The Chemical Synthesis of Tetrodoxin: An Ongoing Quest

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Strategies Explored in Connection with Synthetic Work on TTX
3. The Diels-Alder Route to Tetrodotoxin
4. Syntheses of TTX from Carbohydrates and Congeners
Synthetic Studies toward TTX Based on Carbohydrate Building Blocks and Congeners
5. Other Approaches to TTX through Diels-Alder Reactions or Annulation Processes
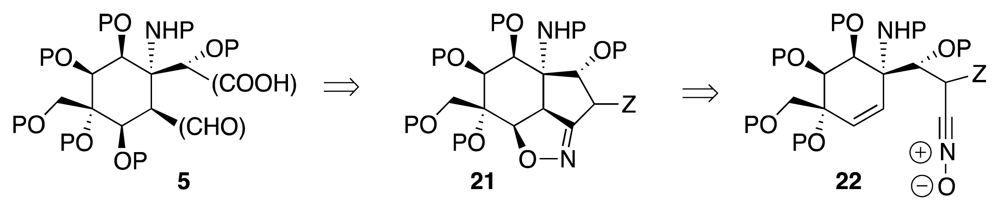
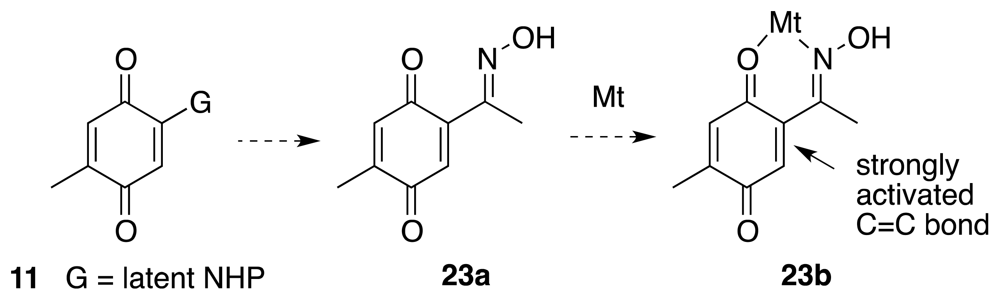
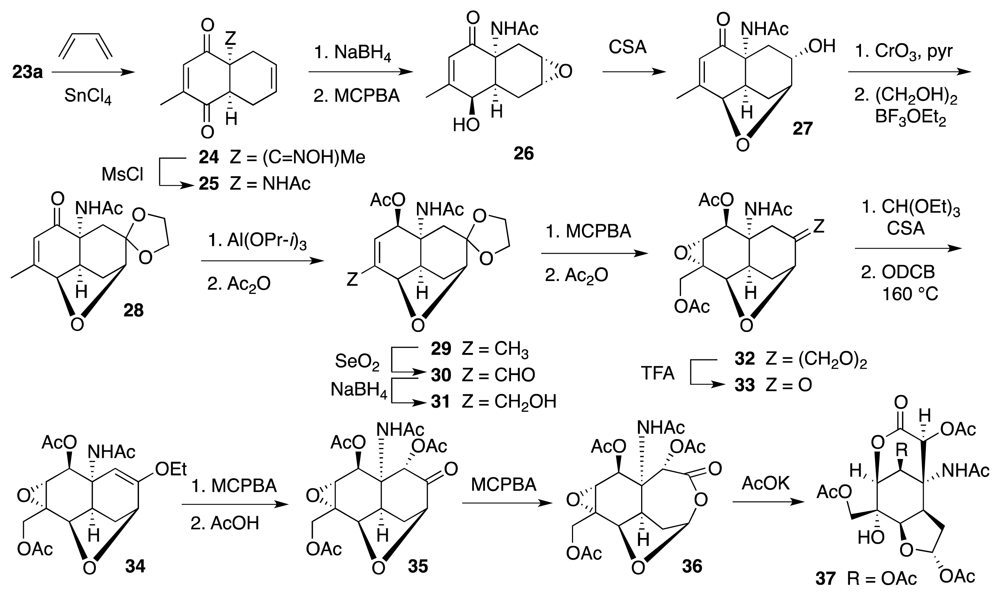
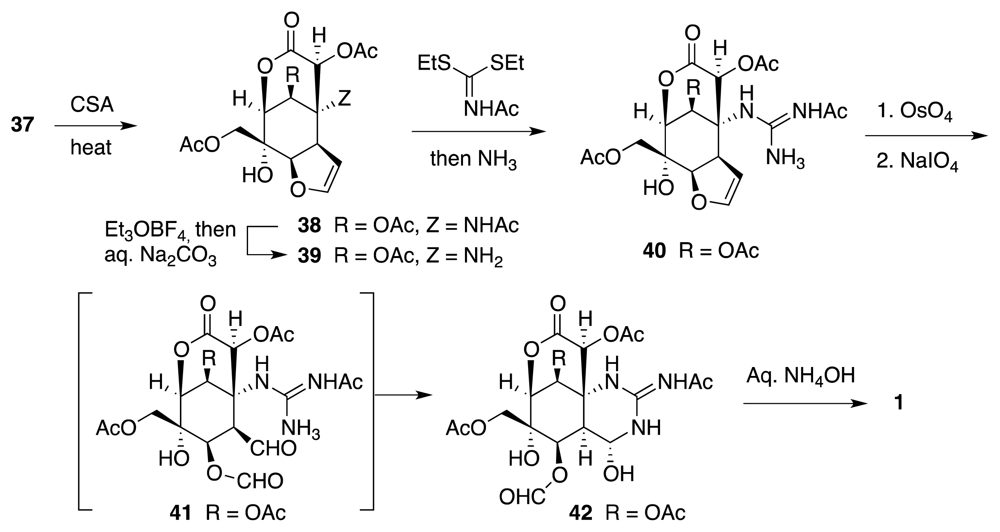
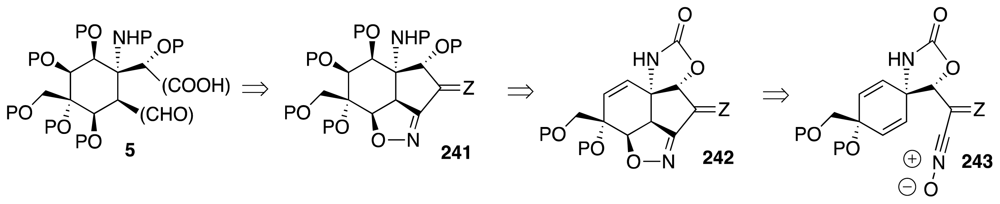
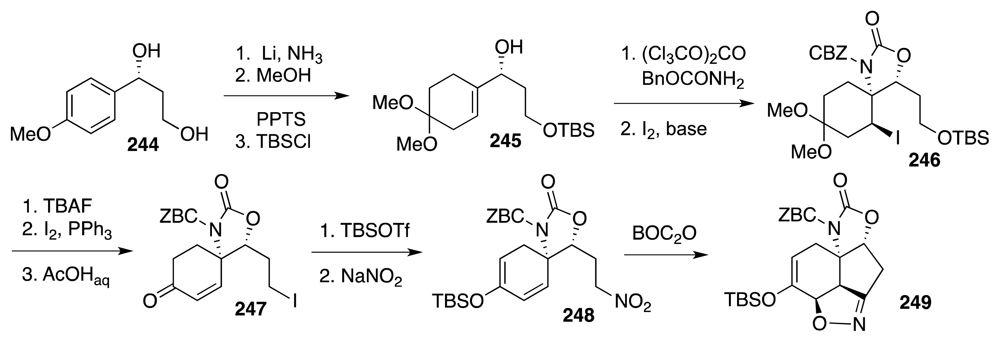
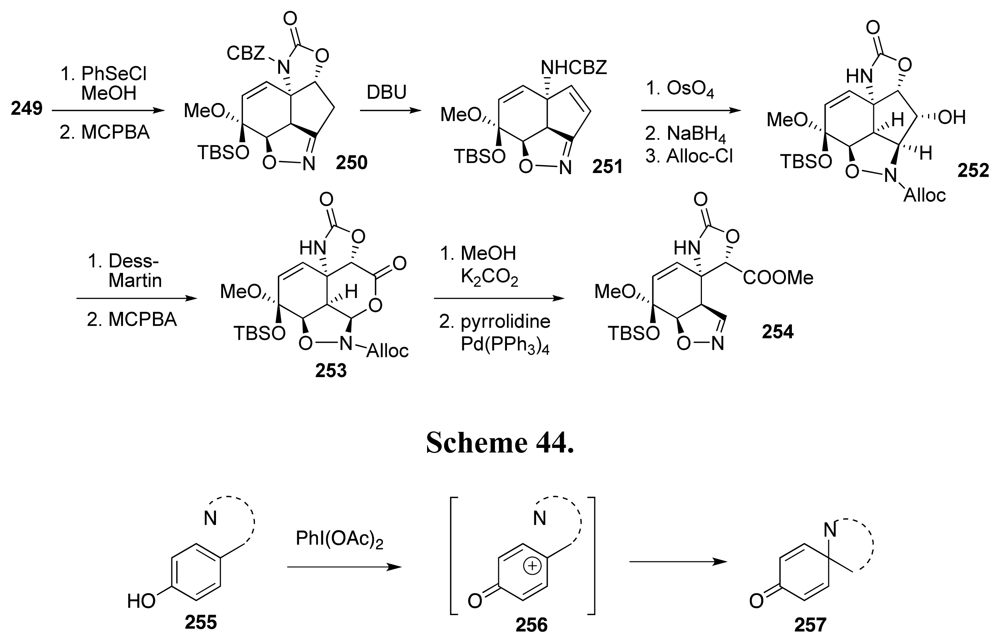
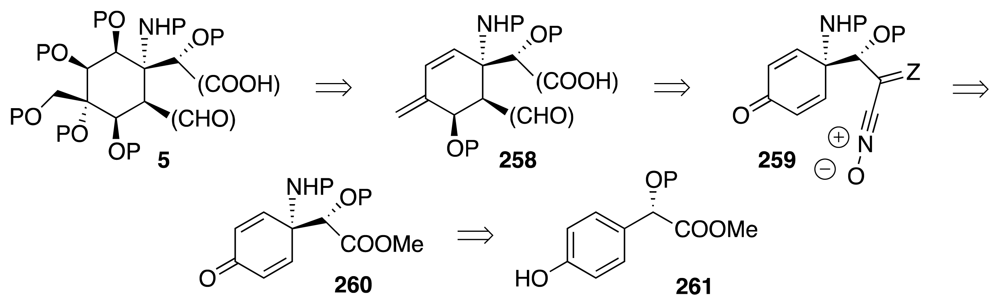
6. Approaches to TTX that Rely on Intramolecular Nitrile Oxide Cycloaddition Reactions
7. Conclusion
Acknowledgments
References
- Koert, U. Syntheses of Tetrodotoxin. Angew. Chem. Int. Ed 2004, 43, 5572–5576. [Google Scholar]
- Woodward, RB. The structure of tetrodotoxin. Pure Appl. Chem 1964, 9, 49–74. [Google Scholar]
- Goto, T; Kishi, Y; Takahashi, S; Hirata, Y. Tetrodotoxin. Tetrahedron 1965, 21, 2059–2088. [Google Scholar]
- Tsuda, K; Ikuma, S; Kawamura, M; Tachikawa, R; Sakai, K; Tamura, C; Amakasu, O. Tetrodotoxin. VII. On the structures of tetrodotoxin and its derivatives. Chem. Pharm. Bull 1964, 12, 1357–1374. [Google Scholar]
- Gutekunst, WR; Baran, PS. C–H functionalization logic in total synthesis. Chem. Soc. Rev 2011, 40, 1976–1991. [Google Scholar]
- Lindley, JM; McRobbie, IM; Meth-Cohn, O; Suschitzsky, H. Competitive cyclisations of singlet and triplet nitrenes. Part 5. Mechanism of cyclisation of 2-nitrenobiphenyls and related systems. J Chem Soc Perkin Trans 1 1977, 2194–2204, and references therein. [Google Scholar]
- Ciufolini, MA; Byrne, NE. The total synthesis of cystodytins. J. Am. Chem. Soc 1991, 113, 8016–8024. [Google Scholar]
- Bishop, MJ; Ciufolini, MA. Total synthesis of kuanoniamines and dercitins. J. Am. Chem. Soc 1992, 114, 10081–10082. [Google Scholar]
- Ciufolini, MA; Bishop, MJ. Studies towards streptonigrinoids: Formal synthesis of lavendamycin methyl ester. J Chem Soc Chem Commun 1993, 1463–1464. [Google Scholar]
