siren, A.; Polvi, A.; Chahine, L.; Labuda, M.; Bourgoin, S.; Anttonen, A.K.; Kousi, M.; Hirvonen, K.; Simola, K.O.; Andermann, E.; et al. Suggestive evidence for a new locus for epilepsy with heterogeneous phenotypes on chromosome 17q. Epilepsy Res. 2010, 88, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Friedman, L.K.; Mancuso, J.; Patel, A.; Kudur, V.; Leheste, J.R.; Iacobas, S.; Botta, J.; Iacobas, D.A.; Spray, D.C. Transcriptome profiling of hippocampal CA1 after early-life seizure-induced preconditioning may elucidate new genetic therapies for epilepsy. Eur. J. Neurosci. 2013, 38, 2139–2152. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.R.; Wen, G.; Lepier, A.; Schroeder, T.; Gotz, M. Par-complex proteins promote proliferative progenitor divisions in the developing mouse cerebral cortex. Development 2008, 135, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Solecki, D.J.; Govek, E.E.; Hatten, M.E. mPar6 alpha controls neuronal migration. J. Neurosci. 2006, 26, 10624–10625. [Google Scholar] [CrossRef] [PubMed]
- Duman, J.G.; Tzeng, C.P.; Tu, Y.K.; Munjal, T.; Schwechter, B.; Ho, T.S.; Tolias, K.F. The adhesion-GPCR BAI1 regulates synaptogenesis by controlling the recruitment of the Par3/Tiam1 polarity complex to synaptic sites. J. Neurosci. 2013, 33, 6964–6978. [Google Scholar] [CrossRef] [PubMed]
- Hur, E.M.; Zhou, F.Q. GSK3 signalling in neural development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, J.; Yang, G.Y.; Wang, Q.J.; Qian, L.; Chen, Y.M.; Chen, F.; Tao, Y.; Hu, H.S.; Wang, T.; et al. Dishevelled promotes axon differentiation by regulating atypical protein kinase C. Nat. Cell Biol. 2007, 9, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Srikakulam, R.; Liu, L.; Winkelmann, D.A. Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS ONE 2008, 3, e2137. [Google Scholar] [CrossRef] [PubMed]
- GTExPortal: The Genotype-Tissue Expression project. Available online: http://www.gtexportal.org/home/ (accessed on 20 July 2016).
- AutismKB: Center for Bioinformatics, PKU. Available online: http://autismkb.cbi.pku.edu.cn/ (accessed on 20 July 2016).
- Xu, L.M.; Li, J.R.; Huang, Y.; Zhao, M.; Tang, X.; Wei, L. AutismKB: An evidence-based knowledgebase of autism genetics. Nucleic Acids Res. 2012, 40, D1016–D1022. [Google Scholar] [CrossRef] [PubMed]
- Bal, R.; Erdogan, S.; Theophilidis, G.; Baydas, G.; Naziroglu, M. Assessing the effects of the neonicotinoid insecticide imidacloprid in the cholinergic synapses of the stellate cells of the mouse cochlear nucleus using whole-cell patch-clamp recording. Neurotoxicology 2010, 31, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Tomizawa, M.; Saito, T.; Miyamoto, T.; Walcott, E.C.; Sumikawa, K. Structural factors contributing to insecticidal and selective actions of neonicotinoids. Arch. Insect Biochem. Physiol. 1998, 37, 24–32. [Google Scholar] [CrossRef]
- Ihara, M.; Okajima, T.; Yamashita, A.; Oda, T.; Asano, T.; Matsui, M.; Sattelle, D.B.; Matsuda, K. Studies on an acetylcholine binding protein identify a basic residue in loop G on the β1 strand as a new structural determinant of neonicotinoid actions. Mol. Pharmacol. 2014, 86, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Fenster, C.P.; Rains, M.F.; Noerager, B.; Quick, M.W.; Lester, R.A. Influence of Subunit Composition on Desensitization of Neuronal Acetylcholine Receptors at Low Concentrations of Nicotine. J. Neurosci. 1997, 17, 5747–5759. [Google Scholar] [PubMed]
- Li, J.; Bo, L.; Zhang, P.; Gao, Q.; Li, L.; Tang, J.; Wu, C.; Li, D.; Xiao, J.; Chen, J.; et al. Exposure to nicotine during pregnancy and altered learning and memory in the rat offspring. Nicotine Tob. Res. 2015, 17, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Wongtrakool, C.; Wang, N.; Hyde, D.M.; Roman, J.; Spindel, E.R. Prenatal nicotine exposure alters lung function and airway geometry through α7 nicotinic receptors. Am. J. Respir. Cell Mol. Biol. 2012, 46, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.X.; Yakel, J.L. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 2009, 30, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Brumwell, C.L.; Johnson, J.L.; Jacob, M.H. Extrasynaptic α7-Nicotinic Acetylcholine Receptor Expression in Developing Neurons Is Regulated by Inputs, Targets, and Activity. J. Neurosci. 2002, 22, 8101–8109. [Google Scholar] [PubMed]
