Neurotoxins from Marine Dinoflagellates: A Brief Review


Abstract
:1. Introduction
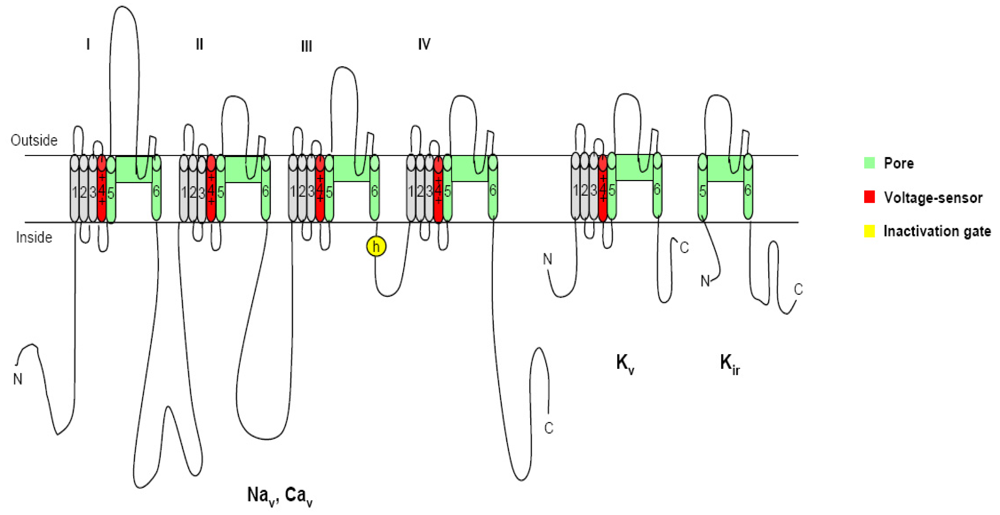
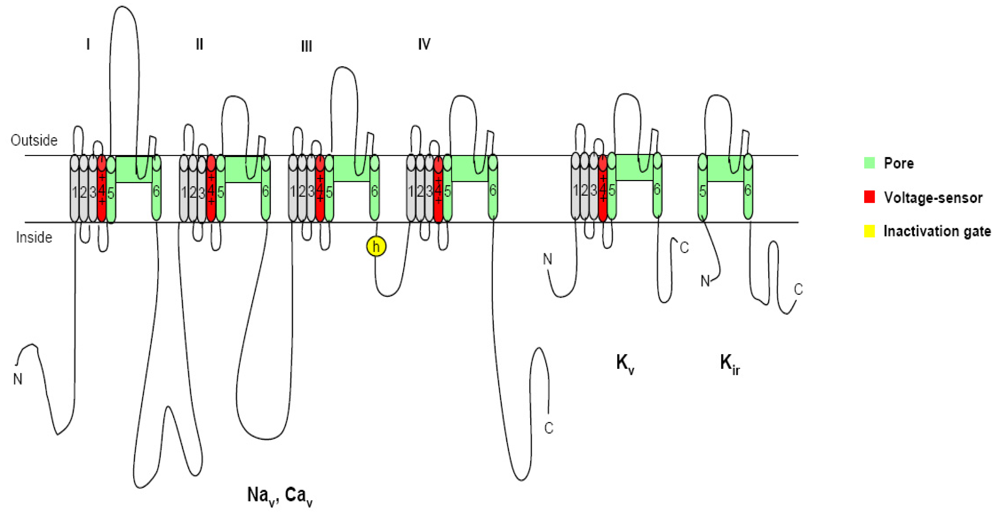
2. Voltage-gated ion channels and neurotoxins
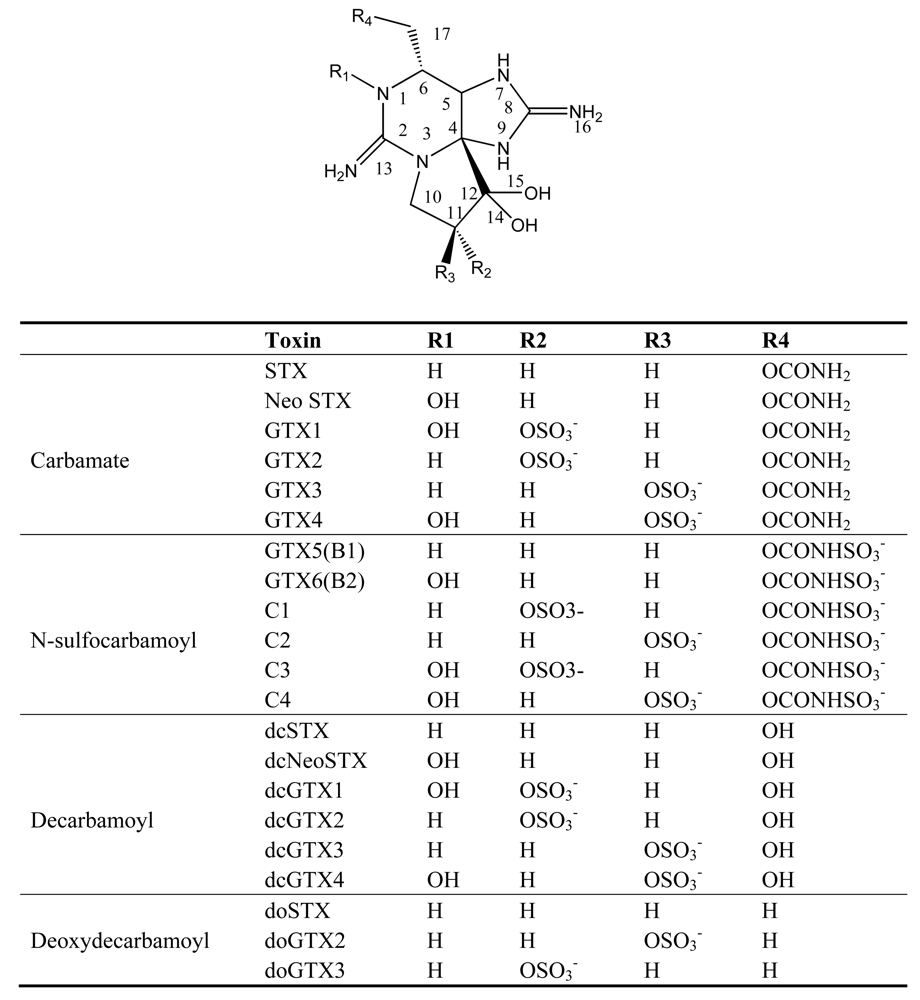
3. Paralytic shellfish poisoning (PSP)
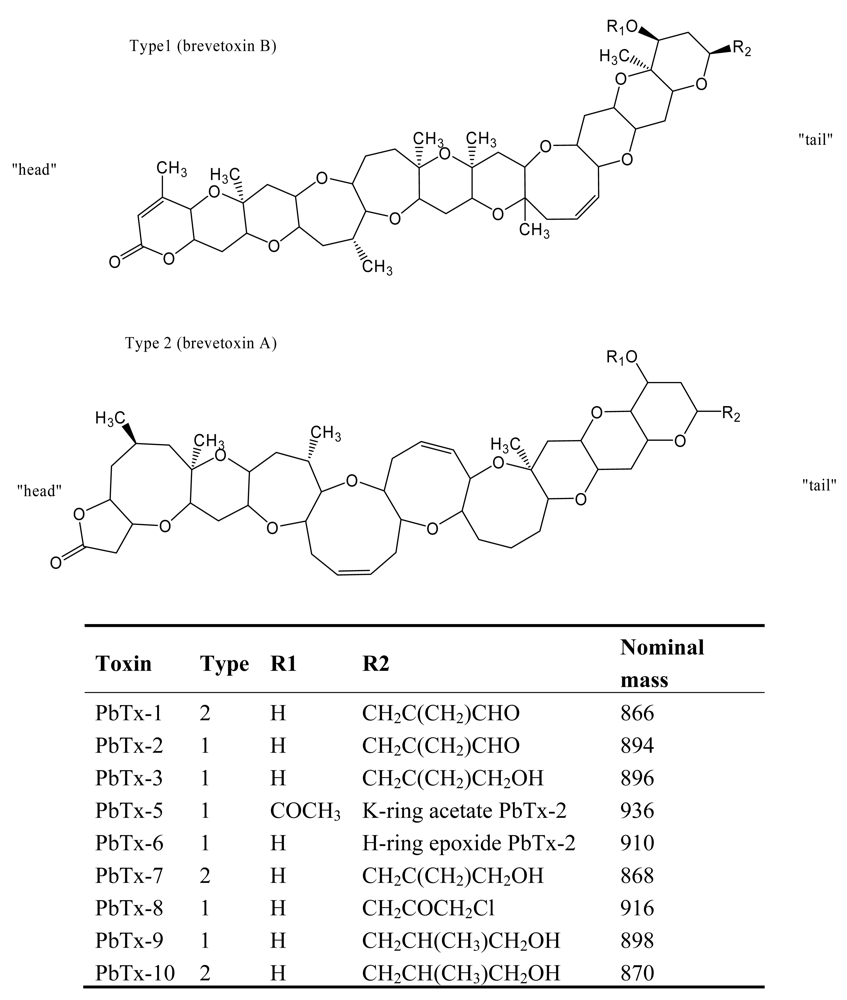
4. Neurotoxic Shellfish Poisoning (NSP)
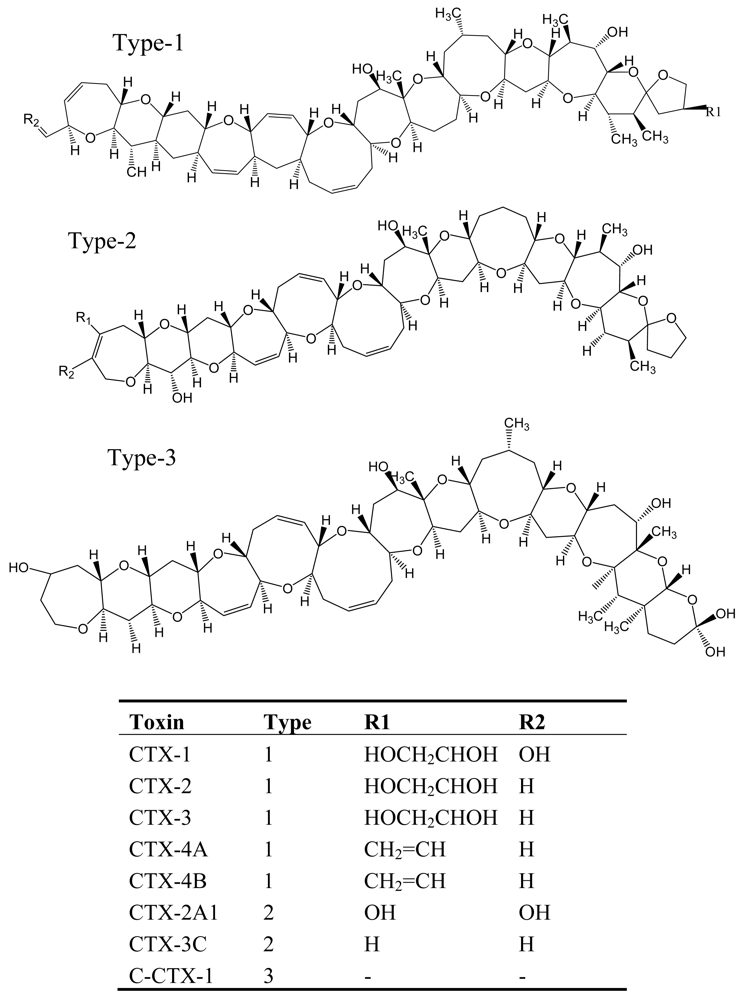
5. Ciguatera Fish Poisoning (CFP)
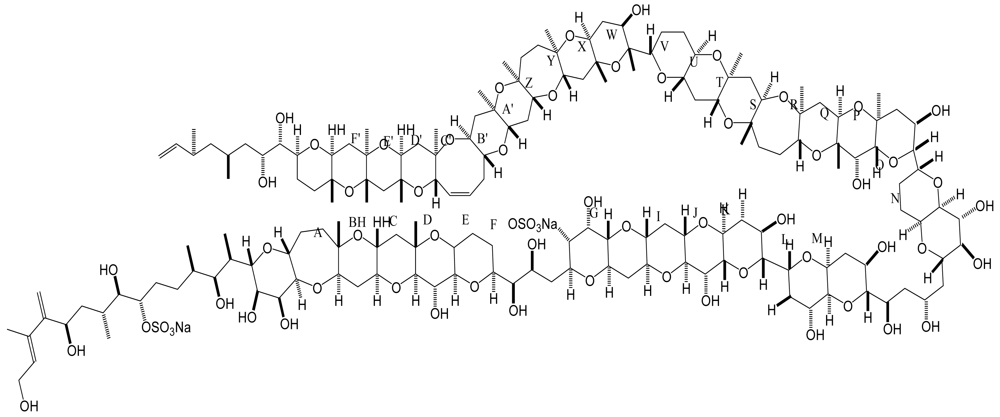
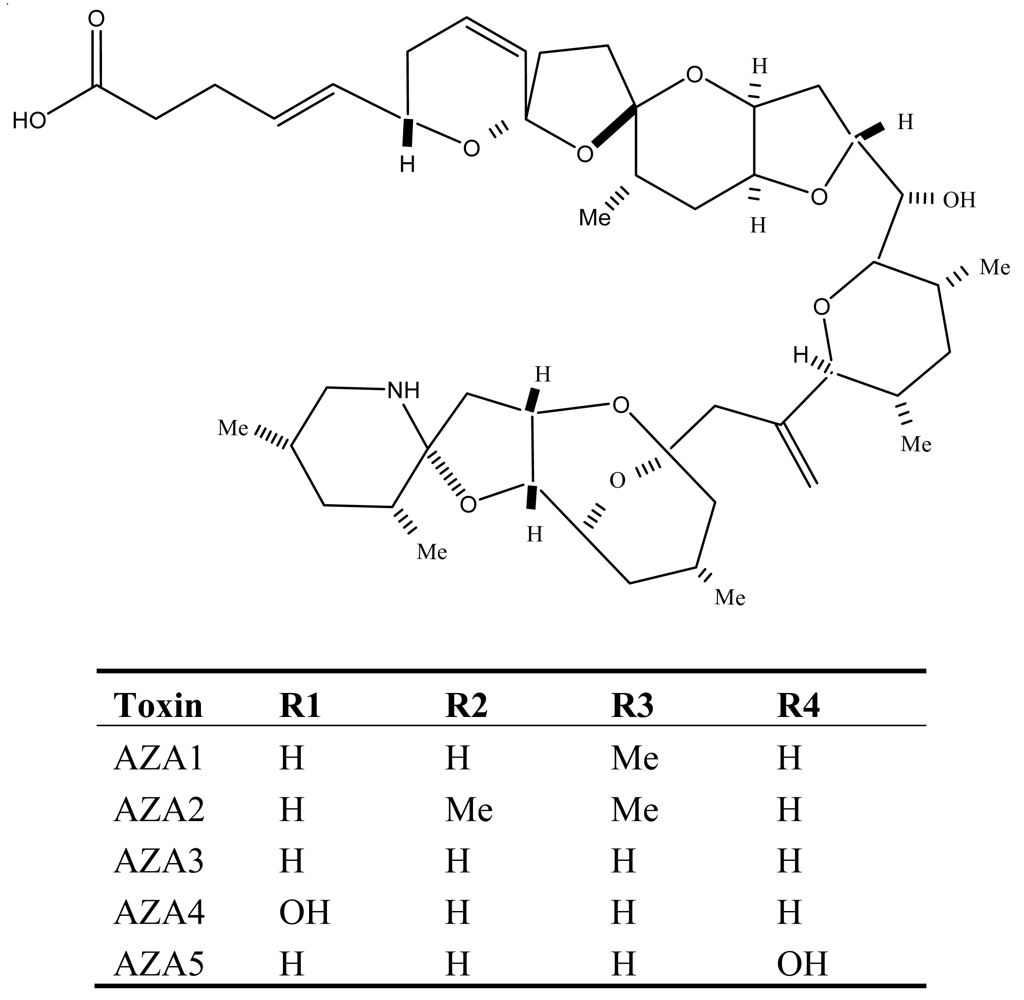
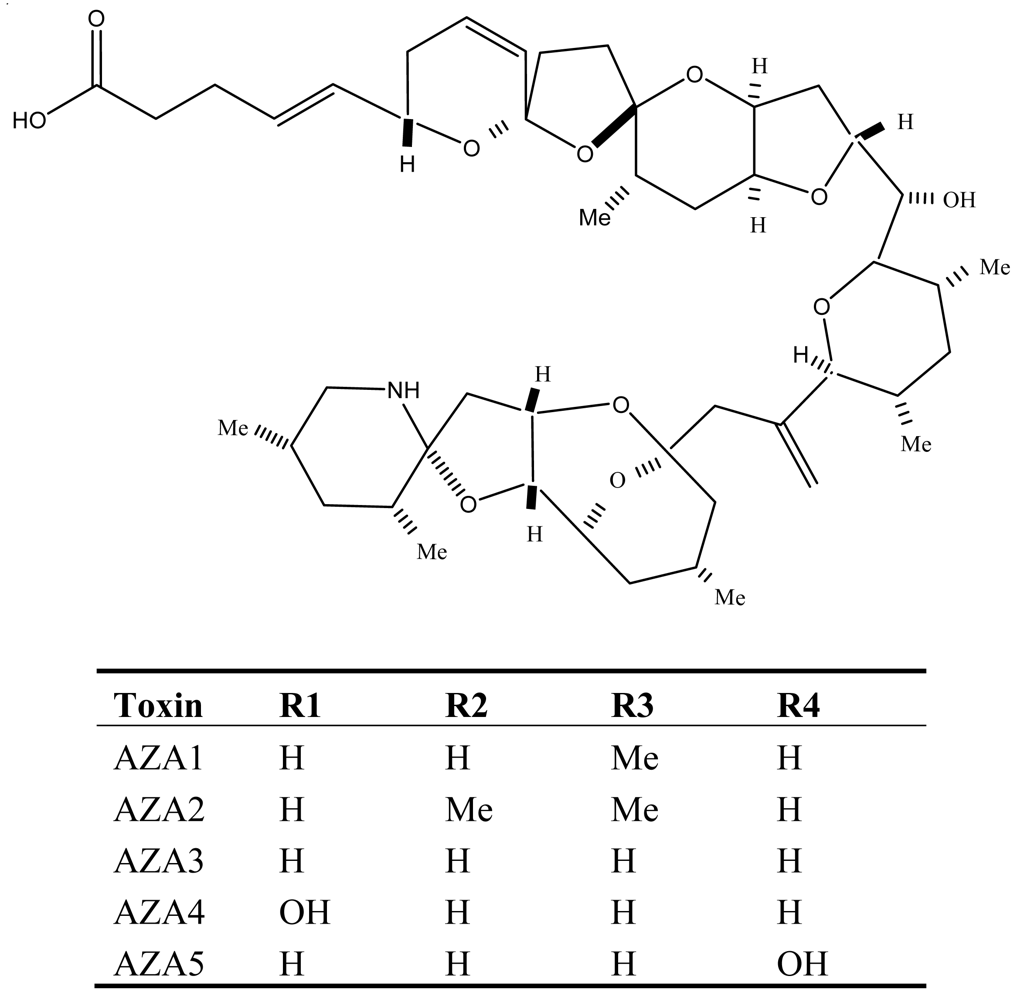
6. Azaspiracid Shellfish Poisoning (AZP)
7. Yessotoxin (YTX)
8. Palytoxin (PTX)
9. Summary
Acknowledgements
References and Notes
- Anderson, DM. Okaichi, T, Anderson, DM, Nemoto, T, Eds.; Toxic algal blooms and red tides: a global perspective. In Red Tides: Biology, Environmental Science and Toxicology; Elsevier: New York, 1989; pp. 11–16. [Google Scholar]
- Smayda, TJ. Graneli, E, Sundstrom, B, Edler, L, Anderson, DM, Eds.; Novel and nuisance phytoplankton blooms in the sea: evidence for a global epidemic. In Toxic Marine Phytoplankton; Elsevier: New York, 1990; pp. 29–40. [Google Scholar]
- Hallegraeff, GM. Hallegraeff, GM, Anderson, DM, Cembella, AD, Eds.; Harmful algal blooms: a Global review. In Manual on Harmful Marine Microalgae; UNESCO: Landais, France, 2005; pp. 25–49. [Google Scholar]
- Van Dolah, FM. Botana, L, Ed.; Diversity of Marine and Freshwater Algal Toxins. In Seafood Toxicology: Pharmacology, Physiology and Detection; Marcel Dekker: New York, 2000; pp. 19–43. [Google Scholar]
- Quod, JP; Turquet, J. Ciguatera fish poisoning in Reunion island (SW Indian Ocean): epidemiology and clinical patterns. Toxicon 1996, 34, 779–785. [Google Scholar]
- Geraci, JR; Anderson, DM; Timperi, RJ; St Aubin, DJ; Early, GA; Prescott, JH; Mayo, CA. Humpback whales (Megaoetera novaeangliae) fatally poisoned by dinoflagellate toxin. Can J Fish Aq Sc 1989, 46, 1895–1898. [Google Scholar]
- Landsberg, JH; Steidinger, K. Reguera, B, Blanco, J, Fernandez, ML, Wyatt, T, Eds.; A historical review of red tide events caused by Gymnodinium breve as related to mass mortalities of the endangered manatee (Trichechus manatus latirostris) in Florida, USA. In Harmful Microalgae; IOC of UNESCO and Xunta de Galicia: Spain, 1998; pp. 97–100. [Google Scholar]