- Ciufolini, MA; Shen, Y-C. Total synthesis of cystodytin J, diplamine and shermilamine B. Tetrahedron Lett 1995, 36, 4709–4712. [Google Scholar]
- Ciufolini, MA; Shen, Y-C; Bishop, MJ. A Unified strategy for the synthesis of sulfur-containing pyridoacridine alkaloids: Antitumor agents of marine origin. J. Am. Chem. Soc 1995, 117, 12460–12469. [Google Scholar]
- Slaughter, RS; Garcia, ML; Kaczorowski, GJ. Ion channels as drug targets in the immune system. Curr. Pharm. Des 1996, 2, 610–623. [Google Scholar]
- Hagen, NA; du Souich, P; Lapointe, B; Ong-Lam, M; Dubuc, B; Walde, D; Love, R; Ngoc, A. Canadian tetrodotoxin study group. J. Pain Symptom Manag 2008, 35, 420–429. [Google Scholar]
- Noheda Marin, P. Synthesis of Tetrodotoxin, its Analogues and Intermediates Thereof. WIPO Patent Application WO 2007/054517, 18 May 2007. [Google Scholar]
- Kishi, Y; Nakatsubo, F; Aratani, M; Goto, T; Inoue, S; Kakoi, H; Sugiura, S. Synthetic approach towards tetrodotoxin. I. Diels-Alder reaction of α-oximinoethylbenzoquinones with butadiene. Tetrahedron Lett 1970, 11, 5127–5128. [Google Scholar]
- Kishi, Y; Nakatsubo, F; Aratani, M; Goto, T; Inoue, S; Kakoi, H. Synthetic approach towards tetrodotoxin. II. A stereospecific synthesis of a compound having the same six chiral centers on the cyclohexane ring as those of tetrodotoxin. Tetrahedron Lett 1970, 11, 5129–5132. [Google Scholar]
- Kishi, Y; Aratani, M; Fukuyama, T; Nakatsubo, F; Goto, T; Inoue, S; Tanino, H; Sugiura, S; Kakoi, H. Synthetic studies on tetrodotoxin and related compounds. III. Stereospecific synthesis of an equivalent of acetylated tetrodamine. J. Am. Chem. Soc 1972, 94, 9217–9219. [Google Scholar]
- Kishi, Y; Fukuyama, T; Aratani, M; Nakatsubo, F; Goto, T; Inoue, S; Tanino, H; Sugiura, S; Kakoi, H. Synthetic studies on tetrodotoxin and related compounds. IV. Stereospecific total syntheses of d,l-tetrodotoxin. J. Am. Chem. Soc 1972, 94, 9219–9221. [Google Scholar]
- Hanessian, S. The Total Synthesis of Natural Products: The Chiron Approach; Pergamon Press: New York, NY, USA, 1983. [Google Scholar]
- Hanessian, S. Approaches to the total synthesis of natural products using “chiral templates” derived from carbohydrates. Acc. Chem. Res 1979, 12, 159–165. [Google Scholar]
