The Structural Basis and Functional Consequences of Interactions Between Tetrodotoxin and Voltage-Gated Sodium Channels

{kind=link}
{kind=link}
{kind=link}
Abstract
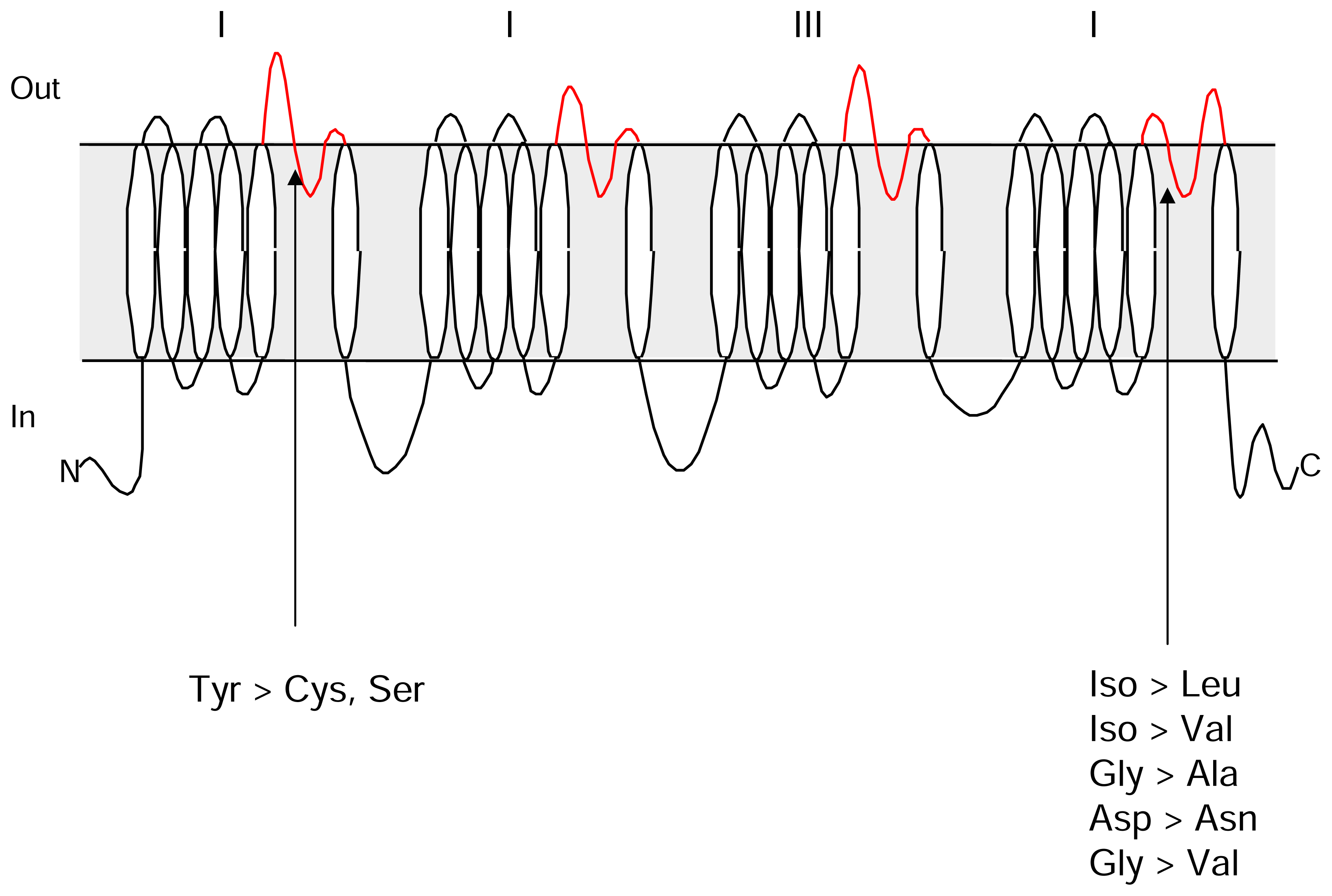
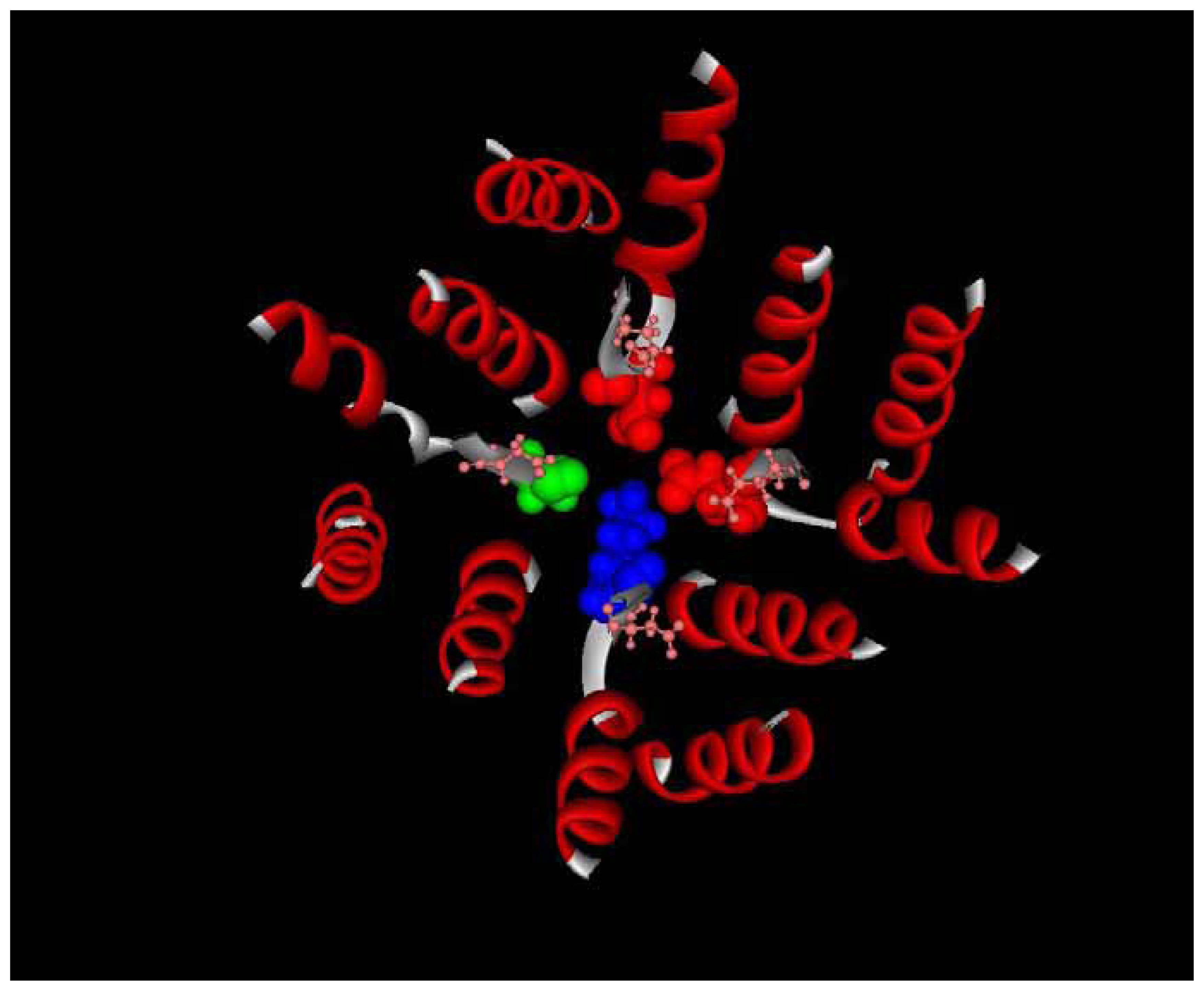
:1. Sodium channel structure and function
2. Sodium channel gene family
3. Sodium channel isoform expression patterns and function
4. Tetrodotoxin binding
5. Physiological effects of TTX
6. Novel mechanisms of TTX resistance
Acknowledgements
Abbreviations
| TTX | tetrodotoxin |
| NaV | voltage-gated sodium channel |
| P-region | pore region |
| S4 | fourth membrane-spanning segment |
- Samples Availability: Available from the authors.