- Scholin, CA; Gulland, F; Doucette, GJ; Benson, S; Busman, M; Chavez, FP; Cordaro, J; DeLong, R; De Vogelaere, A; Harvey, J; Haulena, M; Lefebvre, K; Lipscomb, T; Loscutoff, S; Lowenstine, LJ; Marin, R, III; Miller, PE; McLellan, WA; Moeller, PDR; Powell, CL; Rowles, T; Silvagni, P; Silver, M; Spraker, T; Trainer, V; Van Dolah, FM. Mortality of sea lions along the central California coast linked to a toxic diatom bloom. Nature 2000, 430, 80–84. [Google Scholar]
- Flewelling, LJ; Naar, JP; Abbott, JP; Baden, DG; Barros, NB; Bossart, GD; Bottein, MY; Hammond, DG; Haubold, EM; Heil, CA; Henry, MS; Jacocks, HM; Leighfield, TA; Pierce, RH; Pitchford, TD; Rommel, SA; Scott, PS; Steidinger, KA; Truby, EW; Van Dolah, FM; Landsberg, JH. Brevetoxicosis: red tides and marine mammal mortalities. Nature 2005, 435, 755–756. [Google Scholar]
- Trainer, VL; Baden, DG. High affinity binding of red tide neurotoxins to marine mammal brain. Aquat Toxicol 1999, 46, 139–128. [Google Scholar]
- Botana, LM. Seafood and Freshwater toxins: pharmacology, physiology, and detection; Marcel Dekker: New York, 2000. [Google Scholar]
- Gessner, BD. Botana, LM, Ed.; Neurotoxic toxins. In Seafood and Freshwater toxins: Pharmacology, Physiology and Detection; Marcel Dekker: New York, 2000; pp. 65–90. [Google Scholar]
- Catterall, WA; Cestele, S; Yarov-Yarovoy, V; Yu, FH; Konoki, K; Scheuer, T. Voltage-gated channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar]
- Yu, FH; Catterall, WA. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Science STKE 2004, re 15. [Google Scholar]
- Catterall, WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu Rev Pharmacol Toxicol 1980, 20, 15–43. [Google Scholar]
- Martin-Eauclaire, MF; Couraud, F. Chang, LW, Dyer, RS, Eds.; Scorpion neurotoxins: effects and mechanisms. In Handbook of Neurotoxicology; Marcel-Dekker: New York, USA, 1992; pp. 683–716. [Google Scholar]
- Catterall, WA; Risk, M. Toxin T46 from Ptychodiscus brevis (formerly Gymnodinium breve) enhances activation of voltage-sensitive sodium channels by veratridine. Mol Pharmacol 1981, 19, 345–348. [Google Scholar]
- Poli, MA; Mende, TJ; Baden, DG. Brevetoxins, unique activators of voltage-sensitive sodium channels, bind to specific sites in rat brain synaptosones. Mol Pharmacol 1986, 30, 129–135. [Google Scholar]