- Fraser-Reid, B; Anderson, RC. Carbohydrate derivates in the asymmetric synthesis of natural products. Fort. Chem. Org. Nat 1980, 39, 1–61. [Google Scholar]
- Fraser-Reid, B; Sun, KM; Tam, TF. Carbohydrate derivatives in the asymmetric synthesis of natural products: Some applications of furanose sugars. Bull Soc Chim Fr 1981, 238–246. [Google Scholar]
- Lichtenthaler, FW. Enantiopure Building Blocks from Sugars and their utilization in natural product synthesis. Mod. Synth. Meth 1992, 6, 273–376. [Google Scholar]
- Tadano, K. Natural product synthesis starting with carbohydrates based on the Claisen rearrangement protocol. Stud. Nat. Prod. Chem 1992, 10, 405–455. [Google Scholar]
- Hanessian, S. Reflections on the total synthesis of naturalproducts: Art, craft, logic, and the chiron approach. Pure Appl. Chem 1993, 65, 1189–1204. [Google Scholar]
- Li, J-C; Li, Y-L; Peng, Z-H; Sun, X-L; Wang, Y-F; Wu, W-L; Wu, Y-L; Yao, Z-J. Syntheses of chiral acyclic natural products from sugar. J. Chin. Chem. Soc 1995, 42, 681–689. [Google Scholar]
- Witczak, ZJ. Chiral carbohydrate building blocks with a new perspective: Revisited. ACS Symp. Ser 2003, 841, 1–19. [Google Scholar]
- Lichtenthaler, FW. Sugar-derived building blocks for the synthesis of non-carbohydrate natural products. ACS Symp. Ser 2003, 841, 47–83. [Google Scholar]
- Tatsuta, K. Recent progress in total synthesis and development of natural products using carbohydrates. ACS Symp. Ser 2003, 841, 157–179. [Google Scholar]
- Isobe, M; Ichikawa, Y. Synthesis of natural and unnatural products from sugar synthons. ACS Symp. Ser 2003, 841, 181–193. [Google Scholar]
- Ramesh, NG; Balasubramanian, KK. 2-C-formyl glycals: Emerging chiral synthons in organic synthesis. Eur. J. Org. Chem 2003, 2003, 4477–4487. [Google Scholar]
- Tatsuta, K; Hosokawa, S. Total syntheses of bioactive natural products from carbohydrates. Sci. Technol. Adv. Mater 2006, 7, 397–410. [Google Scholar]
- Jarosz, S. Sugars in the synthesis of natural products and their mimics. Chem. Today 2006, 24, 58–61. [Google Scholar]
- Zhou, J; Wang, G; Zhang, L-H; Ye, X-S. From exocyclic-olefinic carbohydrate derivatives to functionalized carbocyclic compounds. Curr. Org. Chem 2006, 10, 625–642. [Google Scholar]
