
Exploring the Inhibitory Effects of Fucosylated Chondroitin Sulfate (FCS) Oligosaccharide Isolated from Stichopus horrens and the Derivatives on P-Selectin

Abstract
1. Introduction
2. Results
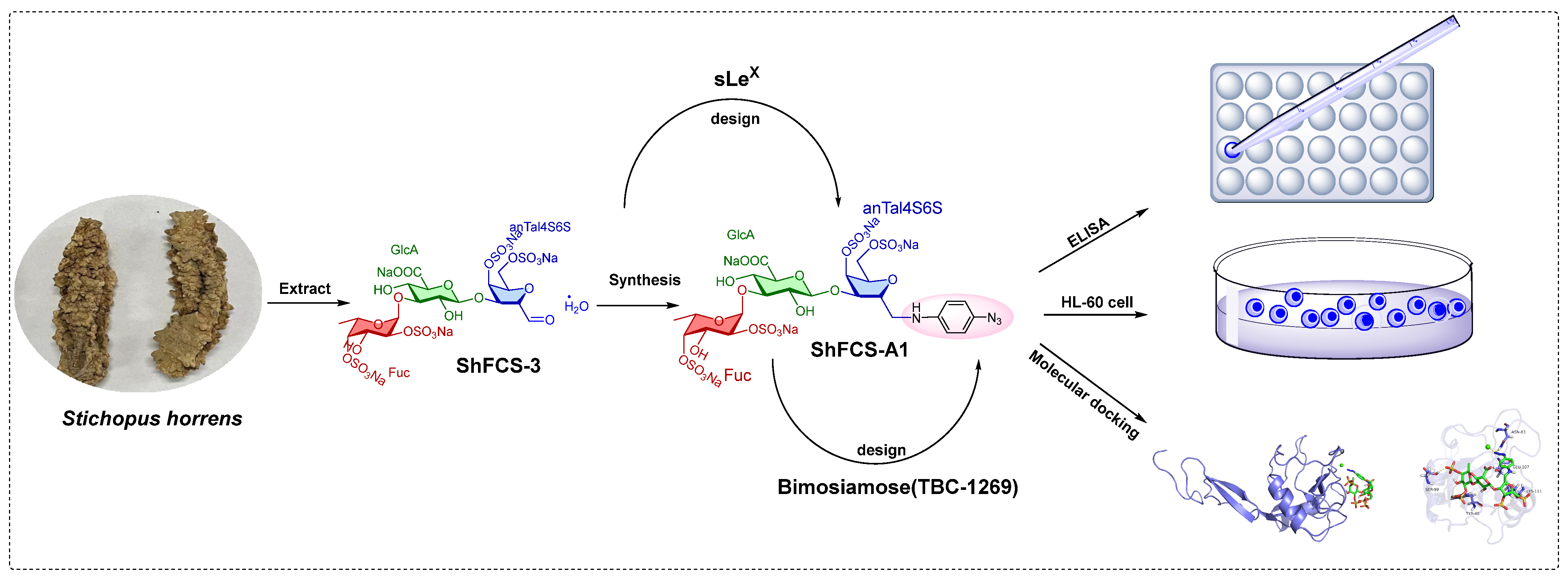