- Lin, H.; Hsu, F.C.; Baumann, B.H.; Coulter, D.A.; Lynch, D.R. Cortical synaptic NMDA receptor deficits in α7 nicotinic acetylcholine receptor gene deletion models: Implications for neuropsychiatric diseases. Neurobiol. Dis. 2014, 63, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Gulisano, W.; Arancio, O.; Palmeri, A. The keystone of Alzheimer pathogenesis might be sought in Abeta physiology. Neuroscience 2015, 307, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, C.P. Nicotinic acetylcholine receptors in human genetic disease. Genet. Med. 2014, 16, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Hsu, F.C.; Baumann, B.H.; Coulter, D.A.; Anderson, S.A.; Lynch, D.R. Cortical parvalbumin GABAergic deficits with α7 nicotinic acetylcholine receptor deletion: Implications for schizophrenia. Mol. Cell. Neurosci. 2014, 61, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Quick, M.W.; Lester, R.A. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002, 53, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.K.; Buckingham, S.D.; Sattelle, D.B. Proteins interacting with nicotinic acetylcholine receptors: Expanding functional and therapeutic horizons. Trends Pharmacol. Sci. 2010, 31, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Tribollet, E.; Bertrand, D.; Raggenbass, M. Role of neuronal nicotinic receptors in the transmission and processing of information in neurons of the central nervous system. Pharmacol. Biochem. Behav. 2001, 70, 457–466. [Google Scholar] [CrossRef]
- Rowell, P.P.; Duggan, D.S. Long-lasting inactivation of nicotinic receptor function in vitro by treatment with high concentrations of nicotine. Neuropharmacology 1998, 37, 103–111. [Google Scholar] [CrossRef]
- Wang, X.; Lippi, G.; Carlson, D.M.; Berg, D.K. Activation of α7-containing nicotinic receptors on astrocytes triggers AMPA receptor recruitment to glutamatergic synapses. J. Neurochem. 2013, 127, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.W.; Gregori, N.Z.; Carlson, N.; Gahring, L.C.; Noble, M. Neuronal nicotinic acetylcholine receptor expression by O2A/oligodendrocyte progenitor cells. Glia 2001, 33, 306–313. [Google Scholar] [CrossRef]
- Meijer, M.; Dingemans, M.M.; van den Berg, M.; Westerink, R.H. Inhibition of voltage-gated calcium channels as common mode of action for (mixtures of) distinct classes of insecticides. Toxicol. Sci. 2014, 141, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Meijer, M.; Brandsema, J.A.; Nieuwenhuis, D.; Wijnolts, F.M.; Dingemans, M.M.; Westerink, R.H. Inhibition of Voltage-Gated Calcium Channels After Subchronic and Repeated Exposure of PC12 Cells to Different Classes of Insecticides. Toxicol. Sci. 2015, 147, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Sheets, L.P.; Li, A.A.; Minnema, D.J.; Collier, R.H.; Creek, M.R.; Peffer, R.C. A critical review of neonicotinoid insecticides for developmental neurotoxicity. Crit. Rev. Toxicol. 2016, 46, 153–190. [Google Scholar] [CrossRef] [PubMed]
- Abou-Donia, M.B.; Khan, W.A.; Dechkovskaia, A.M.; Goldstein, L.B.; Bullman, S.L.; Abdel-Rahman, A. In utero exposure to nicotine and chlorpyrifos alone, and in combination produces persistent sensorimotor deficits and Purkinje neuron loss in the cerebellum of adult offspring rats. Arch. Toxicol. 2006, 80, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Council On Environmental Health. Pesticide exposure in children. Pediatrics 2012, 130, e1757–e1763. [Google Scholar]
- Roberts, J.R.; Karr, C.J.; Council On Environmental Health. Pesticide exposure in children. Pediatrics 2012, 130, e1765–e1788. [Google Scholar] [CrossRef] [PubMed]