- Fraser-Reid, B; Lopez, JC. Unsaturated sugars: A rich platform for methodological and synthetic studies. Curr. Org. Chem 2009, 13, 532–553. [Google Scholar]
- Ohyabu, N; Nishikawa, T; Isobe, M. First asymmetric total synthesis of tetrodotoxin. J. Am. Chem. Soc 2003, 125, 8798–8805. [Google Scholar]
- Nishikawa, T; Urabe, D; Isobe, M. An efficient total synthesis of optically active tetrodotoxin. Angew. Chem. Int. Ed 2004, 43, 4782–4785. [Google Scholar]
- Urabe, T; Nishikawa, T; Isobe, M. An efficient total synthesis of optically active tetrodotoxin from levoglucosenone. Chem. Asian J 2006, 1, 125–135. [Google Scholar]
- Nishikawa, T; Asai, M; Ohyabu, N; Yamamoto, N; Isobe, M. Stereocontrolled synthesis of (−)-5,11-dideoxytetrodotoxin. Angew. Chem. Int. Ed 1999, 38, 3081–3084. [Google Scholar]
- Asai, M; Nishikawa, T; Ohyabu, N; Yamamoto, N; Isobe, M. Stereocontrolled synthesis of (−)-5,11-dideoxoytetrodotoxin. Tetrahedron 2001, 57, 4543–4558. [Google Scholar]
- Nishikawa, T; Asai, M; Ohyabu, N; Yamamoto, N; Fukuda, Y; Isobe, M. Synthesis of a common key intermediate for (−)-tetrodotoxin and its analogs. Tetrahedron 2001, 57, 3875–3883. [Google Scholar]
- Nishikawa, T; Urabe, D; Yoshida, K; Iwabuchi, T; Asai, M; Isobe, M. Stereocontrolled synthesis of 8,11-dideoxytetrodotoxin, unnatural analogue of puffer fish toxin. Org. Lett 2002, 4, 2679–2682. [Google Scholar]
- Nishikawa, T; Asai, M; Isobe, M. Asymmetric total synthesis of 11-deoxytetrodotoxin, a naturally occurring congener. J. Am. Chem. Soc 2002, 124, 7847–7852. [Google Scholar]
- Nishikawa, T; Urabe, D; Yoshida, K; Iwabuchi, T; Asai, M; Isobe, M. Total syntheses of 11-deoxytetrodotoxin and 8,11-dideoxytetrodotoxin. Pure Appl. Chem 2003, 75, 251–257. [Google Scholar]
- Nishikawa, T; Urabe, D; Yoshida, K; Iwabuchi, T; Asai, M; Isobe, M. Stereocontrolled synthesis of 8,11-dideoxytetrodotoxin, an unnatural analogue of puffer fish toxin. Chem. Eur. J 2004, 10, 452–462. [Google Scholar]
- Satake, Y; Nishikawa, T; Hiramatsu, T; Araki, H; Isobe, M. Scalable synthesis of a new dihydroxylated intermediate for tetrodotoxin and its analogues. Synthesis 2010, 12, 1992–1998. [Google Scholar]