- Fainzillber, M; Kofman, O; Zoltkin, E; Gordon, D. A new neurotoxin receptor site on sodium channels is identified by a conotoxin that affects sodium channel inactivation in mollusks and acts as an antagonist in rat brain. J Biol Chem 1994, 269, 2574–2580. [Google Scholar]
- Cestele, S; Catterall, WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar]
- Kodama, M. Botana, L, Ed.; Ecology, classification, and origin. In Seafood and freshwater toxins: Pharmacology, Physiology and Detection; Marcel Dekker: New York, 2000; pp. 125–150. [Google Scholar]
- Sommer, H; Meyer, KF. Paralytic shellfish poisoning. Arch Pathol 1937, 24, 560–598. [Google Scholar]
- Shumway, SE. A review of the effects of algal blooms on shellfish and aquaculture. J World Aquac Soc 1990, 21, 65–104. [Google Scholar]
- Shimizu, Y. Microalgal metabolites: a respective. Ann Rev Microbiol 1996, 50, 431–465. [Google Scholar]
- Halsetead, BW. Poisonous and venomous marine animals of the world; Princeton: Darwin, 1978. [Google Scholar]
- Lagos, N; Andrinolo, D. Botana, LM, Ed.; Paralytic shellfish poisoning (PSP): toxicology and kinetics. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Marcel Dekker: New York, USA, 2000; pp. 203–215. [Google Scholar]
- Kao, CY. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomena. Pharmacol Rev 1966, 18, 997–1049. [Google Scholar]
- Hill, B. The receptor for tetrodotoxin and saxitoxin: a structural hypothesis. Biophys J 1975, 15, 615–619. [Google Scholar]
- Kao, CY; Walkwe, SE. Active groups of saxitoxin and tetrodotoxin as deduced from action of saxitoxin analogs on frog muscle and squid axon. J Physiol 1982, 323, 619–637. [Google Scholar]
- Shimizu, Y. Recent progress in marine toxin research. Pure Appl Chem 1980, 54, 1973–1980. [Google Scholar]
- Numa, S; Noda, M. Molecular structure of sodium channels. Ann NY Acad Sci 1986, 479, 338–355. [Google Scholar]
- Bricelj, VM; Connel, L; Konoki, K; Macquarrie, SP; Scheuer, T; Catterall, WA; Trainer, VL. Sodium channel mutation leading to saxitoxin resistance in clams increase risk of PSP. Nature 2005, 434, 763–767. [Google Scholar]
- Steidinger, KA. Phytoplankton ecology: A conceptual review based on eastern. Gulf of Mexico research. CRC Crit Rev Microbiol 1973, 3, 49–67. [Google Scholar]
