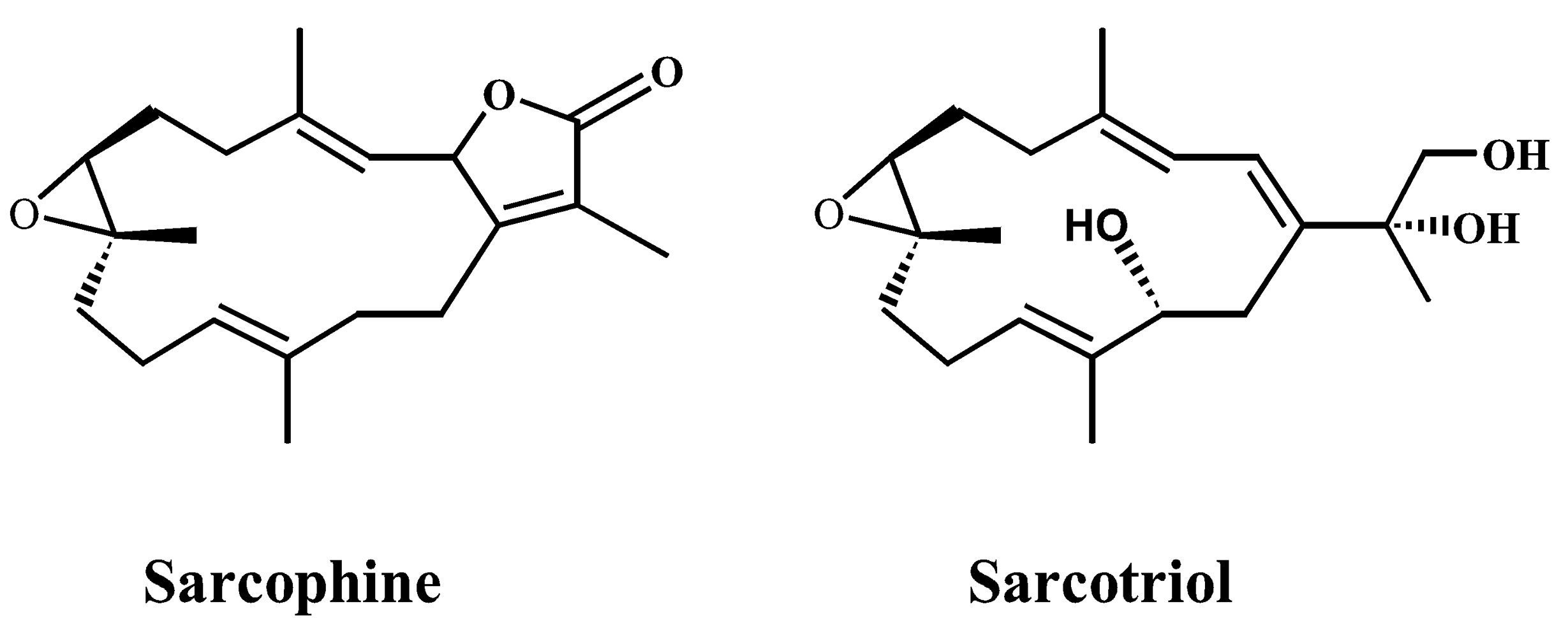
Chemopreventive Effects of Sarcotriol on Ultraviolet B-induced Skin Tumor Development in SKH-1 Hairless Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
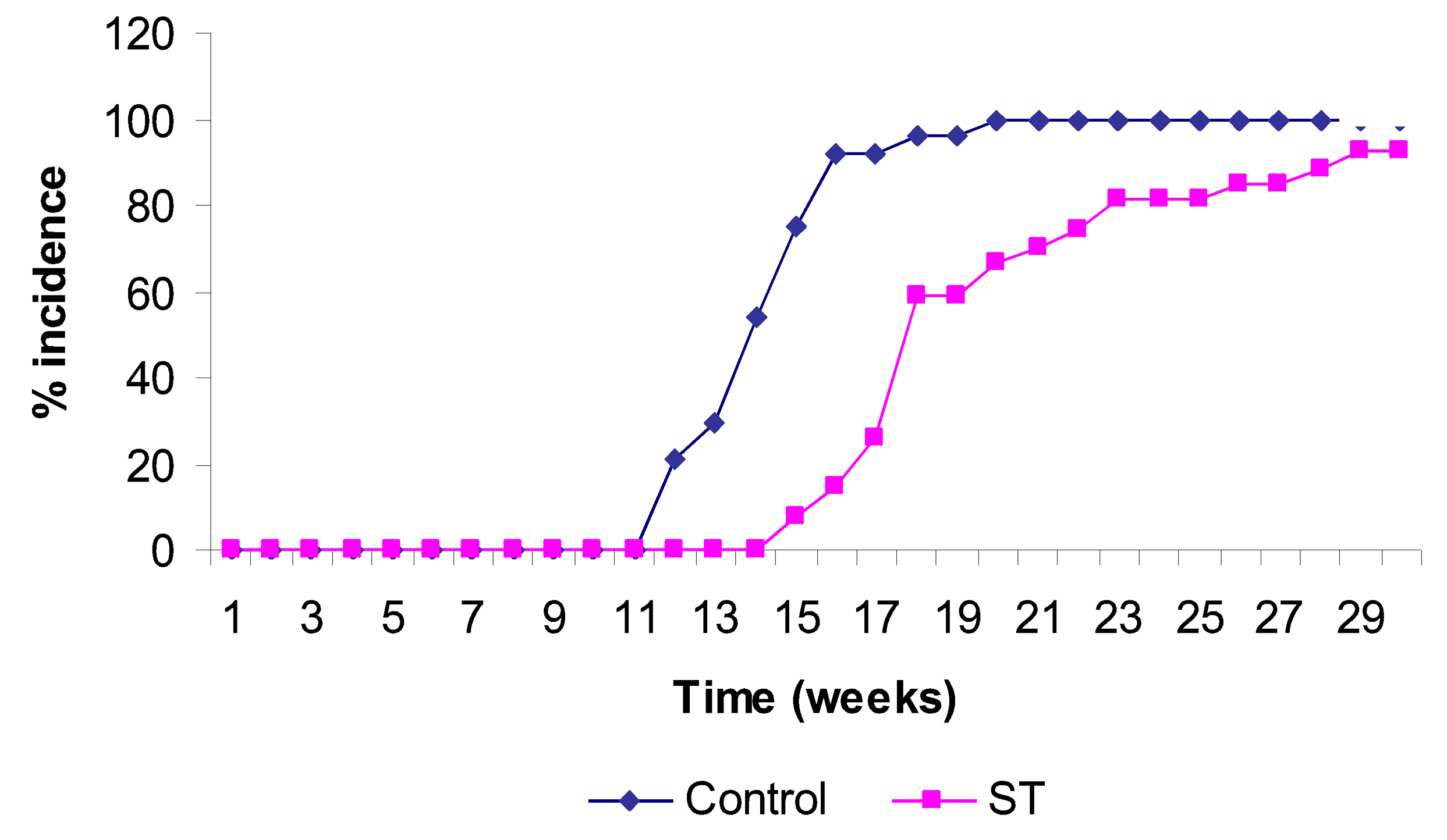
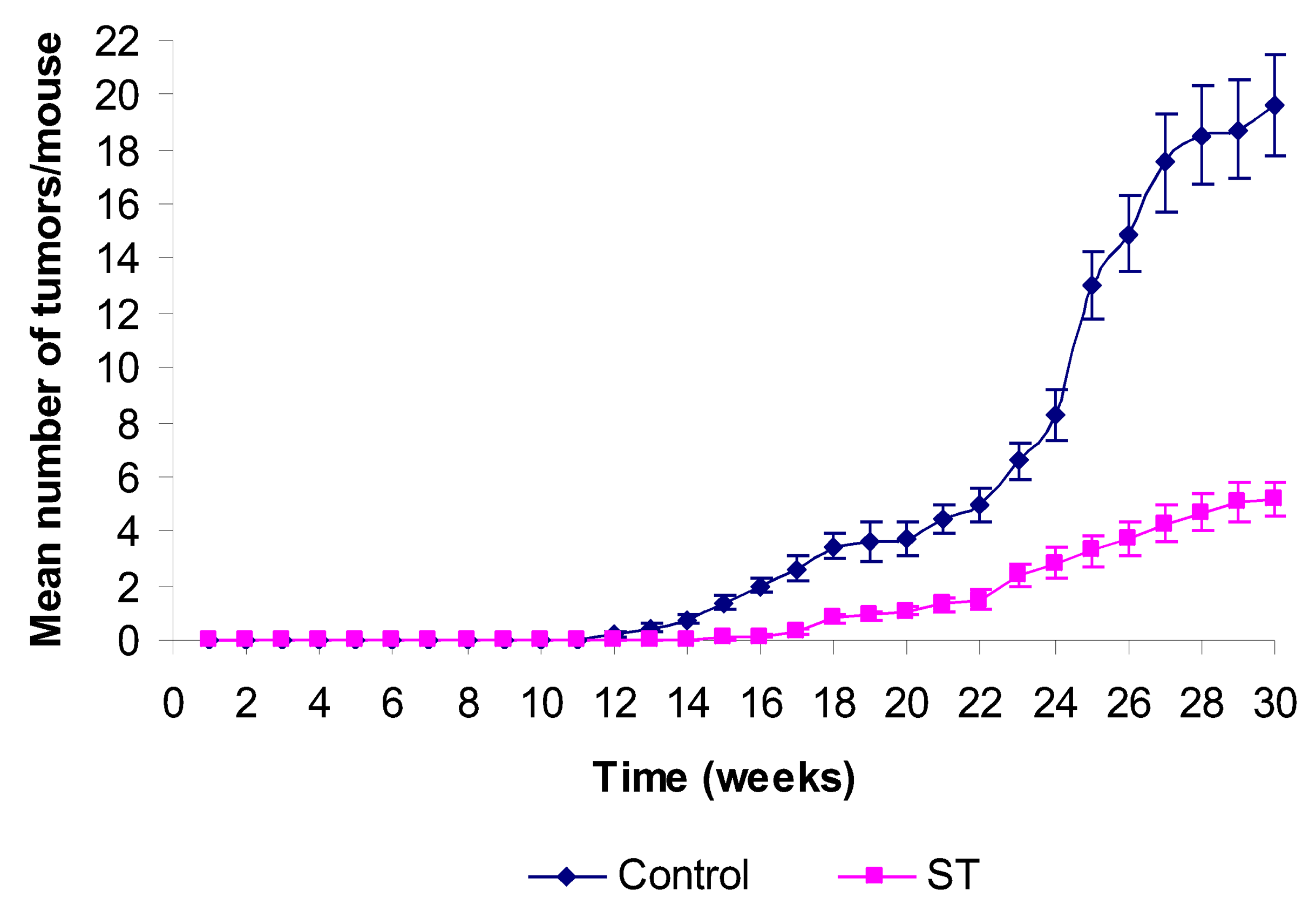
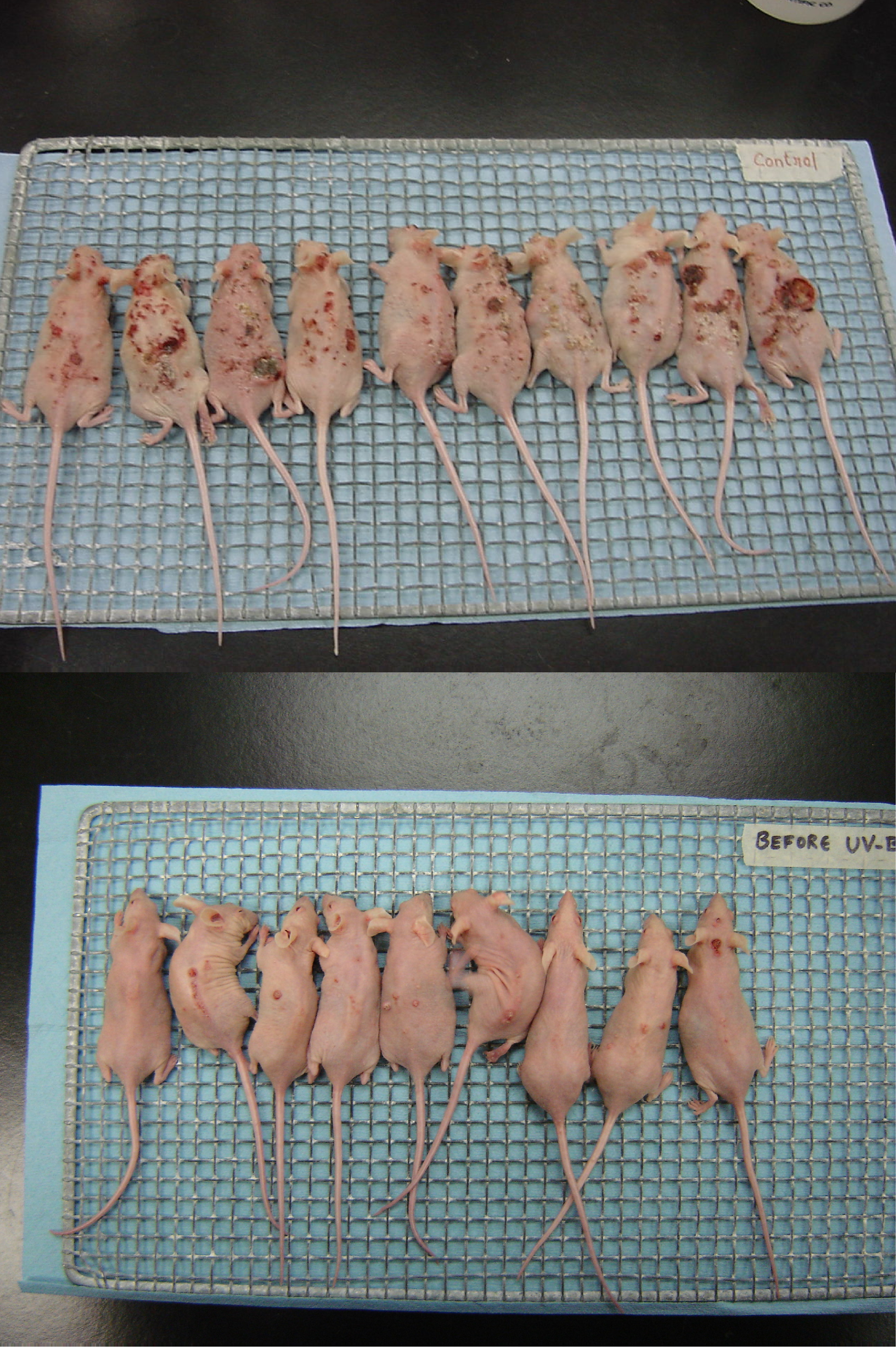
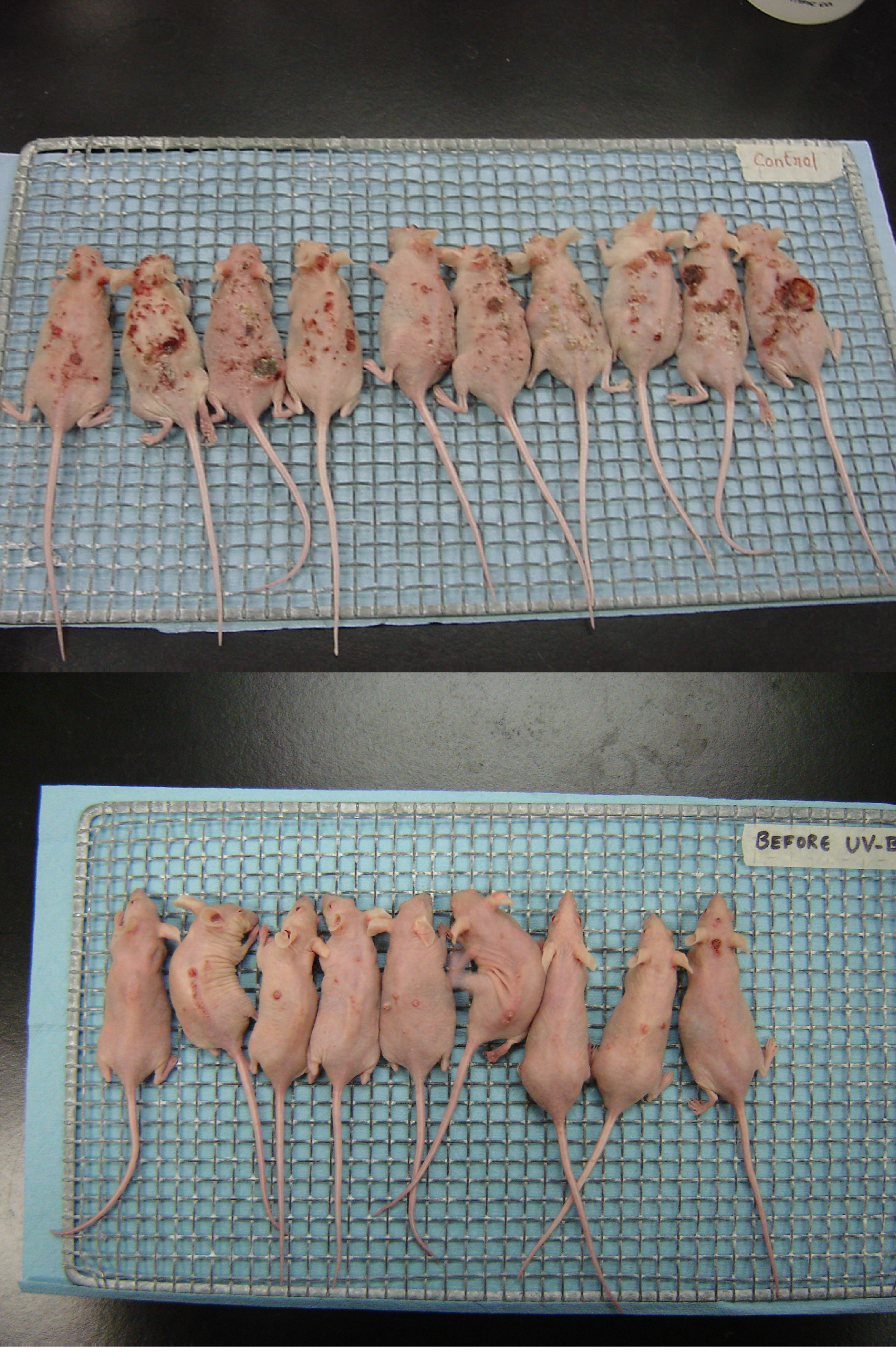
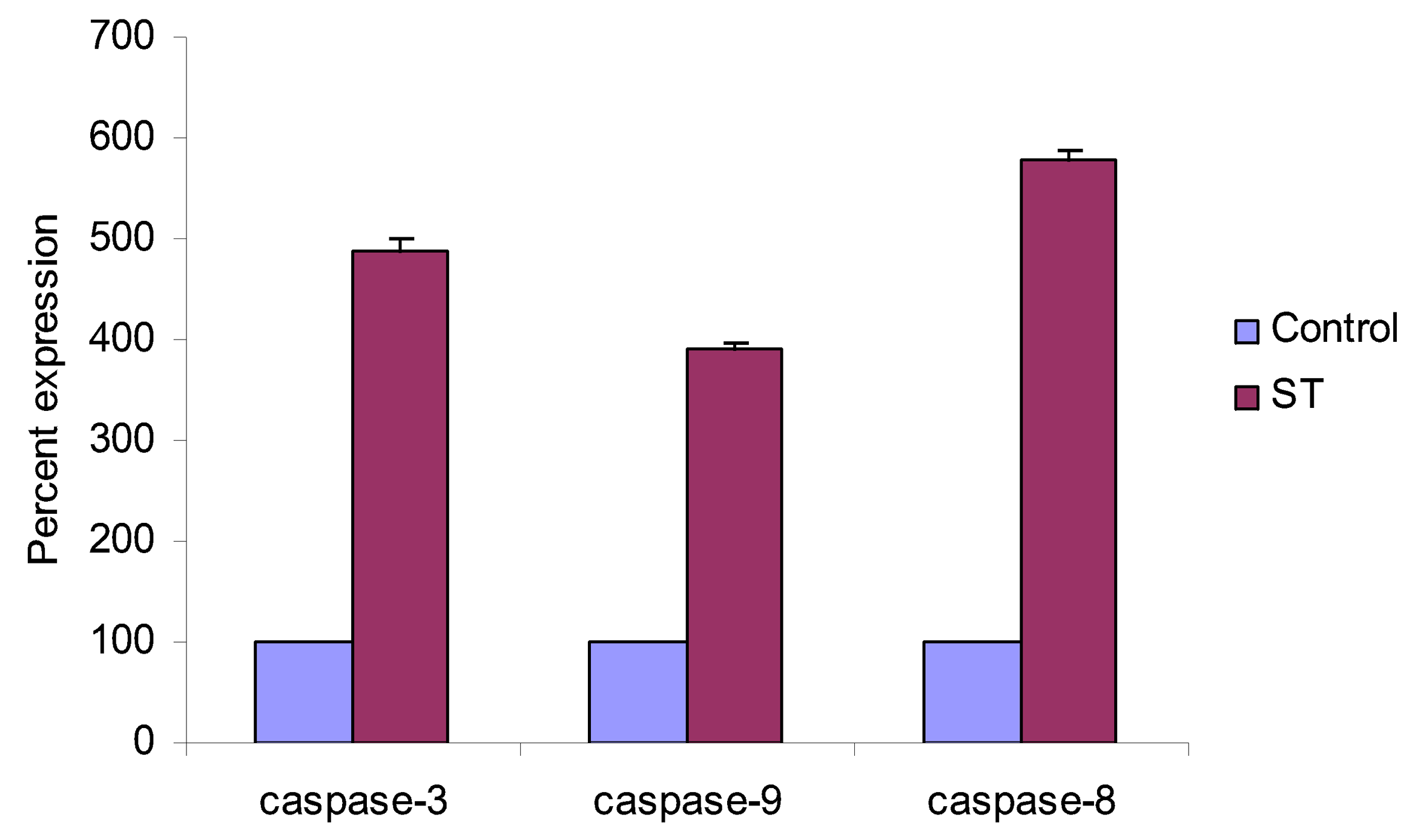
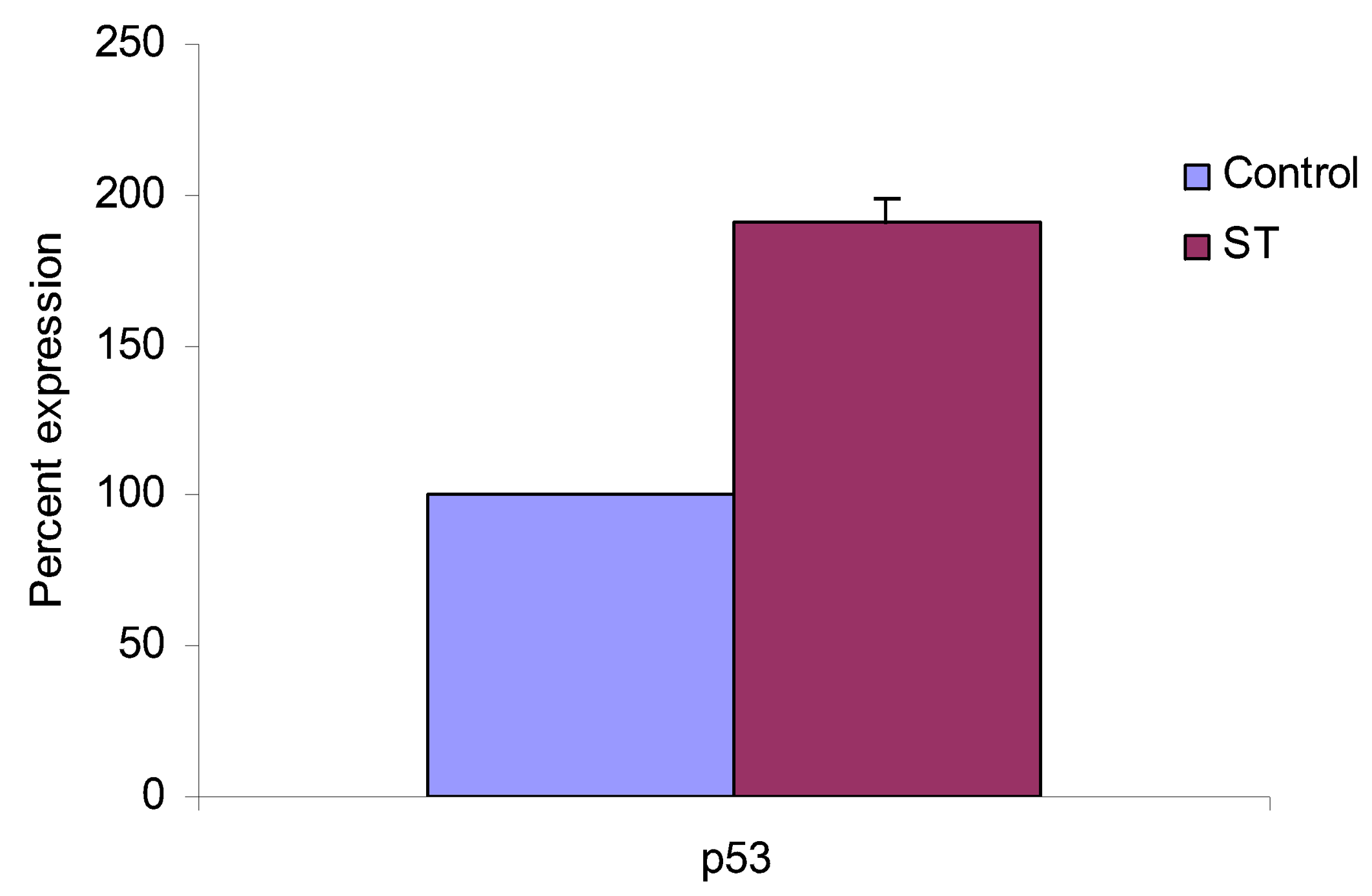
2. Results and Discussion
3. Conclusions
4. Experimental
4.1. General
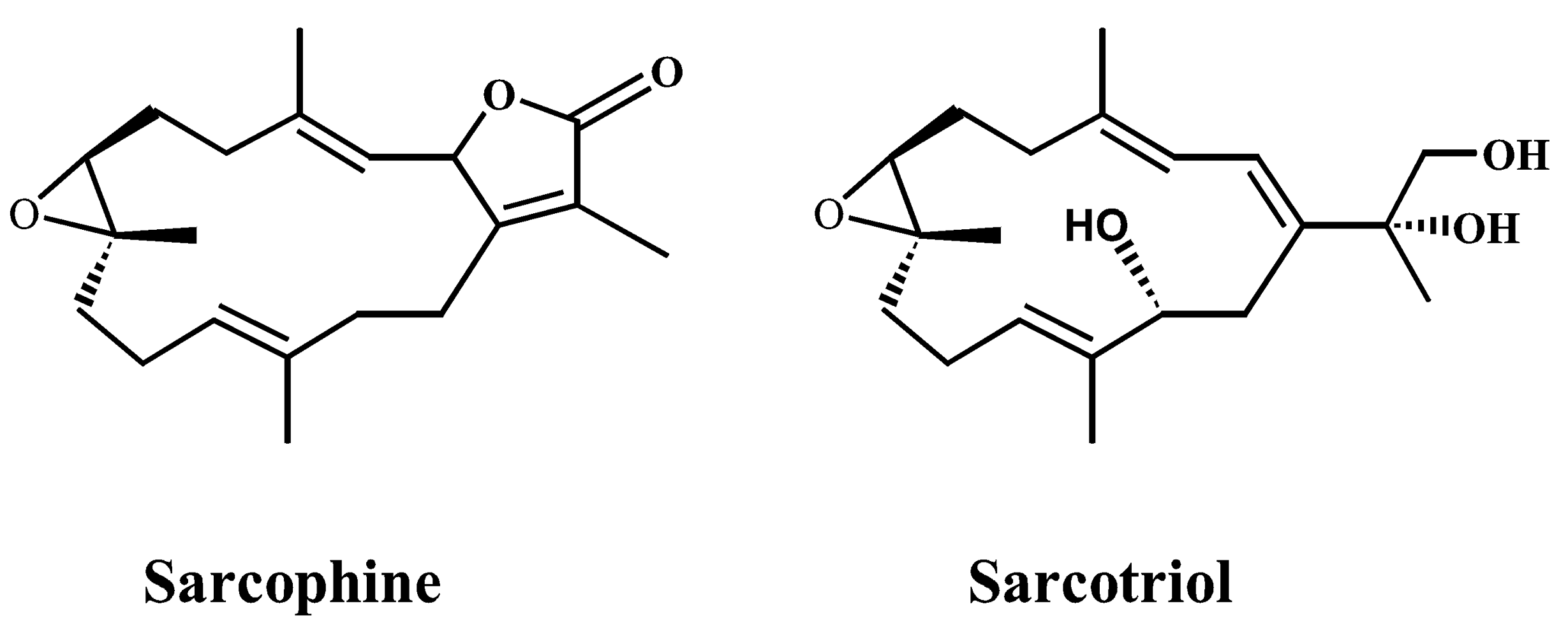
4.2. Materials
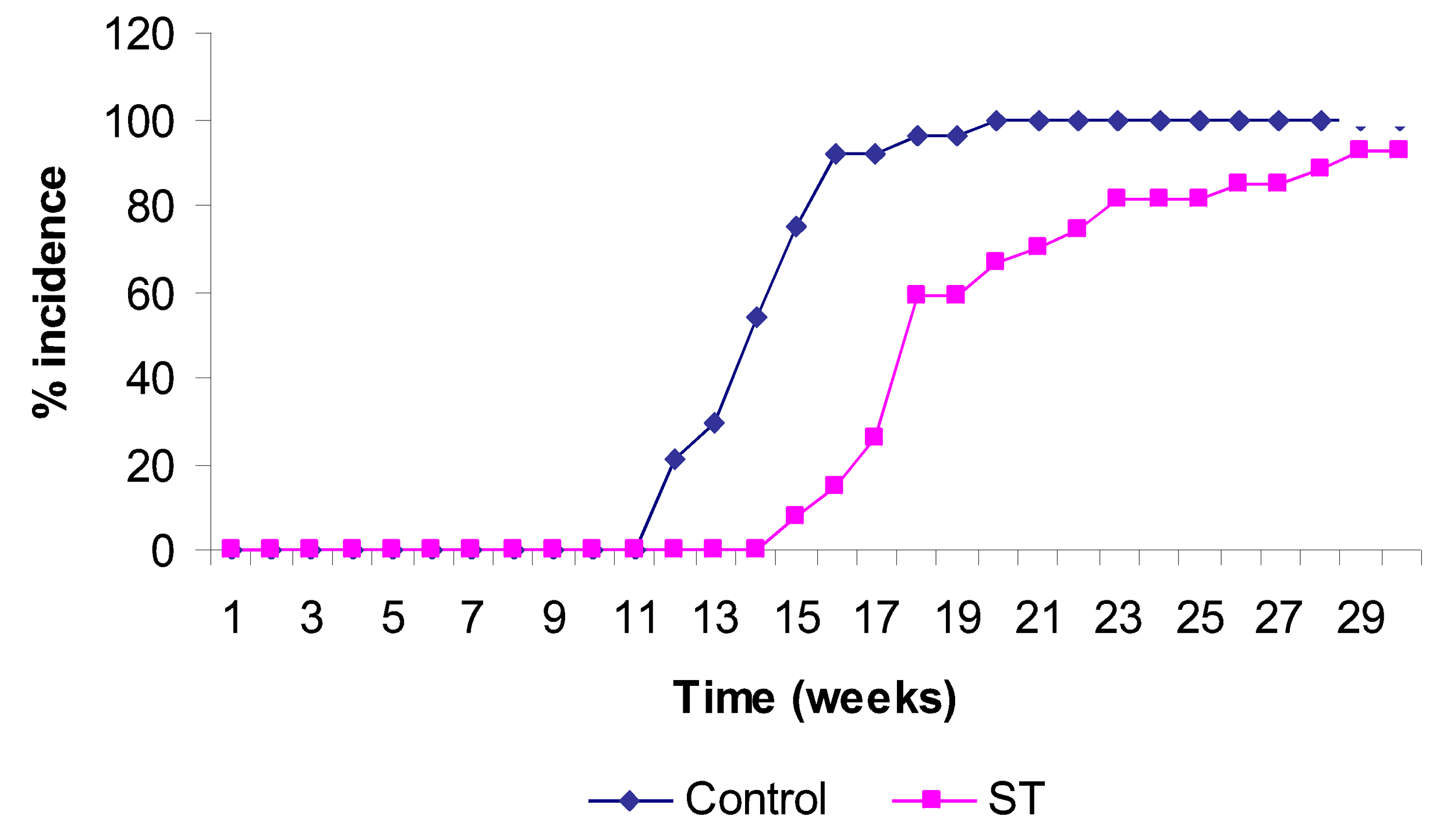
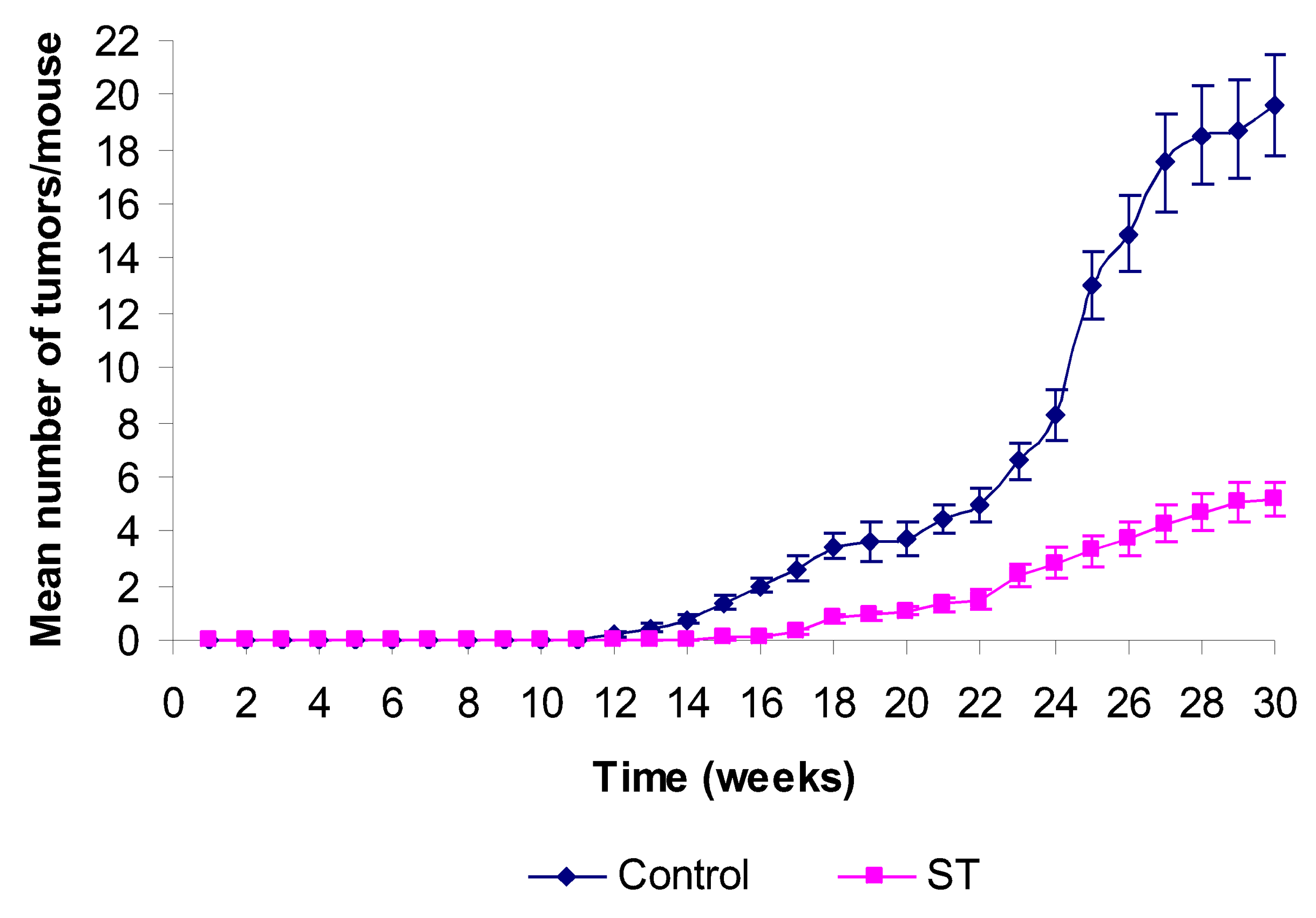
4.3. UVB-induced Tumorigenesis Protocol
4.3.1. UVB Source
4.3.2. Animals
4.3.3. Experiment
4.4 Lysate Preparation
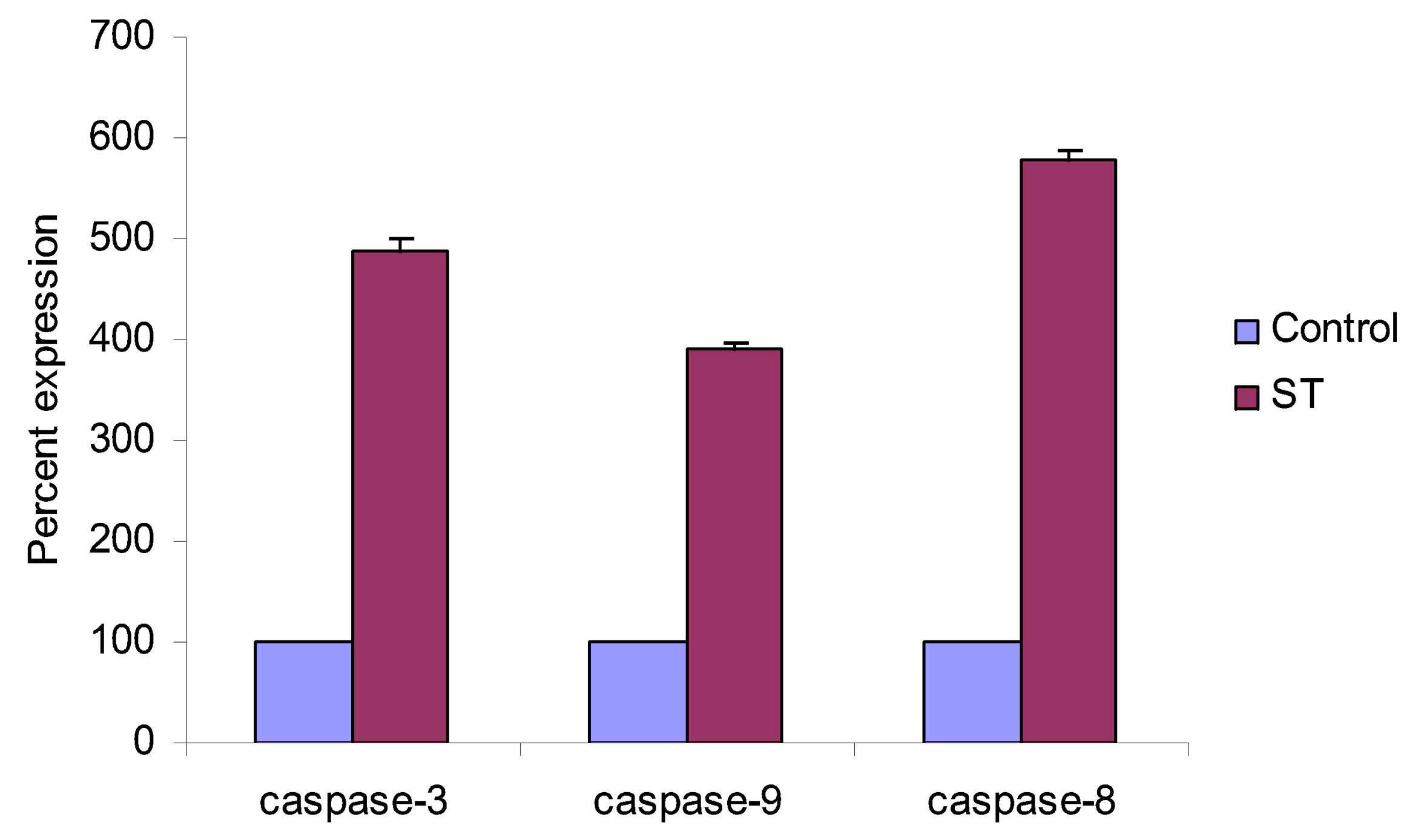
4.5. Western Blot Analysis of Caspase -3, -8, -9 and p53
4.6. Statistical Analysis
Acknowledgements
- Sample Availability: Available from the authors.