- Sato, K; Akai, S; Sugita, N; Ohsawa, T; Kogure, T; Shoji, H; Yoshimura, J. Novel and stereocontrolled synthesis of (±)-tetrodotoxin from myo-inositol. J. Org. Chem 2005, 70, 7496–7504. [Google Scholar]
- Funabashi, M; Wakai, H; Sato, K; Yoshimura, J. Branched-chain sugars. Part 15. Synthesis of 1l-(1,2,3′,4,5/3,6)-3-hydroxymethyl-4,5-O-isopropylidene-3,3′-O-methylene-6-nitro- 2,3,4,5-tetrahydroxycyclohexanecarbaldehyde dimethyl acetal, a potential key compound for total synthesis of optically active tetrodotoxin. J Chem Soc Perkin Trans 1 1980, 14–19. [Google Scholar]
- Sato, KI; Akai, S; Shoji, H; Sugita, N; Yoshida, S; Nagai, Y; Suzuki, K; Nakamura, Y; Kajihara, Y; Funabashi, M; et al. Stereoselective and efficient total synthesis of optically active tetrodotoxin from d-glucose. J. Org. Chem 2008, 73, 1234–1242. [Google Scholar]
- Akai, S; Seki, H; Sugita, N; Kogure, T; Nishizawa, N; Suzuki, K; Nakamura, Y; Kajihara, Y; Yoshimura, J; Sato, K. Total synthesis of (−)-tetrodotoxin from d-glucose: A new route to multi-functionalized cyclitol employing the Ferrier(II) rection toward (−)-tetrodotoxin. Bull. Chem. Soc. Jpn 2010, 83, 279–287. [Google Scholar]
- Hinman, A; Du Bois, J. A stereoselective synthesis of (−)-tetrodotoxin. J. Am. Chem. Soc 2003, 125, 11510–11511. [Google Scholar]
- Taber, DF; Storck, PH. Synthesis of (−)-tetrodotoxin: Preparation of an advanced cyclohexenone intermediate. J. Org. Chem 2003, 68, 7768–7771. [Google Scholar]
- Alonso, RA; Burgey, CS; Venkateswara, RB; Vite, GD; Vollerthun, R; Zottola, MA; Fraser-Reid, B. Carbohydrates to carbocycles: Synthesis of the densely functionalized carbocyclic core of tetrodotoxin by radical cyclization of an anhydro sugar precursor. J. Am. Chem. Soc 1993, 115, 6666–6672. [Google Scholar]
- Burgey, CS; Vollerthun, R; Fraser-Reid, B. Armed/disarmed effects and adamantly expansion of some caged tricyclic acetals en route to tetrodotoxin. J. Org. Chem 1996, 61, 1609–1618. [Google Scholar]
- Fraser-Reid, B; Burgey, CS; Vollerthun, R. Carbohydrates to densely functionalized carbocycles: “Armed and disarmed” effects in an approach to tetrodotoxin. Pure Appl. Chem 1998, 70, 285–288. [Google Scholar]
- Noya, B; Alonso, R. Radical cyclisation onto C-3 of 1,6-anhydro-β-d-mannopyranose derivatives. Applications to the formation of a C8a centre of (−)-tetrodotoxin. Tetrahedron Lett 1997, 38, 2745–2748. [Google Scholar]