- Baden, DG. Marine food-borne dinoflagellate toxins. Int Rev Cytol 1983, 82, 99–150. [Google Scholar]
- Baden, DG; Mende, TJ. Toxicity of two toxins from the Florida red tide marine dinoflagellate Gymnodinium breve. Toxicon 1982, 20, 457–461. [Google Scholar]
- Poli, M; Mende, TJ; Baden, DG. Brevetoxins, unique activators of voltage-sensitive sodium channels bind to specific sites in rat brain synaptosomes. Mol Pharmacol 1986, 30, 129–135. [Google Scholar]
- Baden, D; Fleming, LE; Bean, JA. deWolf, FA, Ed.; Chapter: Marine Toxins. In Handbook of Clinical Neurology: Intoxications of the Nervous System Part H. Natural Toxins and Drugs; Elsevier: Amsterdam, 1995; pp. 141–175. [Google Scholar]
- Baden, DG; Bourdelais, AJ; Jacocks, H; Michelliza, S; Naar, J. Natural and Derivative Brevetoxins: Historical Background, Multiplicity, and Effects. Environ Health Persp 2005, 113. [Google Scholar]
- Sagir Ahmed, MD; Arakawa, O; Onoue, Y. Lassus, P, Arzul, G, Erhard, E, Gentien, P, Marcaillou, C, Eds.; Toxicity of cultured Chatonella marina. In Harmful Marine Algal Blooms; Lavoisier: Paris, 1995; pp. 499–504. [Google Scholar]
- Khan, S; Arakawa, O; Onoue, Y. Neurotoxins in a toxic red tide of Heterosigma akashiwo (Raphidophyceae) in Kagoshima Bay, Japan. Aquacul Res 1997, 28, 9–14. [Google Scholar]
- Hallegraeff, GM; Munday, BL; Baden, DG; Whitney, PL. Reguera, B, Blanco, J, Ferandz, ML, Wyatt, TEd, Eds.; Chatonnella maria raphidophyte bloom associated with mortality of cultured bluefin tuna (Thunnus maccoyii) in south Australia. In Harmful algae; Xunta de Galacia and IOC: Santiago de Compostela, Spain, 1998; pp. 93–96. [Google Scholar]
- Kirkpatrick, B; Fleming, LE; Squicciarini, D; Backer, LC; Clark, R; Abraham, W; Benson, J; Chenge, YS; Johnson, D; Pierce, R; Zaias, J; Bossart, GD; Baden, DG. Literature review of Florida red tide: Implications for human health effects. Harmful algae 2004, 3, 99–115. [Google Scholar]
- Morris, PD; Campbell, DS; Taylor, TJ; Freeman, JI. Clinical and epidemiological features of neurotoxic shellfish poisoning in North Carolina. Am J Public Health 1991, 81(4), 471–474. [Google Scholar]
- Baden, DG; Adams, DJ. Botana, LM, Ed.; Brevetoxins: Chemistry, mechanism of action, and methods of detection. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; 2000; Marcel Dekker: New York; pp. 505–532. [Google Scholar]