- Osaka, A.; Ueyama, J.; Kondo, T.; Nomura, H.; Sugiura, Y.; Saito, I.; Nakane, K.; Takaishi, A.; Ogi, H.; Wakusawa, S.; et al. Exposure characterization of three major insecticide lines in urine of young children in Japan-neonicotinoids, organophosphates, and pyrethroids. Environ. Res. 2016, 147, 89–96. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Probe Name a | Description | CvN Log2FC, p, q | CvA Log2FC, p, q | CvI Log2FC, p, q | |
|---|---|---|---|---|---|---|
| Common to CvN, CvA, CvI | ||||||
| Cacna1h | A_64_P067366 | calcium channel, voltage-dependent, T type, alpha 1H subunit | up | 0.74, 2.8 ×10−2, 1.3 × 10−3 | 0.71, 2.8 × 10−2, 1.6 × 10−3 | 0.64, 4.6 × 10−2, 1.8 × 10−3 |
| Cramp1l | A_64_P047944 | PREDICTED: Crm, cramped-like (Drosophila) | up | 1.22, 4.6 × 10−2, 1.3 × 10−3 | 0.73, 2.8 × 10−2, 1.6 × 10−3 | 0.69, 2.8 × 10−2, 1.8 × 10−3 |
| F2rl2 | A_44_P306344 | coagulation factor II (thrombin) receptor-like 2 | up | 1.02, 4.6 × 10−2, 1.3 × 10−3 | 0.89, 2.8 × 10−2, 1.6 × 10−3 | 0.86, 2.8 × 10−2, 1.8 × 10−3 |
| Tada2b | A_44_P328340 | transcriptional adaptor 2B | up | 0.66, 2.8 × 10−2, 1.3 × 10−3 | 1.19, 2.8 × 10−2, 1.6 × 10−3 | 1.05, 2.8 × 10−2, 1.8 × 10−3 |
| Cacng1 | A_64_P064820 | calcium channel, voltage-dependent, gamma subunit 1 | down | −1.08, 2.8 × 10−2, 1.3 × 10−3 | −0.99, 2.8 × 10−2, 1.6 × 10−3 | −1.04, 2.8 × 10−2, 1.8 × 10−3 |
| Lmod3 | A_64_P103610 | leiomodin 3 (fetal) | down | −2.01, 4.6 × 10−2, 1.3 × 10−3 | −1.75, 4.6 × 10−2, 1.6 × 10−3 | −1.65, 4.6 × 10−2, 1.8 × 10−3 |
| Ndufaf2 | A_43_P22034 | PREDICTED: NADH dehydrogenase (ubiquinone) complex I, assembly factor 2 | down | −0.79, 2.8 × 10−2, 1.3 × 10−3 | −0.69, 2.8 × 10−2, 1.6 × 10−3 | −0.80, 2.8 × 10−2, 1.8 × 10−3 |
| Sdr42e2 | A_64_P077921 | PREDICTED: short chain dehydrogenase/reductase family 42E, member 2 | down | −0.78, 4.6 × 10−2 1.3 × 10−3 | −0.86, 4.6 × 10−2, 1.6 × 10−3 | −0.81, 2.8 × 10−2, 1.8 × 10−3 |
| Unc45b | A_42_P579305 | unc-45 homolog B (C. elegans) | down | −0.75, 4.6 × 10−2, 1.3 × 10−3 | −0.84, 2.8 × 10−2, 1.6 × 10−3 | −0.65, 2.8 × 10−2, 1.8 × 10−3 |
| Common to CvN, CvA | ||||||
| Mcmdc2 | A_64_P000381 | minichromosome maintenance domain containing 2 | up | 0.73, 2.8 × 10−2, 1.3 × 10−3 | 1.17, 4.6 × 10−2, 1.6 × 10−3 | 0.23, 4.6 × 10−1, 5.4 × 10−3 |
| Actrt2 | A_44_P105554 | actin-related protein T2 | down | −0.93, 2.8 × 10−2, 1.3 × 10−3 | −1.08, 4.6 × 10−2, 1.6 × 10−3 | −0.68, 7.5 × 10−1, 7.0 × 10−3 |
| Ankrd60 | A_64_P002697 | Protein Ankrd60 | down | −0.97, 4.6 × 10−2, 1.3 × 10−3 | −0.88, 2.8 × 10−2, 1.6 × 10−3 | −0.87, 1.2 × 10−1, 2.7 × 10−3 |
| Common to CvN, CvI | ||||||
| Celf6 | A_44_P527809 | CUGBP, Elav-like family member 6 | up | 0.72, 4.6 × 10−2, 1.3 × 10−3 | −0.11, 3.5 × 10−1, 3.8 × 10−3 | 0.63, 4.6 × 10−2, 1.8 × 10−3 |
| Pcdhgb7 | A_64_P079069 | protocadherin gamma subfamily B, 7 | up | 0.95, 4.6 × 10−2, 1.3 × 10−3 | 0.27, 4.6 × 10−2, 1.6 × 10−3 | 0.61, 4.6 × 10−2, 1.8 × 10−3 |
| Cyp17a1 | A_44_P508386 | cytochrome P450, family 17, subfamily a, polypeptide 1 | down | −0.59, 2.8 × 10−2, 1.3×10−3 | −0.58, 2.8 × 10−2, 1.6 × 10−3 | −0.73, 4.6 x 10−2, 1.8 × 10−3 |
| Mypn | A_44_P159259 | myopalladin | down | −0.66, 2.8 × 10−2, 1.3 × 10−3 | −0.50, 2.5 × 10−1, 3.3 × 10−3 | −0.71, 2.8 × 10−2, 1.8 × 10−3 |
| Zp3 | A_64_P044241 | zona pellucida glycoprotein 3 (sperm receptor) | down | −0.82, 2.8 × 10−2, 1.3 × 10−3 | −0.54, 4.6 × 10−1, 4.4 × 10−3 | −1.08, 2.8 × 10−2, 1.8 × 10−3 |
| Common to CvA, CvI | ||||||
| Lyn | A_64_P048980 | v-yes-1 Yamaguchi sarcoma viral related oncogene homolog | up | 0.25, 1.2 × 10−1, 1.9 × 10−3 | 1.12, 2.8 × 10−2, 1.6 × 10−3 | 0.73, 2.8 × 10−2, 1.8 × 10−3 |
| Hspb7 | A_43_P15812 | heat shock protein family, member 7 (cardiovascular) | down | −0.26, 7.5 × 10−1, 4.8 × 10−3 | −1.07, 2.8 × 10−2, 1.6 × 10−3 | −1.30, 4.6 × 10−2, 1.8 × 10−3 |
| Mb | A_42_P765066 | myoglobin | down | −0.72, 1.2 × 10−1, 1.9 × 10−3 | −1.1, 2.8 × 10−2, 1.6 × 10−3 | −0.82, 2.8 × 10−2, 1.8 × 10−3 |