References
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed; Sunderland, M. A., Ed.; Sinauer Associates, 2001; pp. 24–82. [Google Scholar]
- Catterall, W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000, 26, 13–25. [Google Scholar]
- Isom, L.L.; De Jongh, K.S.; Patton, D.E.; Reber, B.F.; Offord, J.; Charbonneau, H.; Walsh, K.; Goldin, A.L.; Catterall, W.A. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 1992, 256, 839–842. [Google Scholar]
- Isom, L.L.; Ragsdale, D.S.; De Jongh, K.S.; Westenbroek, R.E.; Reber, B.F.; Scheuer, T.; Catterall, W.A. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 1995, 83, 433–442. [Google Scholar]
- Goldin, A.L.; Snutch, T.; Lubbert, H.; Dowsett, A.; Marshall, J.; Auld, V.; Downey, W.; Fritz, L.C.; Lester, H.A.; Dunn, R. Messenger RNA coding for only the alpha subunit of the rat brain Na channel is sufficient for expression of functional channels in Xenopus oocytes. Proc. Natl. Acad. Sci. USA 1986, 83, 7503–7507. [Google Scholar]
- Noda, M.; Ikeda, T.; Kayano, T.; Suzuki, H.; Takeshima, H.; Kurasaki, M.; Takahashi, H.; Numa, S. Existence of distinct sodium channel messenger RNAs in rat brain. Nature 1986, 320, 188–192. [Google Scholar]
- Noda, M.; Ikeda, T.; Suzuki, H.; Takeshima, H.; Takahashi, T.; Kuno, M.; Numa, S. Expression of functional sodium channels from cloned cDNA. Nature 1986, 322, 826–828. [Google Scholar]
- Guy, H.R.; Seetharamulu, P. Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. USA 1986, 83, 508–512. [Google Scholar]
- Stuhmer, W.; Conti, F.; Suzuki, H.; Wang, X.D.; Noda, M.; Yahagi, N.; Kubo, H.; Numa, S. Structural parts involved in activation and inactivation of the sodium channel. Nature 1989, 339, 597–603. [Google Scholar]
- Kontis, K.J.; Rounaghi, A.; Goldin, A.L. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol 1997, 110, 391–401. [Google Scholar]
- Yang, N.; Horn, R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron 1995, 15, 213–218. [Google Scholar]
- Yang, N.; George, A.L., Jr; Horn, R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron 1996, 16, 113–122. [Google Scholar]
- Armstrong, C.M. Sodium channels and gating currents. Physiol. Rev 1981, 61, 644–483. [Google Scholar]
- Vassilev, P.M.; Scheuer, T.; Catterall, W.A. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 1988, 241, 1658–1661. [Google Scholar]
- Vassilev, P.; Scheuer, T.; Catterall, W.A. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc. Natl. Acad. Sci. USA 1989, 86, 8147–8151. [Google Scholar]
- West, J.W.; Patton, D.E.; Scheuer, T.; Wang, Y.; Goldin, A.L.; Catterall, W.A. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. USA 1992, 89, 10910–10914. [Google Scholar]
- Goldin, A.L. Evolution of voltage-gated Na+ channels. J. Exp. Biol. 2002, 205 Pt 5, 575–584. [Google Scholar]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol 2003, 4, 207:1–207:7. [Google Scholar]
- Goldin, A.L. Resurgence of sodium channel research. Annu. Rev. Physiol 2001, 63, 871–894. [Google Scholar]
- Lopreato, G.F.; Lu, Y.; Southwell, A.; Atkinson, N.S.; Hillis, D.M.; Wilcox, T.P.; Zakon, H.H. Evolution and divergence of sodium channel genes in vertebrates. Proc. Natl. Acad. Sci. USA 2001, 98, 7588–7592. [Google Scholar]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol. Rev 2003, 55, 575–578. [Google Scholar]
- George, A.L., Jr; Knittle, T.J.; Tamkun, M.M. Molecular cloning of an atypical voltage-gated sodium channel expressed in human heart and uterus: evidence for a distinct gene family. Proc. Natl. Acad. Sci. USA 1992, 89, 4893–4897. [Google Scholar]
- Hiyama, T.Y.; Watanabe, E.; Ono, K.; Inenaga, K.; Tamkun, M.M.; Yoshida, S.; Noda, M. Nax channel involved in CNS sodium-level sensing. Nat. Neurosci 2002, 5, 511–512. [Google Scholar]