References
- Cancer Facts & Figures-2007; American Cancer Society Publication; http://www.cancer.org
- Mallikarjuna, DGS; Singh, R; Agarwal, C; Agarwal, R. Silibinin protects against photocarcinogenesis via modulation of cell cycle regulators, mitogen-activated protein kinase, and Akt signaling. Cancer Res 2004, 64, 6349–6356. [Google Scholar]
- Soehnge, H; Ouhtit, A; Ananthaswamy, ON. Mechanisms of induction of skin cancer by UV radiation. Front Biosci 1997, 2, 538–551. [Google Scholar]
- Kraemer, K. Sunlight and skin cancer: another link revealed. Proc Natl Acad Sci 1997, 94, 11–14. [Google Scholar]
- Agarwal, R; Mukhtar, H. Mukhtar, H, Ed.; Cutaneous chemical carcinogens. In Pharmacology of the Skin; CRC Press: Boca Raton, New York, 1991; pp. 371–387. [Google Scholar]
- Tornaletti, S; Pfeifer, GP. UV damage and repair mechanisms in mammalian cells. Bioessays 1996, 18, 221–228. [Google Scholar]
- Pentland, AP. Signal transduction mechanisms in photocarcinogenesis. Photochem Photobiol 1996, 63, 379–380. [Google Scholar]
- Melnikova, VO; Ananthaswamy, HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res 2005, 571, 91–106. [Google Scholar]
- Gu, M; Singh, RP; Dhanalakshmi, S; Agarwal, C; Agarwal, R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res 2007, 67, 3483–3491. [Google Scholar]
- Greenleee, RT; Hill-Harmon, MB; Murray, T; Thun, M. Cancer statistics. CA Cancer J Clin 2001, 51, 15–36. [Google Scholar]
- Gupta, S; Mukhtar, H. Chemoprevention of skin cancer: Current status and future prospects. Cancer Metast Rev 2002, 21, 363–380. [Google Scholar]
- Lamson, DW; Brignall, MS. Natural agents in the prevention of cancer, part two: Preclinical data and chemoprevention for common cancers. Altern Med Rev 2001, 6, 167–187. [Google Scholar]
- Ley, RD; Reeve, VE. Chemoprevention of ultraviolet radiation-induced skin cancer. Environ Health Perspect 1997, 105, 981–984. [Google Scholar] [Green Version]