- Noya, B; Paredes, MD; Ozores, L; Alonso, R. 5-exo Radical cyclization onto 3-alkoxyketimino-1,5-anhydromannopyranosese. Efficient preparation of synthetic intermediates for (−)-tetrodotoxin. J. Org. Chem 2000, 65, 5960–5968. [Google Scholar]
- Torrente, S; Noya, B; Paredes, MD; Alonso, R. Intramolecular 1,3-Dipolar cycloadditions of sugar ketonitrones: A convenient method for stereoselective formation of nitrogenated quaternary centers. J. Org. Chem 1997, 62, 6710–6711. [Google Scholar]
- Torrente, S; Noya, B; Branchadell, V; Alonso, R. Intra- and Intermolecular 1,3-Dipolar Cycloaddition of Sugar Ketonitrones with Mono-, Di-, and Trisubstituted Dipolarophiles. J. Org. Chem 2003, 68, 4772–4783. [Google Scholar]
- Ohtani, Y; Shinada, T; Ohfune, Y. Stereoselective construction of a contiguous tetraol system in tetrodotoxin by means of repetitive operations involving epoxidation and ring-opening reactions of allyl sulfone. Synlett 2003, 2003, 619–622. [Google Scholar]
- Keana, JFW; Kim, CU. Synthetic intermediates potentially useful for the synthesis of tetrodotoxin and derivatives. III. Synthesis of a key lactone intermediate from shikimic acid. J. Org. Chem 1971, 36, 118–127. [Google Scholar]
- Keana, JFW; Boyle, PJ; Erion, M; Hartling, R; Husman, JR; Richman, JE; Roman, RB; Wah, RM. Synthetic intermediates potentially useful for the synthesis of tetrodotoxin derivatives. 8. A series of highly functionalized pyrimidinones. J. Org. Chem 1983, 48, 3621–3626. [Google Scholar]
- Keana, JFW; Bland, JS; Boyle, PJ; Erion, M; Hartling, R; Husman, JR; Roman, RB; Ferguson, G; Parvez, M. Synthetic intermediates potentially useful for the synthesis of tetrodotoxin and derivatives. 9. Hydroquinazolines possessing the carbon skeleton of tetrodotoxin. J. Org. Chem 1983, 48, 3627–3631. [Google Scholar]
- Cagide-Fagin, F; Alonso, R. A cascade annulation based convergent approach to racemic tetrodotoxin. Eur. J. Org. Chem 2010, 2010, 6741–6747. [Google Scholar]
- Kozikowski, AP; Stein, PD. The INOC route to carbocyclics: A formal total synthesis of (+/−)-sarkomycin. J. Am. Chem. Soc 1982, 104, 4023–4024. [Google Scholar]