- Watanabe, E.; Hiyama, T.Y.; Kodama, R.; Noda, M. NaX sodium channel is expressed in non-myelinating Schwann cells and alveolar type II cells in mice. Neurosci. Lett 2002, 330, 109–113. [Google Scholar]
- Plummer, N.W.; Meisler, M.H. Evolution and diversity of mammalian sodium channel genes. Genomics 1999, 57, 323–331. [Google Scholar]
- Trimmer, J.S.; Rhodes, K.J. Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol 2004, 66, 477–519. [Google Scholar]
- Caldwell, J.H.; Schaller, K.L.; Lasher, R.S.; Peles, E.; Levinson, S.R. Sodium channel Nav1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc. Natl. Acad. Sci. USA 2000, 97, 5616–5620. [Google Scholar]
- Boiko, T.; Rasband, M.N.; Levinson, S.R.; Caldwell, J.H.; Mandel, G.; Trimmer, J.S.; Matthews, G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 2001, 30, 91–104. [Google Scholar]
- Kaplan, M.R.; Cho, M.H.; Ullian, E.M.; Isom, L.L.; Levinson, S.R.; Barres, B.A. Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron 2001, 30, 105–119. [Google Scholar]
- Pugsley, M.K.; Goldin, A.L. Effects of bisaramil, a novel class I antiarrhythmic agent, on heart, skeletal muscle and brain Na+ channels. Eur. J. Pharmacol 1998, 342, 93–104. [Google Scholar]
- Zhou, W.; Goldin, A.L. Use-dependent potentiation of the Nav1.6 sodium channel. Biophys. J 2004, 87, 3862–3872. [Google Scholar]
- Vilin, Y.Y.; Ruben, P.C. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem. Biophys 2001, 35, 171–190. [Google Scholar]
- Goldin, A.L. Mechanisms of sodium channel inactivation. Curr. Opin. Neurobiol 2003, 13, 284–290. [Google Scholar]
- Kallen, R.G.; Sheng, Z.H.; Yang, J.; Chen, L.Q.; Rogart, R.B.; Barchi, R.L. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron 1990, 4, 233–242. [Google Scholar]
- Trimmer, J.S.; Cooperman, S.S.; Agnew, W.S.; Mandel, G. Regulation of muscle sodium channel transcripts during development and in response to denervation. Dev. Biol 1990, 142, 360–367. [Google Scholar]
- Ganong, W.F. Review of medical physiology, 22nd ed; New York; McGraw-Hill Medical, 2005; p. p 928. [Google Scholar]
- Jurkat-Rott, K.; Mitrovic, N.; Hang, C.; Kouzmekine, A.; Iaizzo, P.; Herzog, J.; Lerche, H.; Nicole, S.; Vale-Santos, J.; Chauveau, D.; Fontaine, B.; Lehmann-Horn, F. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc. Natl. Acad. Sci. USA 2000, 97, 9549–9554. [Google Scholar]
- Hennig, R.; Lomo, T. Firing patterns of motor units in normal rats. Nature 1985, 314, 164–166. [Google Scholar]
- Richmond, J.E.; Featherstone, D.E.; Hartmann, H.A.; Ruben, P.C. Slow inactivation in human cardiac sodium channels. Biophys. J 1998, 74, 2945–2952. [Google Scholar]
- Purves, D.; Sakmann, B. The effect of contractile activity on fibrillation and extrajunctional acetylcholine-sensitivity in rat muscle maintained in organ culture. J. Physiol 1974, 237, 157–182. [Google Scholar]
- Hayward, L.J.; Brown, R.H., Jr; Cannon, S.C. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys. J. 1997, 72, 1204–1219. [Google Scholar]
- Hayward, L.J.; Sandoval, G.M.; Cannon, S.C. Defective slow inactivation of sodium channels contributes to familial periodic paralysis. Neurology 1999, 52, 1447–1453. [Google Scholar]
- Lipkind, G.M.; Fozzard, H.A. KcsA crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 2000, 39, 8161–8170. [Google Scholar]
- Tikhonov, D.B.; Zhorov, B.S. Modeling P-loops domain of sodium channel: homology with potassium channels and interaction with ligands. Biophys. J 2005, 88, 184–197. [Google Scholar]
- Terlau, H.; Heinemann, S.H.; Stuhmer, W.; Pusch, M.; Conti, F.; Imoto, K.; Numa, S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett 1991, 293, 93–96. [Google Scholar]