- Baden, DG. Marine food-born dinoflagellate toxins. Int Rev Cytol 1983, 82, 99–150. [Google Scholar]
- Rein, KS; Baden, DG; Gawley, RE. Conformational analysis of the sodium channel modulator, brevetoxin A, comparison with brevetoxin B conformations, and a hypothesis about the common pharmacophore of the “site” toxins. J Org Chem 1994, 59, 2101–2106. [Google Scholar]
- Gallagher, P; Shinnick-Gallagher, P. Effect of brevetoxin in the rat phrenic nerve diaphragm preparation. Brit J Pharmacol 1980, 69, 367–372. [Google Scholar]
- Halstead, BW. Poisonous and Venomous Marine Animals of the World; Darwin Press: Princeton, 1988. [Google Scholar]
- Trainer, VL; Thomsen, WJ; Catterall, WA; Baden, DG. Photoaffinity labeling of the brevetoxin receptor on sodium channels in rat brain synaptosomes. Mol Pharmacol 1991, 40, 988–994. [Google Scholar]
- Watters, MR. Organic neurotoxins in seafoods. Clin Neurol Neurosurg 1995, 97, 119–124. [Google Scholar]
- Fleming, LE; Baden, DG. Neurotoxic Shellfish Poisoning: Public Health and Human Health Effects. White Paper for the Proceedings of the Texas Conference on Neurotoxic Shellfish Poisoning, Proceedings of the Texas NSP Conference, Corpus Christi (Texas), April, 1998; pp. 27–34.
- LePage, KT; Baden, DG; Murray, TF. Brevetoxin derivatives act as partial agonists at neurotoxin site 5 on the voltage-gated Na+ channel. Brain Res 2003, 959, 120–127. [Google Scholar]
- Guzman-Perez, SE; Park, DL. Botana, L, Ed.; Ciguatera toxins: Chemistry and diction. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Marcel Dekker: New York, 2000; pp. 401–418. [Google Scholar]
- Terao, K. Botana, L, Ed.; Ciguatera toxins: toxicology. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Marcel Dekker: New York, 2000; pp. 449–472. [Google Scholar]
- Yasumoto, T; Nakajima, I; Bagnis, R; Adachi, R. Finding of a dinoflagellate as a likely culprit of ciguatera. Jpn Soc Sci Fish 1977, 43, 1021–1026. [Google Scholar]
- Legrand, AM. Reguera, B, Blanco, J, Fernandez, M, Wyatt, T, Eds.; 1998; Ciguatera toxins: origin, transfer through the food chain and toxicity to humans. In Harmful Algae, Proceedings of the VIII International Conference on Harmful Algae; Xunta de Galicia and IOC of UNESCO: Vigo, Spain, 1999; pp. 39–43.