| Tnni2 | A_42_P718022 | troponin I type 2 (skeletal, fast) | down | −1.00, 1.2 × 10−1, 1.9 × 10−3 | −0.94, 2.8 × 10−2, 1.6 × 10−3 | −0.85, 4.6 × 10−2, 1.8 × 10−3 |
| Gene Name | Probe Name a | Description | Log2FC, p, q | |
|---|---|---|---|---|
| Atf7 | A_64_P108639 | activating transcription factor 7 | up | 0.66, 4.6 × 10−2, 1.3 × 10−3 |
| Cadm3 | A_44_P454645 | cell adhesion molecule 3 | up | 0.61, 2.8 × 10−2, 1.3 × 10−3 |
| Cd86 | A_44_P222166 | CD86 molecule | up | 0.60, 4.6 × 10−2, 1.3 × 10−3 |
| Gpr83 | A_44_P269457 | G protein-coupled receptor 83 | up | 0.75, 2.8 × 10−2, 1.3 × 10−3 |
| Gramd4 | A_64_P143562 | PREDICTED:GRAM domain containing 4 | up | 0.71, 2.8 × 10−2, 1.3 × 10−3 |
| Ihh | A_43_P15154 | Indian hedgehog | up | 0.91, 4.6 × 10−2, 1.3 × 10−3 |
| Kcnq5 | A_64_P149659 | potassium voltage-gated channel, KQT-like subfamily, member 5 | up | 1.86, 4.6 × 10−2, 1.3 × 10−3 |
| Klhl18 | A_64_P100973 | Protein Klhl18 | up | 0.62, 2.8 × 10−2, 1.3 × 10−3 |
| LOC317356 | A_64_P009658 | PREDICTED: protocadherin alpha-13-like | up | 1.07, 4.6 × 10−2, 1.3 × 10−3 |
| Nt5dc1 | A_44_P440988 | 5’-nucleotidase domain containing 1 | up | 0.66, 2.8 × 10−2, 1.3 × 10−3 |
| Vsig2 | A_64_P117949 | V-set and immunoglobulin domain containing 2 | up | 0.64, 4.6 × 10−2, 1.3 × 10−3 |
| Acta1 | A_44_P255236 | actin, alpha 1, skeletal muscle (Acta1) | down | −1.25, 4.6 × 10−2, 1.3 × 10−3 |
| Car3 | A_44_P244851 | carbonic anhydrase 3 | down | −0.75, 4.6 × 10−2, 1.3 × 10−3 |
| Ms4a12 | A_64_P078240 | PREDICTED: membrane-spanning 4-domains, subfamily A, member 12 | down | −0.59, 2.8 × 10−2, 1.3 × 10−3 |
| Nhlh2 | A_64_P157873 | nescient helix loop helix 2 | down | −0.68, 4.6 × 10−2, 1.3 × 10−3 |
| Olr1051 | A_64_P089733 | olfactory receptor 1051 | down | −1.27, 2.8 × 10−2, 1.3 × 10−3 |
| Phldb3 | A_42_P599116 | pleckstrin homology-like domain, family B, member 3 | down | −0.84, 4.6 × 10−2, 1.3 × 10−3 |
| Gene Name | Probe Name a | Description | Log2FC, p, q | |
|---|---|---|---|---|
| Asap2 | A_43_P20879 | Protein Asap2 | up | 0.64, 2.8 × 10−2, 1.6 × 10−3 |
| B3gnt9 | A_64_P067868 | PREDICTED: UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 9 | up | 1.81, 2.8 × 10-2, 1.6 × 10−3 |
| Dupd1 | A_64_P029907 | dual specificity phosphatase and pro isomerase domain containing 1 | up | 1.84, 2.8 × 10−2, 1.6 × 10−3 |
| Fyb | A_64_P101995 | FYN binding protein | up | 0.59, 2.8 × 10−2, 1.6 × 10−3 |
| Hrh2 | A_64_P030162 | histamine receptor H 2 | up | 1.29, 2.8 × 10−2, 1.6 × 10−3 |
| LOC100363332 | A_44_P421727 | caspase recruitment domain family, member 11 | up | 0.71, 4.6 × 10−2, 1.6 × 10−3 |
| LOC499235 | A_64_P145473 | LRRGT00141; Protein LOC499235 | up | 0.64, 2.8 × 10−2, 1.6 × 10−3 |
| LOC679149 | A_64_P052159 | similar to carboxylesterase 2 (intestine, liver) | up | 0.75, 2.8 × 10−2, 1.6 × 10−3 |
| Mroh1 | A_64_P001622 | maestro heat-like repeat family member 1 | up | 0.60, 2.8 × 10−2, 1.6 × 10−3 |
| Napb | A_44_P333146 | N-ethylmaleimide-sensitive factor attachment protein, beta | up | 0.66, 2.8 × 10−2, 1.6 × 10−3 |
| Ntn1 | A_64_P023527 | netrin 1 | up | 0.69, 2.8 × 10−2, 1.6 × 10−3 |
| Phf21b | A_44_P234547 | PHD finger protein 21B | up | 0.72, 2.8 × 10−2, 1.6 × 10−3 |
| Plac8 | A_44_P212964 | placenta-specific 8 | up | 0.65, 4.6 × 10−2, 1.6 × 10−3 |
| Rasl10b | A_64_P126140 | RAS-like, family 10, member B | up | 0.68, 2.8 × 10−2, 1.6 × 10−3 |
| Slc45a3 | A_43_P10089 | solute carrier family 45, member 3 | up | 1.11, 4.6 × 10−2, 1.6 × 10−3 |
| Zfp498 | A_64_P024399 | zinc finger protein 498 | up | 0.66, 4.6 × 10−2, 1.6 × 10−3 |
| Actc1 | A_64_P078862 | actin, alpha, cardiac muscle 1 | down | −1.14, 4.6 × 10−2, 1.6 × 10−3 |
| Dcdc2 | A_44_P685554 | doublecortin domain containing 2 | down | −0.76, 4.6 × 10−2, 1.6 × 10−3 |
| Des | A_44_P384090 | desmin | down | −1.64, 2.8 × 10−2, 1.6 × 10−3 |
| Dmbt1 | A_64_P237865 | deleted in malignant brain tumors 1 | down | −0.89, 2.8 × 10−2, 1.6 × 10−3 |