- Kelloff, GJ; Sigman, CC; Greenwald, P. Cancer Chemoprevention: Progress and promise. Eur J Cancer 1999, 35, 2031–2038. [Google Scholar] [Green Version]
- Bickers, DR; Athar, M. Novel approaches to chemoprevention of skin cancer. J Dermatol 2000, 27, 691–695. [Google Scholar] [Green Version]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov Today 2003, 8, 536–544. [Google Scholar] [Green Version]
- Fahmy, H; Khalifa, S; Konoshima, T; Zjawiony, JK. An improved synthesis of 7,9-epoxy-1, 3,11-cembratriene-15 R(α),16-diol, a cembranoid of marine origin with apotent cancer chemopreventive activity. Mar Drugs 2004, 2, 1–7. [Google Scholar] [Green Version]
- Katsuyama, I; Fahmy, H; Zjawiony, JK; Khalifa, S; Kilada, RW; Konoshima, T; Takasaki, M; Tokuda, H. Semisynthesis of new sarcophine derivatives with chemopreventive activity. J Nat Prod 2002, 65, 1809–1814. [Google Scholar] [Green Version]
- Ne’eman, A; Fishelson, L; Kashman, Y. Sarcophine-A new Toxin from the soft coral Sarcophyton glaucum (Alcyonaria). Toxicon 1974, 12, 593–598. [Google Scholar] [Green Version]
- Kundoor, V; Zhang, X; Khalifa, S; Fahmy, H; Dwivedi, C. A possible mechanism of action of the chemopreventive effects of sarcotriol on skin tumor development in CD-1 mice. Mar Drugs 2006, 4, 274–285. [Google Scholar] [Green Version]
- Kaur, M; Agarwal, C; Singh, RP; Guan, X; Dwivedi, C; Agarwal, R. Skin cancer chemo-preventive agent, alpha-santalol, induces apoptotic death of human epidermoid carcinoma A431 cells via caspase activation together with dissipation of mitochondrial membrane potential and cytochrome c release. Carcinogenesis 2005, 26, 369–380. [Google Scholar] [Green Version]
- Sun, SY, Jr; Hail, N; Lotan, R. Apoptosis as a novel target for cancer chemoprevention. J Natl Cancer Inst 2004, 96, 662–672. [Google Scholar] [Green Version]