- Heinemann, S.H.; Terlau, H.; Stuhmer, W.; Imoto, K.; Numa, S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 1992, 356, 441–443. [Google Scholar]
- Penzotti, J.L.; Fozzard, H.A.; Lipkind, G.M.; Dudley, S.C., Jr. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys. J. 1998, 75, 2647–2657. [Google Scholar]
- Choudhary, G.; Yotsu-Yamashita, M.; Shang, L.; Yasumoto, T.; Dudley, S.C., Jr. Interactions of the C-11 hydroxyl of tetrodotoxin with the sodium channel outer vestibule. Biophys. J. 2003, 84, 287–294. [Google Scholar]
- Yamagishi, T.; Li, R.A.; Hsu, K.; Marban, E.; Tomaselli, G.F. Molecular architecture of the voltage-dependent Na channel: functional evidence for alpha helices in the pore. J. Gen. Physiol 2001, 118, 171–181. [Google Scholar]
- Patton, D.E.; Goldin, A.L. A voltage-dependent gating transition induces use-dependent block by tetrodotoxin of rat IIA sodium channels expressed in Xenopus oocytes. Neuron 1991, 7, 637–647. [Google Scholar]
- Dumaine, R.; Hartmann, H.A. Two conformational states involved in the use-dependent TTX blockade of human cardiac Na+channel. Am. J. Physiol. 1996, 270 6 Pt 2, H2029–H2037. [Google Scholar]
- Boccaccio, A.; Moran, O.; Imoto, K.; Conti, F. Tonic and phasic tetrodotoxin block of sodium channels with point mutations in the outer pore region. Biophys. J 1999, 77, 229–240. [Google Scholar]
- Moran, O.; Picollo, A.; Conti, F. Tonic and phasic guanidinium toxin-block of skeletal muscle Na channels expressed in mammalian cells. Biophys. J 2003, 84, 2999–3006. [Google Scholar]
- Armstrong, C.M.; Bezanilla, F. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol 1977, 70, 567–590. [Google Scholar]
- Heggeness, S.T.; Starkus, J.G. Saxitoxin and tetrodotoxin. Electrostatic effects on sodium channel gating current in crayfish axons. Biophys. J 1986, 49, 629–643. [Google Scholar]
- Rogart, R.B.; Cribbs, L.L.; Muglia, L.K.; Kephart, D.D.; Kaiser, M.W. Molecular cloning of a putative tetrodotoxin-resistant rat heart Na+ channel isoform. Proc. Natl. Acad. Sci. USA 1989, 86, 8170–8174. [Google Scholar]
- Satin, J.; Kyle, J.W.; Chen, M.; Bell, P.; Cribbs, L.L.; Fozzard, H.A.; Rogart, R.B. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science 1992, 256, 1202–1205. [Google Scholar]
- Backx, P.H.; Yue, D.T.; Lawrence, J.H.; Marban, E.; Tomaselli, G.F. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science 1992, 257, 248–251. [Google Scholar]
- Sangameswaran, L.; Delgado, S.G.; Fish, L.M.; Koch, B.D.; Jakeman, L.B.; Stewart, G.R.; Sze, P.; Hunter, J.C.; Eglen, R.M.; Herman, R.C. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem 1996, 271, 5953–5956. [Google Scholar]
- Dib-Hajj, S.D.; Tyrrell, L.; Black, J.A.; Waxman, S.G. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. USA 1998, 95, 8963–8968. [Google Scholar]
- Kaneko, Y.; Matsumoto, G.; Hanyu, Y. TTX resistivity of Na+ channel in newt retinal neuron. Biochem. Biophys. Res. Commun 1997, 240, 651–656. [Google Scholar]
- Yotsu-Yamashita, M.; Nishimori, K.; Nitanai, Y.; Isemura, M.; Sugimoto, A.; Yasumoto, T. Binding properties of 3H-PbTx-3 and 3H-saxitoxin to brain membranes and to skeletal muscle membranes of puffer fish Fugu pardalis and the primary structure of a voltage-gated Na+ channel α-subunit (fMNa1) from skeletal muscle of F. pardalis. Biochem. Biophys. Res. Commun 2000, 267, 403–412. [Google Scholar]
- Weiss, R.E.; Horn, R. Functional differences between two classes of sodium channels in developing rat skeletal muscle. Science 1986, 233, 361–364. [Google Scholar]
- How, C.K.; Chern, C.H.; Huang, Y.C.; Wang, L.M.; Lee, C.H. Tetrodotoxin poisoning. Am. J. Emerg. Med 2003, 21, 51–54. [Google Scholar]