2.1. Extraction, Isolation, and Purification of ShFCS
2.2. ShFCS Deacetylation
2.3. Deaminative Cleavage of ShFCS
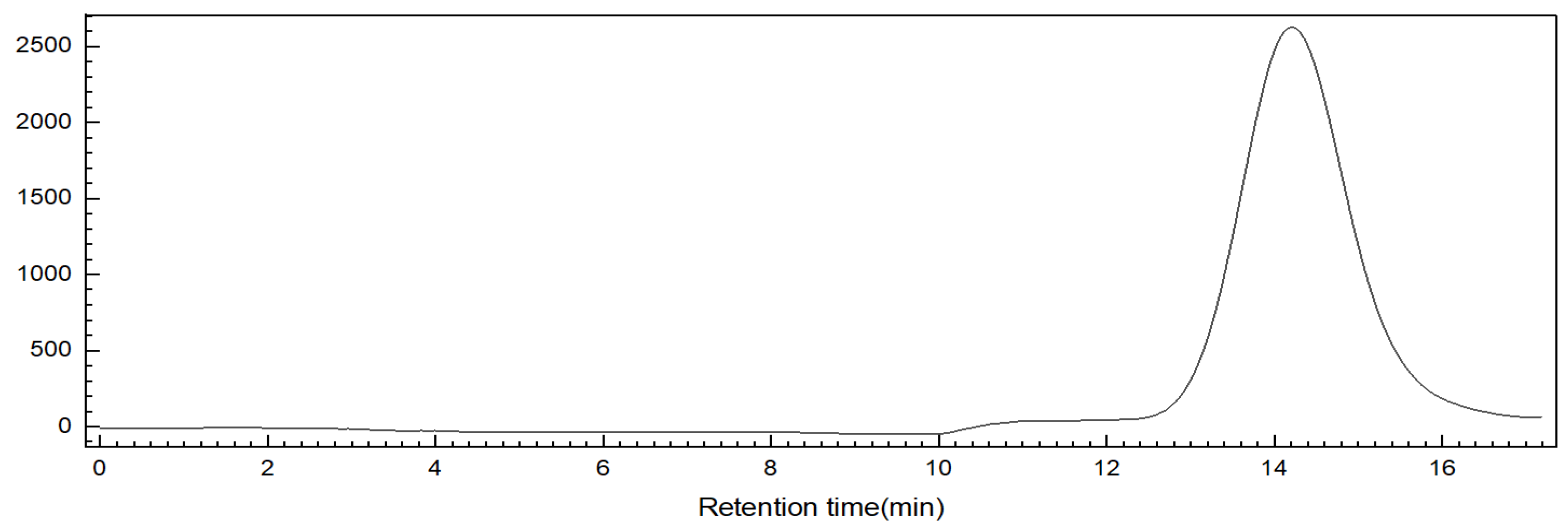
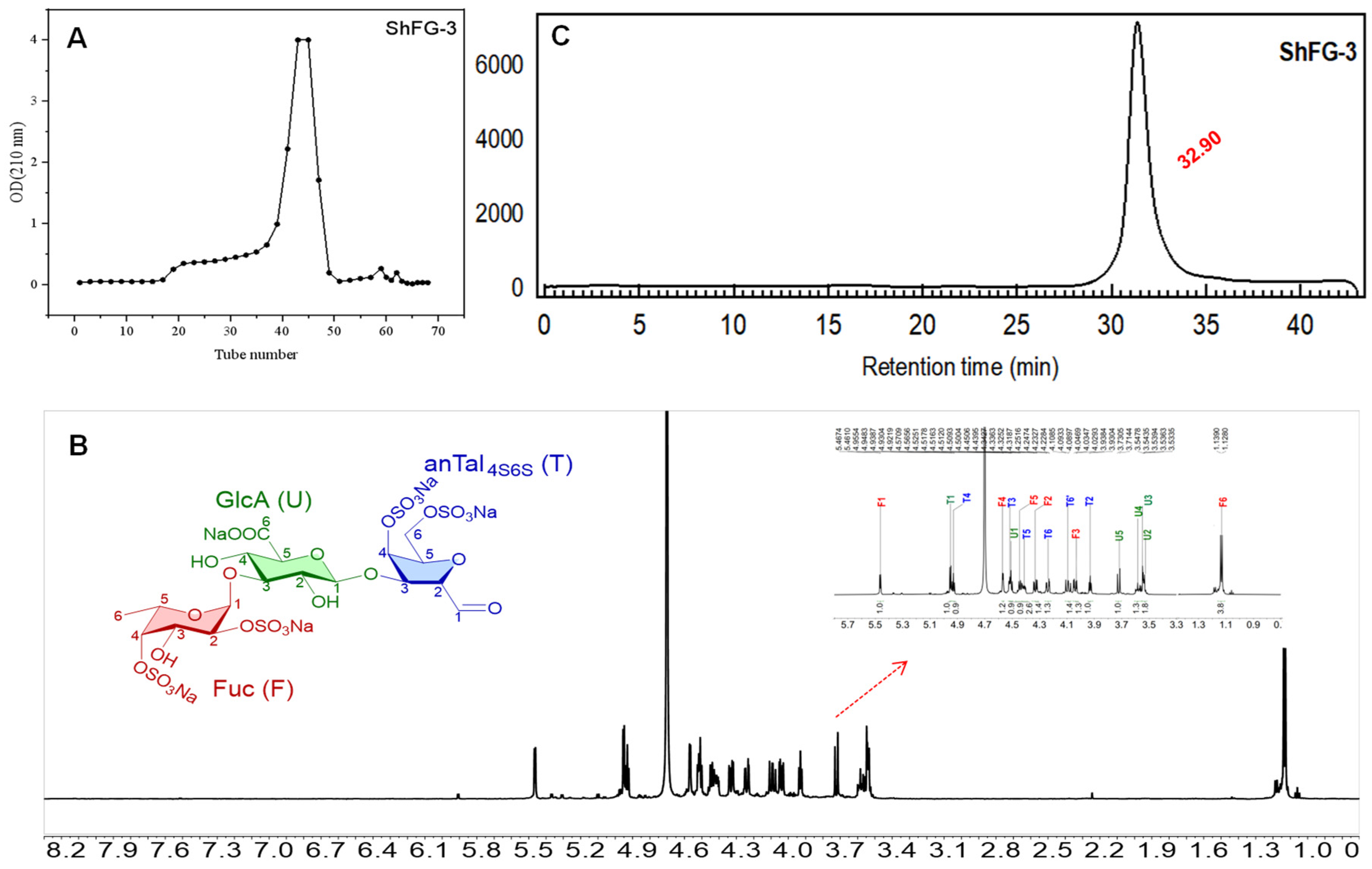