- Curran, DP. Reduction of Δ-2-isoxazolines: A conceptually different approach to the formation of aldol adducts. J. Am. Chem. Soc 1982, 104, 4024–4026. [Google Scholar]
- Itoh, T; Watanabe, M; Fukuyama, T. Synthetic approach to tetrodotoxin. Synlett 2002, 2002, 1323–1325. [Google Scholar]
- Ciufolini, MA; Braun, NA; Canesi, S; Ousmer, M; Chang, J; Chai, D. Oxidative amidation of phenols through the use of hypervalent iodine reagents: Development and applications. Synthesis 2007, 2007, 3759–3772. [Google Scholar]
- Ciufolini, MA; Canesi, S; Ousmer, M; Braun, NA. Synthetic ventures inspired by biosynthetic hypotheses: The evolution of a method for the oxidative amidation of phenols. Tetrahedron 2006, 62, 5318–5337. [Google Scholar]
- Liang, H; Ciufolini, MA. Synthetic aspects of the oxidative amidation of phenols. Tetrahedron 2010, 66, 5884–5892. [Google Scholar]
- Braun, NA; Ciufolini, MA; Peters, K; Peters, E-M. Synthesis of spirolactams from tyrosine amides and related substances. Tetrahedron Lett 1998, 39, 4667–4670. [Google Scholar]
- Braun, NA; Bray, J; Ciufolini, MA. Hypervalent iodine oxidation of indolic 2-oxazolines. Tetrahedron Lett 1999, 40, 4985–4988. [Google Scholar]
- Braun, NA; Bray, J; Ousmer, M; Peters, K; Peters, E-M; Bouchu, D; Ciufolini, MA. New oxidative transformations of phenolic and indolic oxazolines: An avenue to useful azaspirocyclic building blocks. J. Org Chem 2000, 65, 4397–4408. [Google Scholar]
- Canesi, S; Belmont, P; Bouchu, D; Rousset, L; Ciufolini, MA. Efficient oxidative spirocyclization of phenolic sulfonamides. Tetrahedron Lett 2002, 43, 5193–5195. [Google Scholar]
- Liang, H; Ciufolini, MA. Tandem phenolic oxidative amidation–intramolecular Diels-Alder reaction: An approach to the himandrine core. Org. Lett 2010, 12, 1760–1763. [Google Scholar]
- Liang, H; Ciufolini, MA. Oxidative spirocyclization of phenolic sulfonamides: Scope and applications. Chem. Eur. J 2010, 16, 13262–13270. [Google Scholar]
- Canesi, S; Bouchu, D; Ciufolini, MA. Nitrogenous educts through oxidative amidation of phenols: The bimolecular reaction. Org. Lett 2005, 7, 175–177. [Google Scholar]
- Liang, H; Ciufolini, MA. Improved procedure for the bimolecular oxidative amidation of phenols. J. Org. Chem 2008, 73, 4299–4301. [Google Scholar]