- Scheuer, PJ; Takahashi, W; Tsutsumi, J; Yoshida, T. Ciguatoxin: isolation and chemical nature. Science 1967, 155, 1267–1268. [Google Scholar]
- Tachibana, K; Nukina, M; John, YD; Scheuer, PJ. Recent developments in the molecular structure of ciguatoxin. Biol Bull 1987, 172, 122–127. [Google Scholar]
- Murata, M; Legrand, AM; Ishibashi, Y; Yasumoto, T. Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J Am Chem Soc 1990, 112, 4380–4386. [Google Scholar]
- Lewis, RJ; Vernoux, J-P; Brereton, IM. Structure of Caribbean ciguatoxin isolated from Caranx latus. J Am Chem Soc 1998, 120, 5914–5920. [Google Scholar]
- Lewis, RJ; Molgo, J; Adams, DJ. Botana, L, Ed.; Pharmacology of toxins involved in ciguatera and related fish poisonings. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Marcel Dekker, Inc.: New York, 2000; pp. 419–447. [Google Scholar]
- Cameron, J. Effects of ciguatoxin on nerve excitability in rats (part I). J Neurol Sci 1991, 101, 87–92. [Google Scholar]
- Mattei, C; Dechraoui, MY; Molgó, J; Meunier, FA; Legrand, AM; Benoit, E. Neurotoxins targetting receptor site 5 of voltage-dependent sodium channels increase the nodal volume of myelinated axons. J Neurosci Res 1999, 55, 666–673. [Google Scholar]
- Allsop, JL; Martini, L; Lebris, H; Pollard, J; Walsh, J; Hodgkinson, S. Neurologic manifestations of ciguatera. 3 cases with a neurophysiologic study and examination of one nerve biopsy. Rev Neurol 1986, 142, 590–597. [Google Scholar]
- Cameron, J. Electrophysiological studies on ciguatera poisoning in man (part II). J Neurol Sci 1991, 101, 93–97. [Google Scholar]
- Benoit, E. Nodal swelling produced by ciguatoxin-induced selective activation of sodium channels in myelinated nerve fibers. Neuroscience 1996, 71, 1121–1131. [Google Scholar]
- Lehane, L; Lewis, RJ. Ciguatera: recent advances but the risk remains. Int J Food Microbiol 2000, 61, 91–125. [Google Scholar]
- Holmes, MJ; Lewis, RJ. Purification characterization of large and small maitotoxins from cultured Gambierdiscus toxicus. Nat Toxins 1994, 2, 64–72. [Google Scholar]
- Estacion, M. Botana, L, Ed.; Ciguatera toxins: Mechanism of action and pharmacology of maitotoxin. In Seafood and Freshwater Toxins: Pharmacology, Physiology and Detection; Marcel Dekker, Inc.: New York, 2000; pp. 473–504. [Google Scholar]
- Kakizaki, A; Takahashi, M; Akagi, H; Tachikawa, E; Yamamoto, T; Taira, E; Yamakuni, T; Ohizumi, Y. Ca2+ channel activating action of maitotoxin in cultured brainstem neurons. Eur J Pharmacol 2006, 536, 223–231. [Google Scholar]
- Statake, M; Ofuji, K; Naoki, H; James, KJ; Furey, A; McMahon, T. Azaspiracid, a new marine toxin having unique spiro ring assembles, isolated from Irish mussels, Mytilus edulis. J Am Chem Soc 1998, 120, 9967–9968. [Google Scholar]
- Oufji, K; Statake, M; McMahon, T; Silker, J; James, KJ; Naoki, H. Two analogs of azaspiracid isolated from mussels, Mytilus edulis, involved in human intoxication in Ireland. Nat Toxins 1999, 7, 99–102. [Google Scholar]
- James, K; Lehane, M; Moroney, C; Fernandez-Puente, P; Statake, M; Yasumoto, T. Azaspiracid shellfish poisoning: unusual toxin dynamics in shellfish and the increased risk of acute human intoxications. Food Addit Contam 2002, 19, 555–561. [Google Scholar]
- James, K; Sierra, MD; Lehane, M; Brana Magdalena, A; Furey, A. Detection of five newly hydroxyl analogues of azaspiracids in shellfish using multiple tandem mass spectrometry. Toxicon 2003, 41, 277–283. [Google Scholar]
- Ito, E; Statake, M; Ofuji, K; Kurita, N; McMahon, T; James, K. Multiple organ damage caused by a new toxin azaspiracid, isolated from mussels produced in Ireland. Toxicon 2000, 38, 917–930. [Google Scholar]
- Alfonso, A; Roman, Y; Vieytes, MR. Azaspiracid-4 inhibits Ca2+ entry by stored operated channels in human T lymphocytes. Biochem Pharmacol 2005, 69, 1627–1636. [Google Scholar]
- Suganuma, M; Fujiki, H; Suguri, H; Yoshizawa, S; Hirota, M; Nakayasu, M; Ojika, M; Wakamatsu, K; Yamada, K; Sugimura, T. Specific binding of okadaic acid, a new tumor promoter. Proc Natl Acad Sci USA 1988, 85, 1768–1771. [Google Scholar]
- Ito, E; Statake, M; Ofuji, K; Higashi, M; Harigaya, K; McMahon, T. Chronic effects in mice caused by oral administration of sublethal doses of azaspiracid, a new marine toxin isolated from mussels. Toxicon 2002, 40, 193–202. [Google Scholar]
- Murata, M; Kumagai, M; Lee, JS; Yasumoto, T. Isolation and structure of yessotoxin, a novel polyether compound implicated in diarrheic shellfish poisoning. Tetrahedron Lett 1987, 28, 5869–5872. [Google Scholar]
- Draisci, R; Ferretti, E; Palleschi, L; Marchiafava, C; Poletti, R; Milandri, A; Ceredi, A; Pompei, M. High levels of yessotoxin in mussels and presence of yessotoxin and homoyessotoxin in dinoflagellates of the Adriatic Sea. Toxicon 1999, 37, 1187–1193. [Google Scholar]
- Paz, B; Riobó, P; Luisa Fernández, M; Fraga, S; Franco, JM. Production and release of yessotoxins by the dinoflagellates Protoceratium reticulatum and Lingulodinium polyedrum in culture. Toxicon 2004, 44, 251–258. [Google Scholar]