- Wattenberg, LW. Watterberg, L, Lipkin, M, Boone, C, Kelloff, G, Eds.; Chemoprevention of cancer by naturally occurring and synthetic compounds. In Cancer Prevention; CRC Press: Boca Raton FL, 1992; pp. 19–39. [Google Scholar]
- Nicholson, DW; Thornberry, NA. Caspase: Killer proteases. Trends Biochem Sci 1997, 22, 299–306. [Google Scholar] [Green Version]
- Ghobrial, IM; Witzig, TE; Adjei, AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 2005, 55, 178–194. [Google Scholar] [Green Version]
- Li, P; Nijhawan, D; Budihardjo, L; Srinivasula, SM; Ahmad, M; Alnemri, ES; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [Green Version]
- Stenback, F; Makinen, M; Jussila, T. p53 expression in skin carcinogenesis and its relationship to cell proliferation and tumor growth. Eur J Cancer 1998, 34, 1415–1424. [Google Scholar] [Green Version]
- Ruggeri, B; Caamano, J; Goodrow, T. Alterations of the p53 tumor suppressor gene during mouse skin tumor progression. Cancer Res 1991, 51, 6615–6621. [Google Scholar] [Green Version]
- Ruggeri, B; DiRado, M; Zhang, SY; Bauer, B; Goodrow, T; Kleinszanto, AJ. Benzo (a) pyrene-induced murine skin tumors exihibit frequent and characteristic G to T mutations in the p53 gene. Proc Natl Acad Sci 1993, 90, 1013–1017. [Google Scholar] [Green Version]
- Shen, Y; White, E. p53-dependent apoptosis pathways. Adv Cancer Res 2001, 82, 55–84. [Google Scholar] [Green Version]
- Dwivedi, C; Valluri, HB; Guan, X; Agarwal, R. Chemopreventive effects of alpha-santalol on ultraviolet B radiation-induced skin tumor development in SKH-1 hairless mice. Carcinogenesis 2006, 27, 1917–1922. [Google Scholar] [Green Version]
- Zhang, X; Kundoor, V; Khalifa, S; Zeman, D; Fahmy, H; Dwivedi, C. Chemopreventive Effects of Sarcophine-diol on Skin Tumor Development in CD-1 Mice. Cancer Lett 2007, 253, 53–59. [Google Scholar] [Green Version]
Share and Cite