- Ahasan, H.A.; Mamun, A.A.; Karim, S.R.; Bakar, M.A.; Gazi, E.A.; Bala, C.S. Paralytic complications of puffer fish (tetrodotoxin) poisoning. Singapore Med. J 2004, 45, 73–74. [Google Scholar]
- Isbister, G.K.; Kiernan, M.C. Neurotoxic marine poisoning. Lancet Neurol 2005, 4, 219–228. [Google Scholar]
- Berra-Romani, R.; Blaustein, M.P.; Matteson, D.R. TTX-sensitive voltage-gated Na+ channels are expressed in mesenteric artery smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H137–H145. [Google Scholar]
- Maier, S.K.; Westenbroek, R.E.; Schenkman, K.A.; Feigl, E.O.; Scheuer, T.; Catterall, W.A. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. USA 2002, 99, 4073–4078. [Google Scholar]
- Lei, M.; Jones, S.A.; Liu, J.; Lancaster, M.K.; Fung, S.S.; Dobrzynski, H.; Camelliti, P.; Maier, S.K.; Noble, D.; Boyett, M.R. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J. Physiol. 2004, 559 Pt 3, 835–848. [Google Scholar]
- Brodie, E.D., Jr. Investigations on the skin toxin of the adult rough-skinned newt Taricha granulosa. Copeia 1968, 2, 307–313. [Google Scholar]
- Brodie, E.D., Jr; Ridenhour, B.J.; Brodie, E.D., III. The evolutionary response of predators to dangerous prey: hotspots and coldspots in the geographic mosaic of coevolution between garter snakes and newts. Evolution Int. J. Org. Evolution 2002, 56, 2067–2082. [Google Scholar]
- Janzen, F.J.; Krenz, J.G.; Haselkorn, T.S.; Brodie, E.D.; Brodie, E.D. Molecular phylogeography of common garter snakes (Thamnophis sirtalis) in western North America: implications for regional historical forces. Mol. Ecol 2002, 11, 1739–1751. [Google Scholar]
- Geffeney, S.; Brodie, E.D., Jr; Ruben, P.C.; Brodie, E.D., III. Mechanisms of adaptation in a predator-prey arms race: TTX-resistant sodium channels. Science 2002, 297, 1336–1339. [Google Scholar]
- Geffeney, S.L.; Fujimoto, E.; Brodie, E.D., 3rd; Brodie, E.D., Jr; Ruben, P.C. Evolutionary diversification of TTX-resistant sodium channels in a predator-prey interaction. Nature 2005, 434, 759–763. [Google Scholar]
- Chiamvimonvat, N.; Perez-Garcia, M.T.; Tomaselli, G.F.; Marban, E. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J. Physiol. 1996, 491 Pt 1, 51–59. [Google Scholar]
- Tsushima, R.G.; Li, R.A.; Backx, P.H. Altered ionic selectivity of the sodium channel revealed by cysteine mutations within the pore. J. Gen. Physiol 1997, 109, 463–475. [Google Scholar]
- Brodie, E.D., III; Brodie, E.D., Jr. Costs of exploiting poisonous prey: evolutionary trade-offs in a predator-prey arms race. Evolution 1999, 53, 626–631. [Google Scholar]
- Al-Sabi, A.; McArthur, J.; Ostroumov, V.; French, R.J. Marine toxins that target voltage-gated sodium channels. Mar. Drugs 2006, 4, 157–192. [Google Scholar]



© 2006 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Geffeney, S.L.; Ruben, C. The Structural Basis and Functional Consequences of Interactions Between Tetrodotoxin and Voltage-Gated Sodium Channels. Mar. Drugs 2006, 4, 143-156. https://doi.org/10.3390/md403143
Geffeney SL, Ruben C. The Structural Basis and Functional Consequences of Interactions Between Tetrodotoxin and Voltage-Gated Sodium Channels. Marine Drugs. 2006; 4(3):143-156. https://doi.org/10.3390/md403143
Chicago/Turabian StyleGeffeney, Shana L., and C. Ruben. 2006. "The Structural Basis and Functional Consequences of Interactions Between Tetrodotoxin and Voltage-Gated Sodium Channels" Marine Drugs 4, no. 3: 143-156. https://doi.org/10.3390/md403143
APA StyleGeffeney, S. L., & Ruben, C. (2006). The Structural Basis and Functional Consequences of Interactions Between Tetrodotoxin and Voltage-Gated Sodium Channels. Marine Drugs, 4(3), 143-156. https://doi.org/10.3390/md403143