2.4. Purification of the Oligosaccharide ShFCS-3
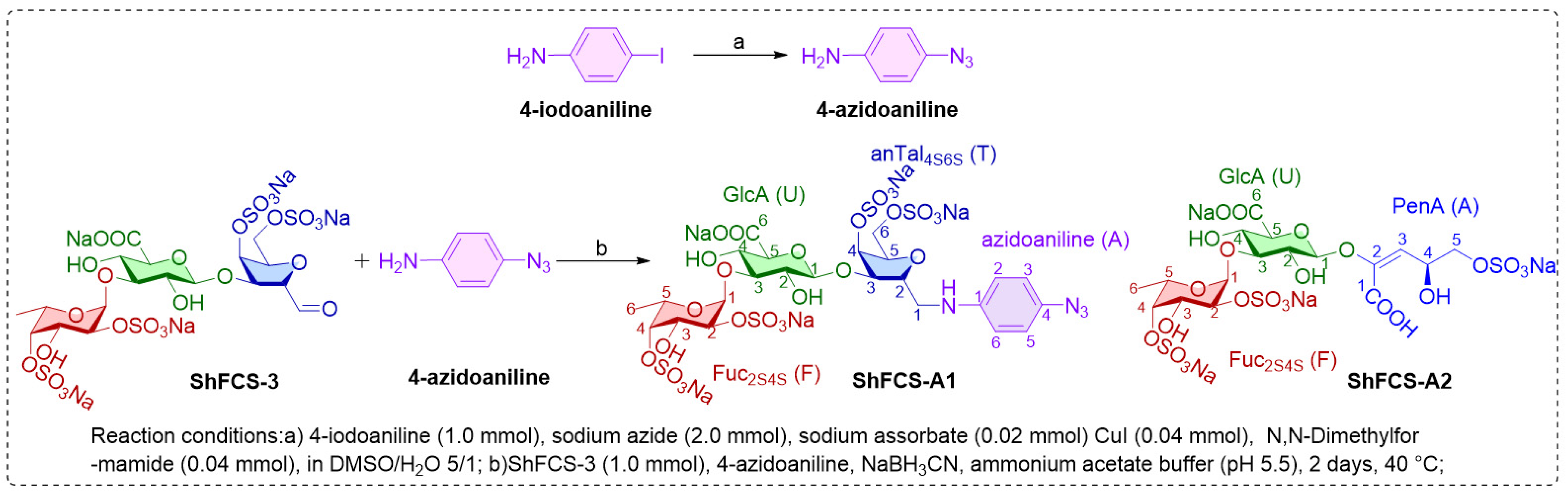
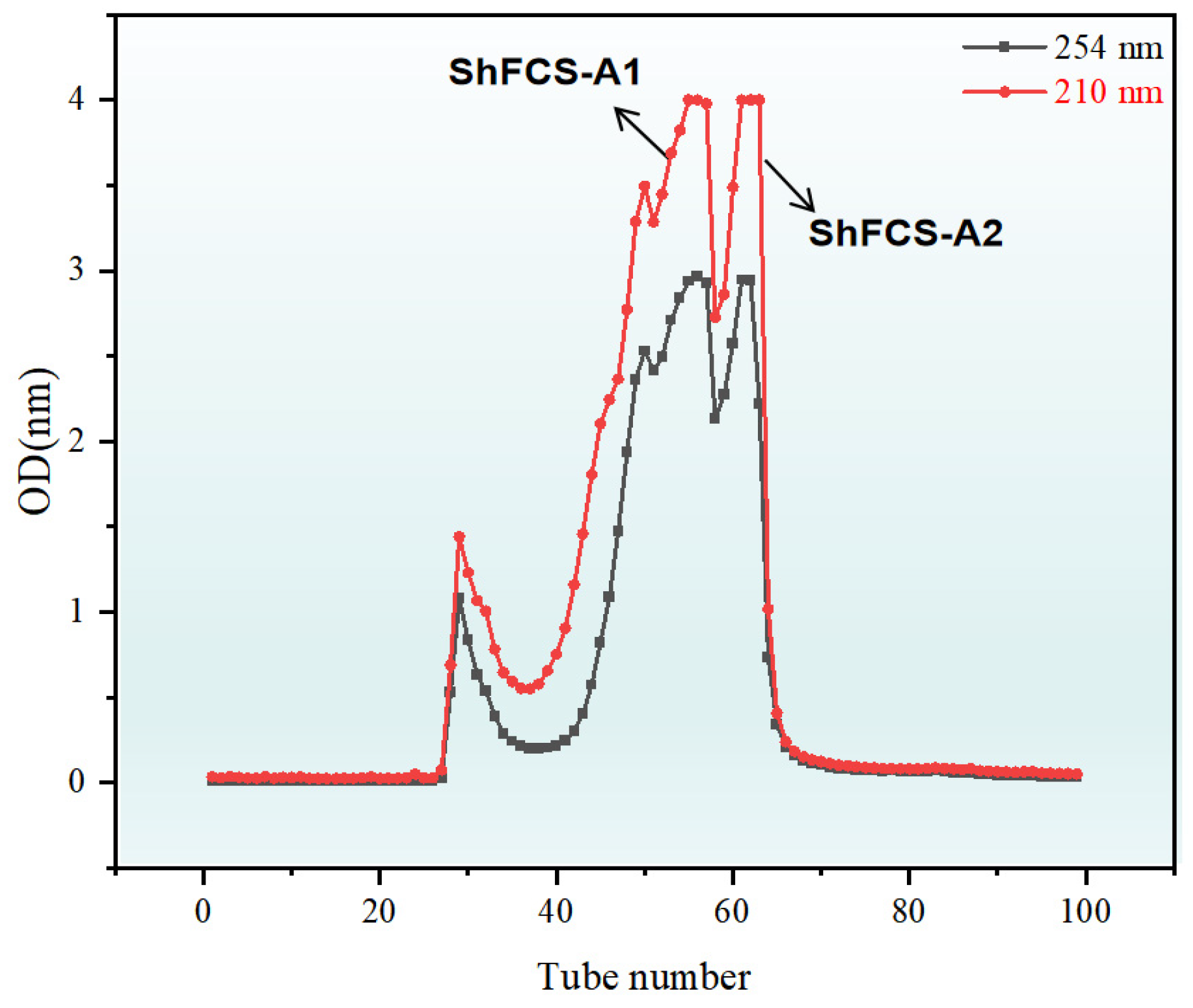