| Hsd3b1 | A_64_P123655 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 | down | −0.91, 2.8 × 10−2, 1.6 × 10−3 |
| Iqcf1 | A_44_P744940 | IQ motif containing F1 | down | −0.60, 2.8 × 10−2, 1.6 × 10−3 |
| Lmod2 | A_44_P1003728 | leiomodin 2 (cardiac) | down | −1.63, 2.8 × 10−2, 1.6 × 10−3 |
| LOC684762 | A_42_P755367 | PREDICTED: histone H3.2-like | down | −1.49, 4.6 × 10−2, 1.6 × 10−3 |
| Myog | A_64_P056293 | myogenin | down | −0.74, 2.8 × 10−2, 1.6 × 10−3 |
| Nags | A_42_P803406 | N-acetylglutamate synthase | down | −1.05, 4.6 × 10−2, 1.6 × 10−3 |
| Olr1714 | A_64_P043808 | olfactory receptor 1714 | down | −0.80, 2.8 × 10−2, 1.6 × 10−3 |
| Olr825 | A_44_P130360 | olfactory receptor 825 | down | −0.60, 2.8 × 10−2, 1.6 × 10−3 |
| Olr94 | A_44_P285601 | olfactory receptor 94 | down | −0.70, 4.6 × 10−2, 1.6 × 10−3 |
| Slc5a5 | A_64_P080233 | solute carrier family 5 (sodium/iodide cotransporter), member 5 | down | −0.68, 4.6 × 10−2, 1.6 × 10−3 |
| Slco1b2 | A_44_P328097 | solute carrier organic anion transporter family, member 1B2 | down | −1.41, 2.8 × 10−2, 1.6 × 10−3 |
| Vom1r73 | A_64_P097332 | vomeronasal 1 receptor 73 | down | −0.59, 2.8 × 10−2, 1.6 × 10−3 |
| Gene Symbol | Probe Name a | Description | Log2FC, p, q | |
|---|---|---|---|---|
| Ccdc81 | A_44_P499041 | coiled-coil domain containing 81 | up | 0.62, 4.6 × 10−2, 1.8 × 10−3 |
| Cdc6 | A_64_P052665 | cell division cycle 6 | up | 0.68, 4.6 × 10−2, 1.8 × 10−3 |
| Crb3 | A_44_P1043302 | crumbs homolog 3 (Drosophila) | up | 0.89, 2.8 × 10−2, 1.8 × 10−3 |
| Cxcl17 | A_64_P053431 | chemokine (C-X-C motif) ligand 17 | up | 0.70, 4.6 × 10−2, 1.8 × 10−3 |
| Efna3 | A_64_P087380 | PREDICTED: ephrin A3 (Efna3) | up | 0.70, 2.8 × 10−2, 1.8 × 10−3 |
| Galnt7 | A_44_P406169 | UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 7 (GalNAc-T7) | up | 0.66, 4.6 × 10−2, 1.8 × 10−3 |
| Grb7 | A_64_P135843 | growth factor receptor bound protein 7 | up | 0.77, 4.6 × 10−2, 1.8 × 10−3 |
| Htr2c | A_44_P792784 | 5-hydroxytryptamine (serotonin) receptor 2C | up | 1.61, 2.8 × 10−2, 1.8 × 10−3 |
| Iqsec3 | A_64_P037666 | IQ motif and Sec7 domain 3 | up | 0.62, 2.8 × 10−2, 1.8 × 10−3 |
| Lbx1 | A_64_P009892 | ladybird homeobox 1 | up | 1.10, 2.8 × 10−2, 1.8 × 10−3 |
| Magel2 | A_64_P065185 | PREDICTED: MAGE-like 2 | up | 0.63, 2.8 × 10−2, 1.8 × 10−3 |
| Mybpc2 | A_44_P201295 | myosin binding protein C, fast-type | up | 0.64, 2.8 × 10−2, 1.8 × 10−3 |
| Oasl | A_44_P492025 | 2’-5’-oligoadenylate synthetase-like | up | 0.77, 4.6 × 10−2, 1.8 × 10−3 |
| Plekha2 | A_64_P136831 | PREDICTED: pleckstrin homology domain-containing, family A (phosphoinositide binding specific) member 2 | up | 0.75, 4.6 × 10−2, 1.8 × 10−3 |
| Rbfox2 | A_44_P403410 | RNA binding protein, fox-1 homolog (C. elegans) 2 | up | 0.67, 2.8 × 10−2, 1.8 × 10−3 |
| S100pbp | A_64_P084038 | S100P binding protein | up | 0.61, 2.8 × 10−2, 1.8 × 10−3 |
| Slc2a3 | A_44_P292510 | solute carrier family 2 (facilitated glucose transporter), member 3 | up | 0.74, 4.6 × 10−2, 1.8 × 10−3 |
| Syne4 | A_64_P006878 | spectrin repeat containing, nuclear envelope family member 4 | up | 1.01, 4.6 × 10-2, 1.8 × 10−3 |
| Tacr3 | A_43_P11794 | tachykinin receptor 3 | up | 0.86, 2.8 × 10−2, 1.8 × 10−3 |
| Txk | A_64_P100138 | TXK tyrosine kinase | up | 0.71, 2.8 × 10−2, 1.8 × 10−3 |
| Amer3 | A_64_P009815 | PREDICTED:APC membrane recruitment protein 3 | down | −0.72, 4.6 × 10−2, 1.8 × 10−3 |
| Ampd1 | A_44_P276087 | adenosine monophosphate deaminase 1 | down | −0.83, 2.8 × 10−2, 1.8 × 10−3 |
| Bmp8b | A_64_P039949 | PREDICTED: bone morphogenetic protein 8b | down | −0.67, 4.6 × 10−2, 1.8 × 10−3 |
| C2cd4c | A_64_P000514 | C2 calcium-dependent domain containing 4C | down | −0.64, 2.8 × 10−2, 1.8 × 10−3 |
| Cyp4a1 | A_64_P040870 | cytochrome P450, family 4, subfamily a, polypeptide 1 | down | −0.65, 4.6 × 10−2, 1.8 × 10−3 |
| Egr3 | A_64_P128219 | early growth response 3 | down | −0.77, 4.6 × 10−2, 1.8 × 10−3 |