- Satake, M; Ichimura, T; Sekiguchi, K; Yoshimatsu, S; Oshima, Y. Confirmation of yessotoxin and 45, 46, 47-trinoryessotoxin production by Protoceratium reticulatum collected in Japan. Nat Toxins 1999, 7, 147–150. [Google Scholar]
- Rhodes, L; McNabb, P; de Salas, M; Briggs, L; Beuzenberg, V; Gladstone, M. Yessotoxin production by Gonyaulax spinifera. Harmful Algae 2006, 5, 148–155. [Google Scholar]
- Satake, M; Terasawa, K; Kadowaki, Y; Yasumoto, T. Relative configuration of yessotoxin and isolation of two new analogs from toxic scallops. Tetrahedron Lett 1996, 37, 5955–5958. [Google Scholar]
- Satake, M; Viviani, R; Yasumoto, T. Yessotoxin in mussels of the northern Adriatic Sea. Toxicon 1997, 35, 177–183. [Google Scholar]
- Ciminiello, P; Fattorusso, E; Forino, M; Poletti, R. 42,43,44,45,46,47,55- Heptanor-41-oxohomoyessotoxin, a new biotoxin from mussels of the northern Adriatic sea. Chem Res Toxicol 2001, 14, 596–599. [Google Scholar]
- Ciminiello, P; Dell’Aversano, C; Fattorusso, E; Forino, M; Magno, S; Poletti, R. Direct detection of yessotoxin and its analogues by liquid chromatography coupled with electrospray ion trap mass spectrometry. J Chromatogr A 2002, 968, 61–69. [Google Scholar]
- Tarao, K; Ito, E; Oarada, M; Murata, M; Yasumoto, T. Histopathiological studies on experimental marine toxin poisoning-5. The effects in mice of yessotoxin isolated from Patinopecten yessoessis and a desulfated derivative. Toxicon 1990, 28, 1095–1104. [Google Scholar]
- Aune, T; Sørby, R; Yasumoto, T; Ramstad, H; Landsverk, T. Comparison of oral and intraperitoneal toxicity of yessotoxin towards mice. Toxicon 2002, 40, 77–82. [Google Scholar]
- Tubaro, A; Sosa, S; Carbonatto, M; Altinier, G; Vita, F; Melato, M; Satake, M; Yasumoto, T. Oral and intraperitoneal acute toxicity studies of yessotoxin and homoyessotoxins in mice. Toxicon 2003, 41, 783–792. [Google Scholar]
- Tubaro, A; Sosa, S; Altinier, G; Soranzo, MR; Satake, M; Della Loggia, R; Yasumoto, T. Short-term toxicity of homoyessotoxins, yessotoxin and okadaic acid in mice. Toxicon 2004, 43, 439–445. [Google Scholar]
- Wolf, LW; Laregina, MC; Tolbert, DL. A behavioural study of the development of hereditary cerebellar ataxia in the shaker rat mutant. Behav Brain Res 1996, 75, 67–81. [Google Scholar]
- Franchini, A; Marchesini, E; Poletti, R; Ottaviani, E. Acute toxic effect of the algal yessotoxin on Purkinje cells from the cerebellum of Swiss CDq mice. Toxicon 2004, 43, 347–352. [Google Scholar]
- Perez-Gomez, A; Ferrero-Gutierrez, A; Novelli, A; Franco, JM; Paz, B; Fernandez-Sanchze, MT. Potent neurotoxic action of the shellfish biotoxin yessotoxin on cultured cerebellar neurons. Toxicol Sci 2006, 90, 168–177. [Google Scholar]
- De la Rosa, LA; Alfonso, A; Vieytes, MR; Botana, LM. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem Pharmacol 2001, 61, 827–833. [Google Scholar]
- Moore, RE; Scheuer, PJ. Palytoxin: a new marine toxin from a coelenterate. Science 1971, 172, 495–498. [Google Scholar]
- Penna, A; Vila, M; Fraga, S; Giacobbe, MG; Andreoni, F; Riobó, P; Veronesi, C. Characterization of Ostreopsis and Coolia (Dinophyceae) isolates in the western Mediterranean Sea based on morphology, toxicity, and internal transcribed spacer 5.8S rDNA sequences. J Phycol 2005, 41, 212–225. [Google Scholar]
- Gallitelli, M; Ungaro, N; Addante, LM; Gentiloni, N; Sabbà, C. Respiratory illness as a reaction to tropical algal blooms occurring in a temperate climate. JAMA 2005, 293, 2599–2600. [Google Scholar]
- Sansoni, G; Borghini, B; Camici, G; Casotti, M; Righini, P; Rustighi, C. Fioriture algali di Ostreopsis Ovata (Gonyaulacales: Dinophyceae): Unproblema emergente. Biol Ambientale 2003, 17, 17–23. [Google Scholar]
- Ciminiello, P; Dell’Aversano, C; Fattorusso, E; Forino, M; Magno, GS; Tartaglione, L; Grillo, C; Melchiorre, N. The Genoa 2005 outbreak: Determination of putative palytoxin in Mediterranean Ostreopsis ovata by a new liquid chromatography tandem mass spectrometry method. Anal Chem 2006, 78, 6153–6159. [Google Scholar]
- Riobó, P; Paz, B; Franco, JM. Analysis of palytox-in- like in Ostreopsis cultures by liquid chromatography with precolumn derivatization and fluorescence detection. Anal Chim Acta 2006, 566, 217–223. [Google Scholar]
- Monti, M; Minocci, M; Beran, A; Ivena, L. First record of Ostreopsis cfr. Ovata on macroalgae in the northern Adriatic. Sea, Mar Pol Bull 2007, 54, 598–601. [Google Scholar]
- Alcala, AC; Alcala, LC; Garth, JS; Yasumura, D; Yasumoto, T. Human fatality due to ingestion of the crab Demania reynaudii contained a palytoxin-like toxin. Toxicon 1998, 26, 105–107. [Google Scholar]
- Granéli, E; Ferreira, CEL; Yasumoto, T; Rodrigues, E; Neves, MHB. Sea urchins poisoning by the benthic dinoflagellate Ostreopsis ovata on the Brazilian coast. In Book of Abstracts of Xth International Conference on Harmful Algae; Florida, 2002. [Google Scholar]