Kundoor, V.; Zhang, X.; Bommareddy, A.; Khalifa, S.; Fahmy, H.; Dwivedi, C. Chemopreventive Effects of Sarcotriol on Ultraviolet B-induced Skin Tumor Development in SKH-1 Hairless Mice. Mar. Drugs 2007, 5, 197-207. https://doi.org/10.3390/md504197
Kundoor V, Zhang X, Bommareddy A, Khalifa S, Fahmy H, Dwivedi C. Chemopreventive Effects of Sarcotriol on Ultraviolet B-induced Skin Tumor Development in SKH-1 Hairless Mice. Marine Drugs. 2007; 5(4):197-207. https://doi.org/10.3390/md504197
Chicago/Turabian StyleKundoor, Vipra, Xiaoying Zhang, Ajay Bommareddy, Sherief Khalifa, Hesham Fahmy, and Chandradhar Dwivedi. 2007. "Chemopreventive Effects of Sarcotriol on Ultraviolet B-induced Skin Tumor Development in SKH-1 Hairless Mice" Marine Drugs 5, no. 4: 197-207. https://doi.org/10.3390/md504197
APA StyleKundoor, V., Zhang, X., Bommareddy, A., Khalifa, S., Fahmy, H., & Dwivedi, C. (2007). Chemopreventive Effects of Sarcotriol on Ultraviolet B-induced Skin Tumor Development in SKH-1 Hairless Mice. Marine Drugs, 5(4), 197-207. https://doi.org/10.3390/md504197