| Gapt | A_64_P093787 | Grb2-binding adaptor protein, transmembrane | down | −0.71, 2.8 × 10−2, 1.8 × 10−3 |
| Hao1 | A_44_P340172 | hydroxyacid oxidase (glycolate oxidase) 1 | down | −0.80, 2.8 × 10−2, 1.8 × 10−3 |
| Hfe2 | A_44_P140248 | hemochromatosis type 2 (juvenile) | down | −1.67, 4.6 × 10−2, 1.8 × 10−3 |
| Hlcs | A_64_P059834 | PREDICTED: holocarboxylase synthetase (biotin-(proprionyl-CoA-carboxylase (ATP-hydrolysing)) ligase) | down | −0.59, 2.8 × 10−2, 1.8 × 10−3 |
| Itpripl1 | A_44_P943876 | inositol 1,4,5-trisphosphate receptor interacting protein-like 1 | down | −0.79, 4.6 × 10−2, 1.8 × 10−3 |
| Klk1c7 | A_44_P399414 | kallikrein 1-related peptidase C7 | down | −1.22, 4.6 × 10−2, 1.8 × 10−3 |
| Krt4 | A_64_P105389 | keratin 4 | down | −0.83, 2.8 × 10−2, 1.8 × 10−3 |
| Krt5 | A_44_P461130 | keratin 5 | down | −0.66, 2.8 × 10−2, 1.8 × 10−3 |
| LOC100364862 | A_64_P078448 | PREDICTED: hypothetical protein LOC100364862 | down | −0.88, 4.6 × 10−2, 1.8 × 10−3 |
| LOC100912563 | A_64_P007208 | PREDICTED: insulinoma-associated protein 1-like | down | −1.19, 2.8 × 10−2, 1.8 × 10−3 |
| LOC294497 | A_64_P000312 | similar to double homeobox, 4; double homeobox protein 4 | down | −0.61, 2.8 × 10−2, 1.8 × 10−3 |
| LOC685406 | A_64_P009947 | LRRGT00062 | down | −0.76, 2.8 × 10−2, 1.8 × 10−3 |
| Mrgprb4 | A_64_P090883 | MAS-related GPR, member B4 | down | −0.65, 2.8 × 10−2, 1.8 × 10−3 |
| Neurog3 | A_44_P353797 | neurogenin 3 | down | −0.75, 4.6 × 10−2, 1.8 × 10−3 |
| Pitx3 | A_64_P072653 | paired-like homeodomain 3 | down | −0.76, 4.6 × 10−2, 1.8 × 10−3 |
| Prr16 | A_64_P026928 | proline rich 16 | down | −0.89, 2.8 × 10−2, 1.8 × 10−3 |
| RGD1561114 | A_64_P012158 | PREDICTED: ral guanine nucleotide dissociation stimulator-like | down | −0.62, 4.6 × 10−2, 1.8 × 10−3 |
| RGD1564095 | A_64_P043524 | PREDICTED: 60S acidic ribosomal protein P2-like | down | −0.88, 2.8 × 10−2, 1.8 × 10−3 |
| RGD1564571 | A_64_P159245 | PREDICTED: CD209 antigen-like protein A-like | down | −0.61, 2.8 × 10−2, 1.8 × 10−3 |
| RGD1566248 | A_64_P157099 | PREDICTED: necdin-like | down | −1.17, 4.6 × 10−2, 1.8 × 10−3 |
| Scnn1b | A_64_P068913 | sodium channel, non-voltage-gated 1, beta subunit | down | −1.02, 2.8 × 10−2, 1.8 × 10−3 |
| Slc38a8 | A_64_P056897 | solute carrier family 38, member 8 | down | −0.77, 2.8 × 10−2, 1.8 × 10−3 |
| Tmem52b | A_64_P114148 | transmembrane protein 52B | down | −0.70, 4.6 × 10−2, 1.8 × 10−3 |
| Enrichment Categories | p-Value | FDR | Related Genes |
|---|---|---|---|
| GO processes | |||
| Calcium ion transmembrane transport | 7.3 × 10−7 | 1.5 × 10−4 | Cacna1h, Cacng1, Ms4a12, Zp3 |
| Calcium ion transport | 8.2 × 10−7 | 1.6 × 10−4 | Cacna1h, Cacng1, Ms4a12, Zp3, F2rl2 |
| Cellular component assembly involved in morphogenesis | 1.3 × 10−6 | 2.1 × 10−4 | Lmod3, Acta1, Ihh, Mypn, |
| Glucocorticoid biosynthetic process | 1.4 × 10−6 | 2.1 × 10−4 | Cacna1h, Cyp17a1 |
| Divalent metal ion transport | 2.5 × 10−6 | 3.0 × 10−4 | Cacna1h, Cacng1, Ms4a12, Zp3, F2rl2 |
| Divalent inorganic cation transport | 2.6 × 10−6 | 3.0 × 10−4 | Cacna1h, Cacng1, Ms4a12, Zp3, F2rl2 |
| Negative regulation of astrocyte differentiation | 4.6 × 10−6 | 4.3 × 10−4 | Atf7, F2rl2 |
| Response to steroid hormone | 1.0 × 10−5 | 7.0 × 10−4 | Acta1, Ihh, Atf7, Cyp17a1, Cacna1h, F2rl2, Gpr83 |
| Cell-cell adhesion | 1.1 × 10−5 | 7.1 × 10−4 | Cd86, Ihh, Pcdhgb7, Atf7, Acta1, F2rl2, Cadm3 |
| Cellular component morphogenesis | 1.4 × 10−5 | 8.4 × 10−4 | Lmod3, Acta1, Ihh, Mypn, Cacna1h, Atf7, F2rl2 |
| Diseases (in biomarkers) | |||
| Epilepsy, absence | 8.9 × 10−5 | 4.2 × 10−3 | Cacna1h, Cacng1 |
| Movement disorders | 4.7 × 10−4 | 1.2 × 10−2 | Acta1, Ihh, Cyp17a1, Atf7, F2rl2 |
| Epilepsy, generalized | 5.5 × 10−4 | 1.3 × 10−2 | Cacna1h, Cacng1 |
| Neurodegenerative diseases | 1.4 × 10−3 | 2.0 × 10−2 | Cd86, Lmod3, Ihh, Cyp17a1, Cacng1, Acta1, Atf7, F2rl2 |