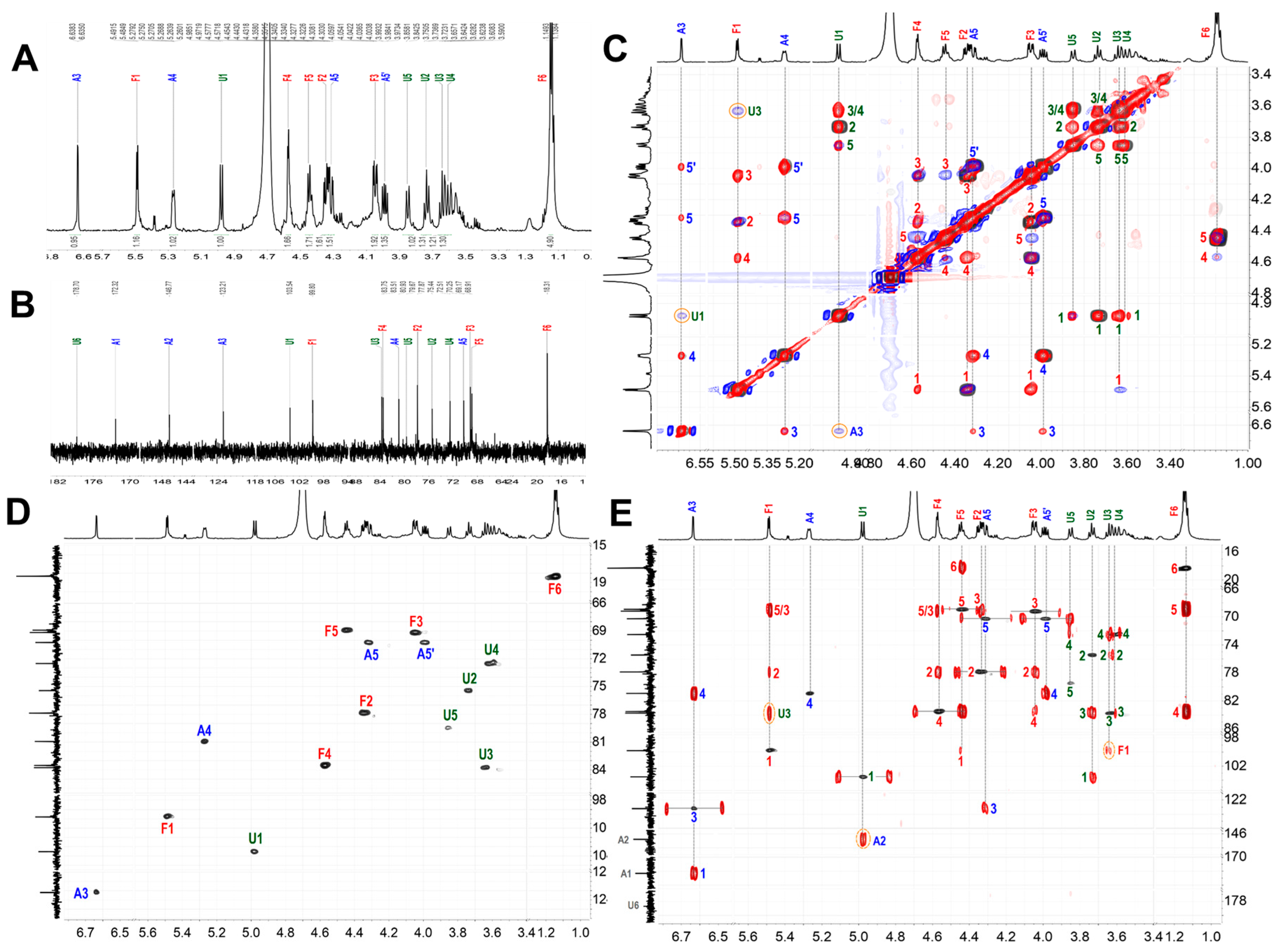
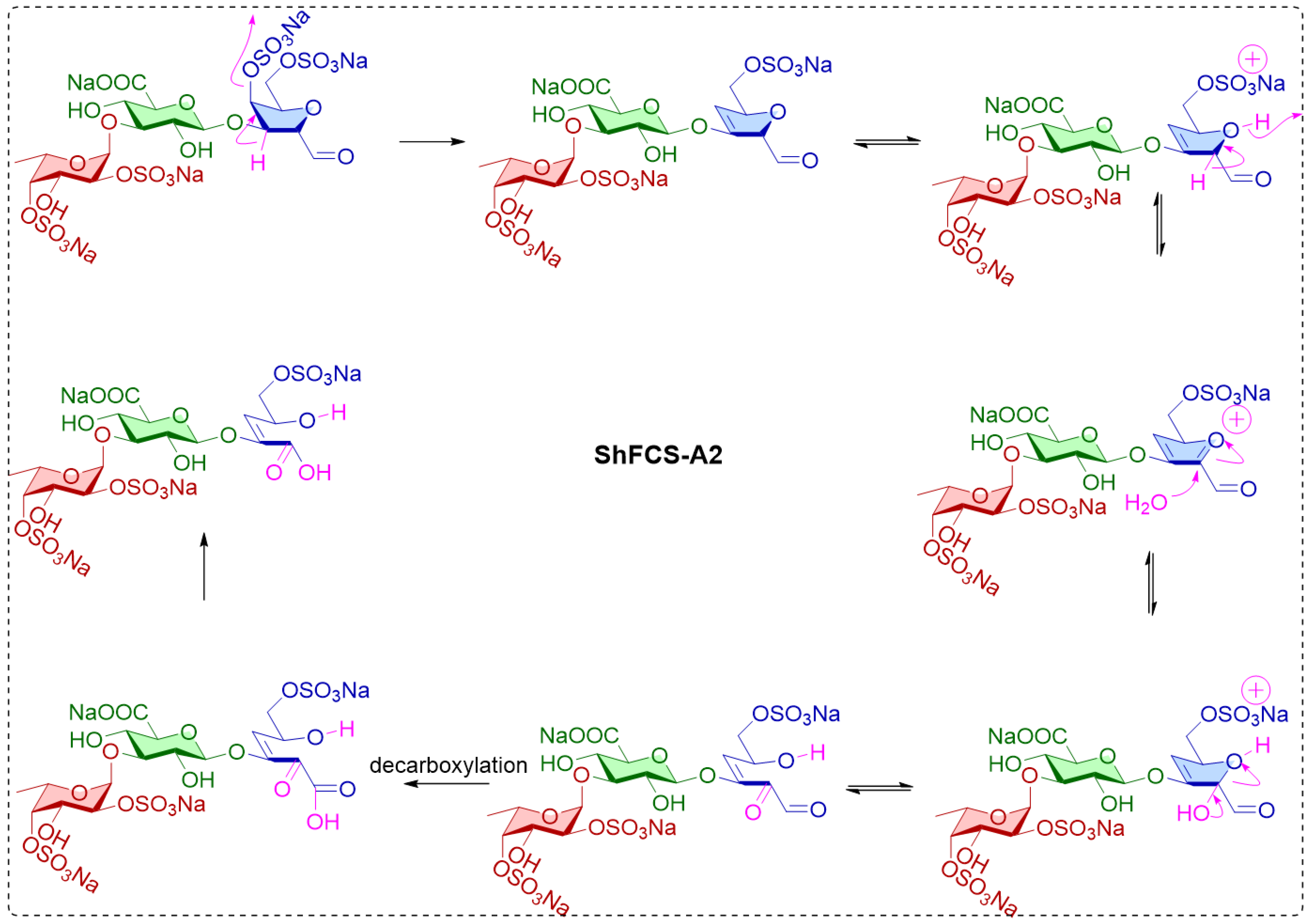
2.5. Synthesis, Purification, and