- Ousmer, M; Braun, NA; Ciufolini, MA. Total synthesis of FR-901483. Org. Lett 2001, 3, 765–767. [Google Scholar]
- Ousmer, M; Braun, NA; Bavoux, C; Perrin, M; Ciufolini, MA. Total synthesis of tricyclic azaspirane derivatives of tyrosine: FR-901483 and TAN 1251C. J. Am. Chem. Soc 2001, 123, 7534–7538. [Google Scholar]
- Canesi, S; Bouchu, D; Ciufolini, MA. Fully stereocontrolled syntheses of (−)-cylindricine C and (−)-2-epicylindricine C: A departure in sulfonamide chemistry. Angew. Chem. Int. Ed 2004, 43, 4336–4338. [Google Scholar]
- Canesi, S. Dissertation, Université Claude Bernard Lyon 1, Villeurbanne, France, 2004.
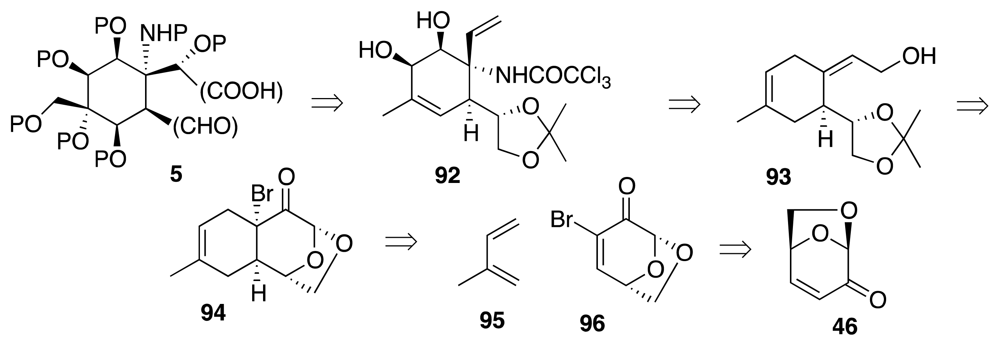
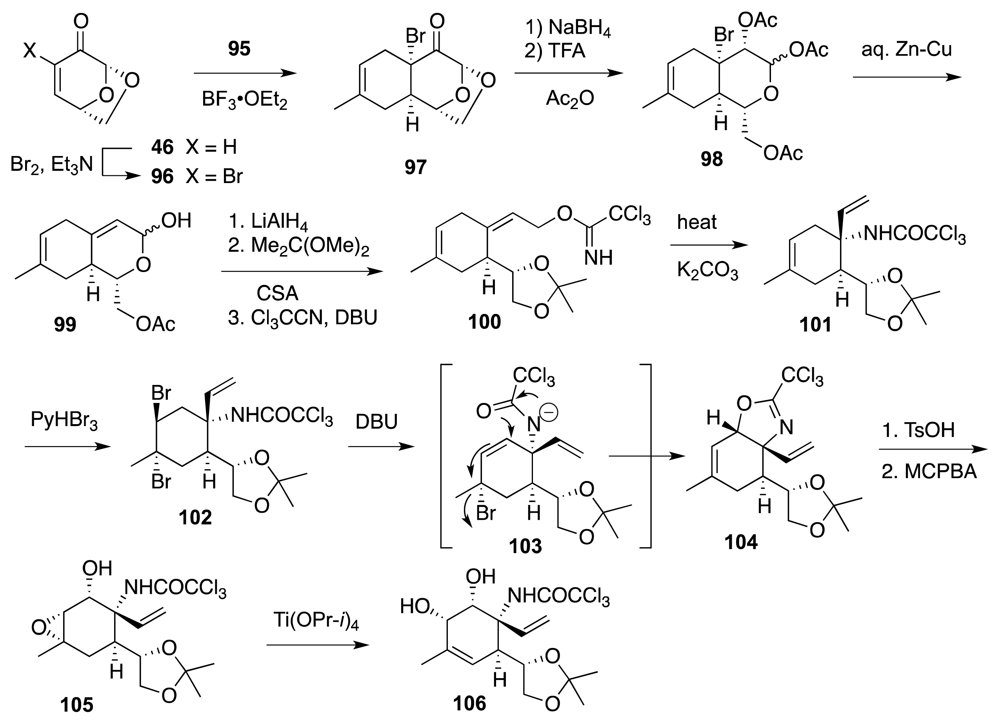
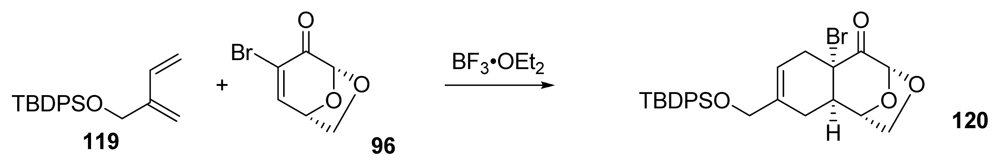
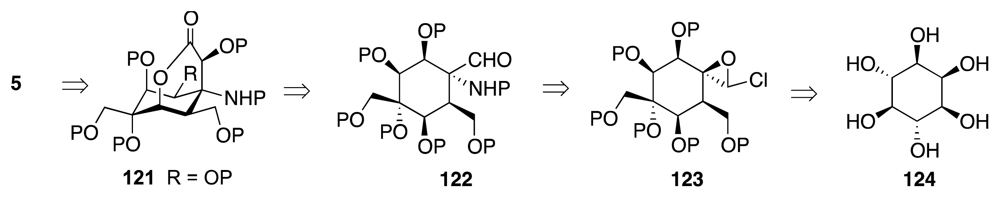
- Mendelsohn, BA; Ciufolini, MA. Approach to tetrodotoxin via the oxidative amidation of a phenol. Org. Lett 2009, 11, 4736–4739. [Google Scholar]
- Mendelsohn, B; Lee, S; Kim, S; Teyssier, F; Aulakh, VS; Ciufolini, MA. Oxidation of oximes to nitrile oxides with hypervalent iodine reagents. Org. Lett 2009, 11, 1539–1542. [Google Scholar]
- Jen, T; Mendelsohn, B; Ciufolini, MA. Oxidation of α-oxo-oximes to nitrile oxides with hypervalent iodine reagents. J. Org. Chem 2011, 76, 728–731. [Google Scholar]
- Turner, CD; Ciufolini, MA. Oxidation of oximes with hypervalent iodine reagents: Opportunities, development, and applications. ARKIVOC 2011, 2011, 410–428. [Google Scholar]
















© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chau, J.; Ciufolini, M.A. The Chemical Synthesis of Tetrodoxin: An Ongoing Quest. Mar. Drugs 2011, 9, 2046-2074. https://doi.org/10.3390/md9102046
Chau J, Ciufolini MA. The Chemical Synthesis of Tetrodoxin: An Ongoing Quest. Marine Drugs. 2011; 9(10):2046-2074. https://doi.org/10.3390/md9102046
Chicago/Turabian StyleChau, Jaclyn, and Marco A. Ciufolini. 2011. "The Chemical Synthesis of Tetrodoxin: An Ongoing Quest" Marine Drugs 9, no. 10: 2046-2074. https://doi.org/10.3390/md9102046
APA StyleChau, J., & Ciufolini, M. A. (2011). The Chemical Synthesis of Tetrodoxin: An Ongoing Quest. Marine Drugs, 9(10), 2046-2074. https://doi.org/10.3390/md9102046