| Brain diseases | 1.4 × 10−3 | 2.0 × 10−2 | Cd86, Lmod3, Ihh, Cyp17a1, Cacng1, Cacna1h, Acta1, Atf7, F2rl2 |
| Demyelinating autoimmune diseases, CNS | 1.5 × 10−3 | 2.0 × 10−2 | Cd86, Ihh, Cacng1, Acta1, F2rl2 |
| Seizures | 1.7 × 10−3 | 2.0 × 10−2 | Cacna1h, Cacng1 |
| Autoimmune diseases of the nervous system | 2.0 × 10−3 | 2.3 × 10−2 | Cd86, Ihh, Cacng1, Acta1, F2rl2 |
| Autistic disorder | 2.0 × 10−3 | 2.3 × 10−2 | Cacna1h, F2rl2 |
| Child development disorders, pervasive | 2.1 × 10−3 | 2.3 × 10−2 | Cacna1h, F2rl2 |
| Enrichment Categories | p-Value | FDR | Related Genes |
|---|---|---|---|
| GO processes | |||
| Negative regulation of eicosanoid secretion | 7.5 × 10−9 | 3.0 × 10−6 | Hrh2, F2rl2 |
| Glucocorticoid biosynthetic process | 1.6 × 10−8 | 3.8 × 10−6 | Cacna1h, Hsd3b1 |
| Negative regulation of fatty acid transport | 4.1 × 10−8 | 7.6 × 10−6 | Hrh2, F2rl2 |
| System process | 4.5 × 10−7 | 4.5 × 10−5 | Actc1, Lmod3, Cacng1, Des, Dcdc2, Tnni2, Hrh2, Myog, Cacna1h, Lmod2, F2rl2, Mb, Rasl10b, Olr1714 |
| Response to hormone | 1.1 × 10−6 | 8.5 × 10−5 | Actc1, Lyn, Slco1b2 Dmbt1, Hsd3b1, Myog, Cacna1h, Hrh2, Myb, F2rl2 |
| Cellular component morphogenesis | 1.2 × 10−6 | 8.9 × 10−5 | Actc1, Cacna1h, Hrh2, F2rl2, Lyn, Dcdc2, Lmod2, Lmod3, Ntn1 |
| Regulation of neurotransmitter secretion | 2.5 × 10−6 | 1.5 × 10−4 | Cacna1h, Cacng1, Hrh2 |
| Calcium ion transmembrane transport | 3.6 × 10−6 | 2.0 × 10−4 | Cacna1h, Cacng1, Hrh2 |
| Positive regulation of gamma-aminobutyric acid secretion | 3.9 × 10−6 | 2.1 × 10−4 | Hrh2, F2rl2 |
| Calcium ion transport | 5.1 × 10−6 | 2.5 × 10−4 | Cacna1h, Cacng1, F2rl2, Hrh2 |
| Diseases (in biomarkers) | |||
| Autistic disorder | 1.5 × 10−5 | 8.9 × 10−4 | Cacna1h, Hrh2, F2rl2 |
| Child development disorders, pervasive | 1.6 × 10−5 | 9.0 × 10−4 | Cacna1h, Hrh2, F2rl2 |
| Communication disorders | 6.1 × 10−5 | 2.3 × 10−3 | Dcdc2, Hrh2 |
| Language disorders | 6.1 × 10−5 | 2.3 × 10−3 | Dcdc2, Hrh2 |
| Mental disorders diagnosed in childhood | 8.8 × 10−5 | 3.0 × 10−3 | Cacna1h, Dcdc2, Hrh2 |
| Tourette syndrome | 1.3 × 10−4 | 4.1 × 10−3 | Hrh2, F2rl2 |
| Tic disorders | 1.3 × 10−4 | 4.2 × 10−3 | Hrh2, F2rl2 |
| Epilepsy, absence | 1.7 × 10−4 | 4.8 × 10−3 | Cacna1h, Cacng1 |
| Neurotoxicity syndromes | 2.1 × 10−4 | 4.8 × 10−3 | Hrh2, F2rl2 |
| Pathological conditions, signs and symptoms | 5.6 × 10−4 | 9.1 × 10−3 | Des, Dcdc2, Ntn1, Cacng1, Cacna1h, Hsd3b1, Tnni2, F2rl2, Mb, Dmbt1, Actc1, Nags, Napb |
| Enrichment Categories | p-Value | FDR | Related Genes |
|---|---|---|---|
| GO processes | |||
| Positive regulation of phosphatidylinositol biosynthetic process | 7.5 × 10−10 | 1.1 × 10−6 | Htr2c, ZP3 |
| Positive regulation of acetylcholine secretion, neurotransmission | 3.5 × 10−9 | 3.3 × 10−6 | Htr2c, F2rl2, Tacr3 |
| Regulation of dopamine metabolic process | 1.2 × 10−8 | 5.7 × 10−6 | Htr2c, F2rl2, Tacr3 |
| Regulation of catecholamine metabolic process | 1.4 × 10−8 | 5.9 × 10−6 | Htr2c, F2rl2, Tacr3 |
| Positive regulation of gamma-aminobutyric acid secretion | 4.9 × 10−8 | 1.6 × 10−5 | Htr2c, F2rl2, Tacr3 |
| Regulation of acetylcholine secretion, neurotransmission | 1.2 × 10−7 | 2.8 × 10−5 | Htr2c, F2rl2, Tacr3 |
| Calcium ion transport | 1.3 × 10−7 | 2.8 × 10−5 | Cacna1h, Cacng1, Htr2c, F2rl2, Tacr3, ZP3 |
| Positive regulation of synaptic transmission, cholinergic | 1.5 × 10−7 | 2.8 × 10−5 | Htr2c, F2rl2, Tacr3 |
| Regulation of gamma-aminobutyric acid secretion | 1.5 × 10−7 | 2.8 × 10−5 | Htr2c, F2rl2, Tacr3 |
| Cation transport | 3.7 × 10−6 | 2.6 × 10−4 | Cacng1, Htr2c, Lyn, Cacna1h, F2rl2, Tacr3, Slc2a3, Slc38a8, Scnn1b, Zp3 |
| Diseases (in biomarkers) | |||
| Dyskinesia, drug-induced | 2.0 × 10−7 | 1.2 × 10−4 | Htr2c, Cyp17a1 |
| Neurotoxicity syndromes | 2.7 × 10−7 | 1.2 × 10−4 | Htr2c, F2rl2, Tacr3, Cyp17a1 |
| Borderline personality disorder | 5.1 × 10−6 | 9.2 × 10−4 | Htr2c, F2rl2, Tacr3 |
| Mental disorders diagnosed in childhood | 1.7 × 10−5 | 1.5 × 10−3 | Cacna1h, Htr2c, F2rl2, Tacr3 |