- Fukui, M; Murata, M; Inoue, A; Gawel, M; Yasumoto, T. Occurrence of palytoxin in the Trigger fish Melichtys vidua. Toxicon 1987, 25, 1121–1124. [Google Scholar]
- Onuma, Y; Satake, M; Ukena, T; Roux, J; Chanteau, S; Rasolofonirina, N; Ratsimaloto, N; Naoki, H; Yasumoto, T. Identification of putative palytoxin as the cause of clupeotoxism. Toxicon 1999, 37, 55–65. [Google Scholar]
- Taniyama, S; Arakawa, O; Terada, M; Nishio, S; Takatani, T; Mahmud, Y; Noguchi, T. Ostreopsis sp., a possible origin of palytoxin (PTX) in parrotfish Scarus ovifrons. Toxicon 2003, 42, 29–33. [Google Scholar]
- Moore, RE; Bartolini, G; Barchi, J; Bothmer-By, AA; Dadok, J; Ford, J. Absolute stereochemistry of palytoxin. J Am Chem Soc 1982, 104, 3776–3779. [Google Scholar]
- Bottinger, H; Beress, L; Habermann, E. Involvement of (Na+, K+-ATPase) in binding and actions of palytoxin on human erythrocytes. Biochim Biophys Acta 1986, 861, 164–176. [Google Scholar]
- Habermann, E. Palytoxin acts through the Na+, K+-ATPase. Toxicon 1989, 27, 1171–1187. [Google Scholar]
- Kim, SY; Marx, KA; Wu, CH. Involvement of the Na+, K+-ATPase in the introduction of ion channels by palytoxin. Naunyn-Schmiedeberg’s Arch Pharmacol 1995, 351, 542– 554. [Google Scholar]
- Hirsh, JK; Wu, CH. Palytoxin-induced single-channel currents from the sodium pump synthesized by in vitro expression. Toxicon 1997, 35, 169–176. [Google Scholar]
- Scheiner-Bobis, G; Meyer zu Heringdorf, D; Christ, M; Habermann, E. Palytoxin induces K+ efflux from yeast cells expressing the mammalian sodium pump. Mol Pharmacol 1994, 45, 1132– 1136. [Google Scholar]
- Habermann, E; Laux, M. Depolarization increases inositol phosphate production in a particulate preparation from rat brain. Naunyn-Schmiederberg’s Arch Pharmac 1986, 334, l–15. [Google Scholar]
- Levine, L; Fujiki, H. Stimulation of arachidonic acid metabolism by different types of tumor promoters. Carcinogenesis 1985, 6, 1631–1635. [Google Scholar]
- Frelin, C; Vigne, P; Breittmayer, JP. Mechanism of the cardiotoxic action of palytoxin. Mol Pharmacol 1991, 38, 904–909. [Google Scholar]
- Yoshizumi, M; Houchi, H; Ishimura, Y; Masuda, Y; Morita, K; Oka, M. Mechanism of palytoxin induced Na+ influx into cultured bovine adrenal chromaffin cells: possible involvement of Na+/H+ exchange system. Neurosci Left 1991, 130, 103–l 06. [Google Scholar]
- Satoh, E; Nakazato, Y. Mode of action of palytoxin on the release of acetylcholine from rat cerebrocortical synaptosomes. J Neurochem 1991, 57, 1276–1280. [Google Scholar]
- Tsumuraya, T; Fujii, I; Inoue, M; Tatami, A; Miyazaki, K; Hirama, M. Production of monoclonal antibodies for sandwich immunoassay detection of ciguatoxin 51-hydroxy CTX3C. Toxicon 2006, 48, 287–294. [Google Scholar]
- Ishikawa, Y; Nishiyama, S. Synthesis of the BCD ring system of azaspiracid: construction of the trispiro ring structure by the thioether approach. Tetrahedron Lett 2004, 45, 351–354. [Google Scholar]
- Miles, CO; Samdal, IA; Aasen, JAG; Jensen, DJ; Quilliam, MA; Petersen, D; Briggs, LM; Wilkins, AL; Rise, F; Cooney, JM; MacKenzie, AL. Evidence for numerous analogs of yessotoxin in Protoceratium reticulatum. Harmful Algae 2005, 6, 1075–1091. [Google Scholar]
- Cha, JK; Christ, WJ; Finan, JM; Fujiko, H; Kishi, Y; Klein, LL; Ko, SS; Leder, J; McWhorter, WW, Jr; Pfaff, KP; Yonaga, M; Uemura, D; Hirata, Y. Stereochemistry of palytoxin. Complete structure. J Am Chem Soc 1982, 104, 7369–7371. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of poisoning | Toxins | Sources of toxins | Primary vector | Action target | Ref. |
|---|---|---|---|---|---|
| PSP | Saxitoxins and gonyautoxins | Alexandrium spp., Gymnodinium spp., Pyrodinium spp. | Shellfish | Voltage-gated sodium channel 1 | 23, 27–29 |
| NSP | Brevetoxins | Kerenia brevis, Chatonella marina, C. antiqua, Fibrocapsa japonica, Heterosigma akashiwo | Shellfish | Voltage-gated sodium channel 5 | 33, 39–41, 46, 52 |
| Yessotoxins | Protoceratium reticulatum, Lingulodinium polyedrum Gonyaulax spinifera | Shellfish | Voltage-gated calcium/sodium channel? | 94–95 | |
| CFP | Ciguatoxins | Gambierdiscus toxicus | Coral reef fish | Voltage-gated sodium channel 5 | 55, 62, 63 |
| CFP | Maitotoxins | Gambierdiscus toxicus | Coral reef fish | Voltage-gated calcium channel | 69, 70 |
| AZP | Azaspiracids | Protoperidinium crassipes | Shellfish | Voltage-gated calcium channel | 72, 76 |
| Palytoxin poisoning | Palytoxins | Ostrepsis siamensis | Shellfish | Na+-K+ ATPase | 97, 109–111 |
Share and Cite
Wang, D.-Z. Neurotoxins from Marine Dinoflagellates: A Brief Review. Mar. Drugs 2008, 6, 349-371. https://doi.org/10.3390/md6020349
Wang D-Z. Neurotoxins from Marine Dinoflagellates: A Brief Review. Marine Drugs. 2008; 6(2):349-371. https://doi.org/10.3390/md6020349
Chicago/Turabian StyleWang, Da-Zhi. 2008. "Neurotoxins from Marine Dinoflagellates: A Brief Review" Marine Drugs 6, no. 2: 349-371. https://doi.org/10.3390/md6020349
APA StyleWang, D.-Z. (2008). Neurotoxins from Marine Dinoflagellates: A Brief Review. Marine Drugs, 6(2), 349-371. https://doi.org/10.3390/md6020349