| Mutism | 1.8 × 10−5 | 1.5 × 10−3 | Htr2c |
| Autistic disorder | 3.6 × 10−5 | 1.7 × 10−3 | Cacna1h, Htr2c, F2rl2, Tacr3 |
| Child development disorders, pervasive | 3.9 × 10−5 | 1.7 × 10−3 | Cacna1h, Htr2c, F2rl2, Tacr3 |
| Medulloblastoma | 2.1 × 10−4 | 5.6 × 10−3 | Htr2c, Mypn, Slc38a8, Slc2a3, Cacna1h, Txk, Syne4, F2rl2, Tacr3, Pcdhgb7, Celf6, Hfe2, Efna3, Ampd1 |
| Tourette syndrome | 2.2 × 10−4 | 5.7 × 10−3 | Htr2c, F2rl2, Tacr3 |
| Epilepsy, absence | 2.9 × 10−4 | 6.9 × 10−3 | Cacna1h, Cacng1 |
| Enrichment Categories | p-Value | FDR | Related Common Genes | Other Related Genes |
|---|---|---|---|---|
| GO processes | ||||
| Calcium ion transport | 1.4 × 10−7 | 6.9 × 10−5 | Cacna1h, Cacng1, F2rl2 | Hrh2 a, Htr2c b, Zp3 b,c, Tacr3 b Msa4a12 c |
| Divalent metal ion transport | 3.2 × 10−7 | 1.1 × 10−4 | Cacna1h, Cacng1, F2rl2 | Hrh2, Htr2c, Zp3, Tacr3, Msa4a12 |
| Calcium ion transmembrane transport | 1.1 × 10−6 | 3.1 × 10−4 | Cacna1h, Cacng1 | Hrh2, Htr2c, Zp3, Msa4a12 |
| Regulation of ion transmembrane transport | 1.9 × 10−6 | 4.2 × 10−4 | Cacna1h, Cacng1, F2rl2 | Hrh2, Htr2c, Zp3, Tacr3, Kcnq5 c |
| Cellular response to potassium ion | 1.1 × 10−5 | 1.1 × 10−3 | Cacna1h | |
| Metal ion transport | 1.2 × 10−5 | 1.1 × 10−3 | Cacna1h, Cacng1, F2rl2 | Hrh2, Htr2c, Zp3, Tacr3, Kcnq5, Msa4a12, Scnn1b b, Slc38a8 b, Slc5a5 a |
| Thrombin receptor signaling pathway | 1.3 × 10−5 | 1.1 × 10−3 | F2rl2 | |
| Positive regulation of exocytosis | 1.7 × 10−5 | 1.3 × 10−3 | Cacna1h, F2rl2 | Zp3 |
| Response to potassium ion | 3.8 × 10−5 | 2.3 × 10−3 | Cacna1h | |
| Membrane depolarization | 6.6 × 10−5 | 3.3 × 10−3 | Cacna1h, Cacng1 | Hrh2 |
| Diseases (in biomarkers) | ||||
| Epilepsy, absence | 2.0 × 10−6 | 1.5 × 10−3 | Cacna1h, Cacng1 | |
| Epilepsy, generalized | 1.3 × 10−5 | 4.7 × 10−3 | Cacna1h, Cacng1 | |
| Seizures | 4.0 × 10−5 | 7.7 × 10−3 | Cacna1h, Cacng1 | |
| Autistic disorder | 4.9 × 10−5 | 7.7 × 10−3 | Cacna1h, F2rl2 | Hrh2, Htr2c, Tacr3 |
| Child development disorders, pervasive | 5.2 × 10−5 | 7.7 × 10−3 | Cacna1h, F2rl2 | Hrh2, Htr2c, Tacr3 |
| Mental disorders diagnosed in childhood | 1.5 × 10−4 | 1.8 × 10−2 | Cacna1h, F2rl2 | Hrh2, Htr2c, Tacr3 |
| Epilepsy | 1.2 × 10−3 | 6.9 × 10−2 | Cacna1h, Cacng1 | Hrh2, Htr2c, Tacr3, Ihh c |
| Mitochondrial complex I deficiency | 2.0 × 10−3 | 7.8 × 10−2 | Ndufaf2 | |
| Delirium | 3.0 × 10−3 | 8.0 × 10−2 | F2rl2 | |
| Neurologic manifestations | 4.8 × 10−3 | 8.4 × 10−2 | Cacna1h, Cacng1, F2rl2 | Hrh2, Htr2c, Dcdc2 a, Magel2 b, Pitx3 b, Acta1 c, Actc1 a |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimura-Kuroda, J.; Nishito, Y.; Yanagisawa, H.; Kuroda, Y.; Komuta, Y.; Kawano, H.; Hayashi, M. Neonicotinoid Insecticides Alter the Gene Expression Profile of Neuron-Enriched Cultures from Neonatal Rat Cerebellum. Int. J. Environ. Res. Public Health 2016, 13, 987. https://doi.org/10.3390/ijerph13100987
Kimura-Kuroda J, Nishito Y, Yanagisawa H, Kuroda Y, Komuta Y, Kawano H, Hayashi M. Neonicotinoid Insecticides Alter the Gene Expression Profile of Neuron-Enriched Cultures from Neonatal Rat Cerebellum. International Journal of Environmental Research and Public Health. 2016; 13(10):987. https://doi.org/10.3390/ijerph13100987
Chicago/Turabian StyleKimura-Kuroda, Junko, Yasumasa Nishito, Hiroko Yanagisawa, Yoichiro Kuroda, Yukari Komuta, Hitoshi Kawano, and Masaharu Hayashi. 2016. "Neonicotinoid Insecticides Alter the Gene Expression Profile of Neuron-Enriched Cultures from Neonatal Rat Cerebellum" International Journal of Environmental Research and Public Health 13, no. 10: 987. https://doi.org/10.3390/ijerph13100987
APA StyleKimura-Kuroda, J., Nishito, Y., Yanagisawa, H., Kuroda, Y., Komuta, Y., Kawano, H., & Hayashi, M. (2016). Neonicotinoid Insecticides Alter the Gene Expression Profile of Neuron-Enriched Cultures from Neonatal Rat Cerebellum. International Journal of Environmental Research and Public Health, 13(10), 987. https://doi.org/10.3390/ijerph13100987