Structural Characterization of ShFCS-A1 and ShFCS-A2
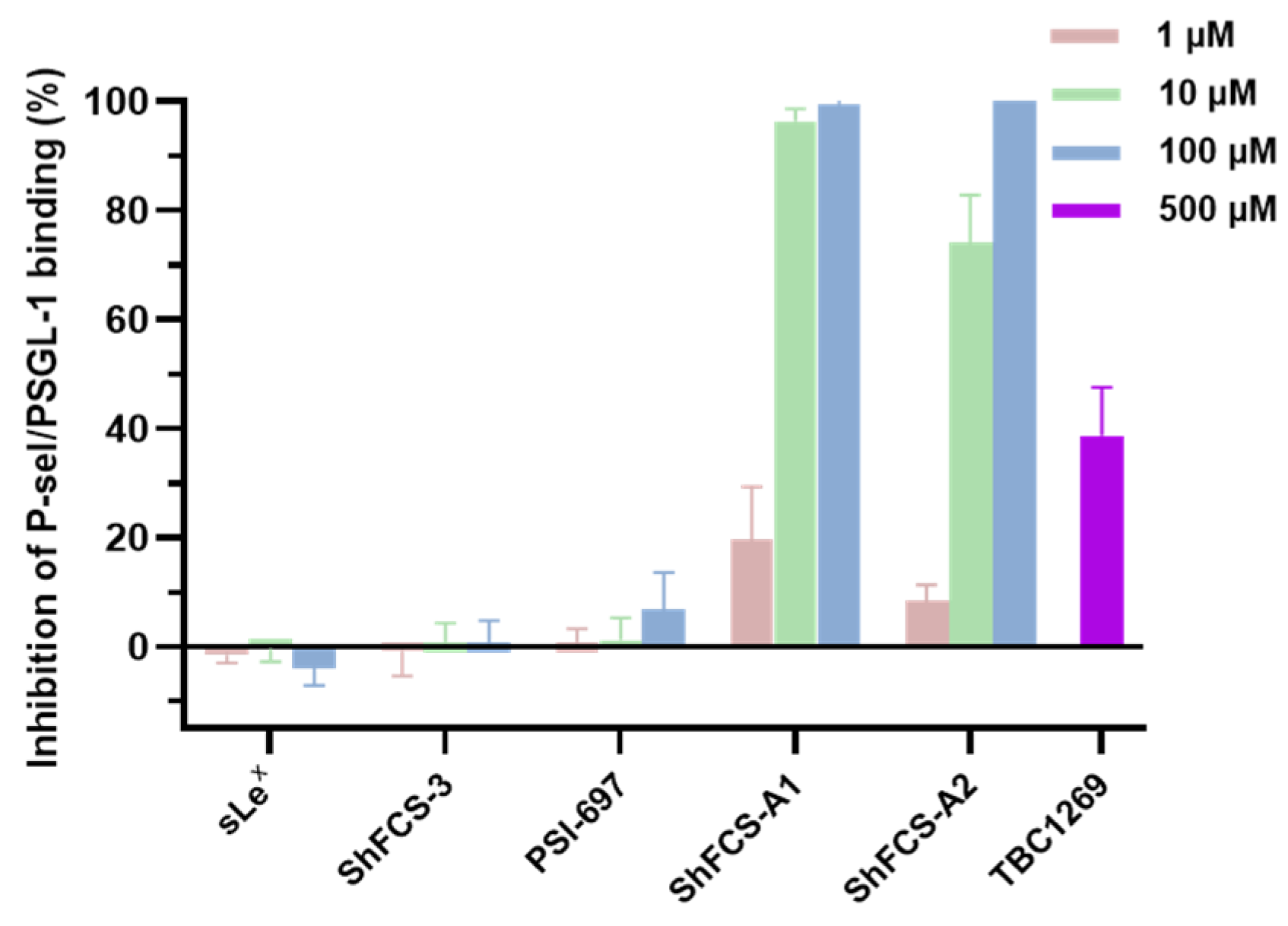
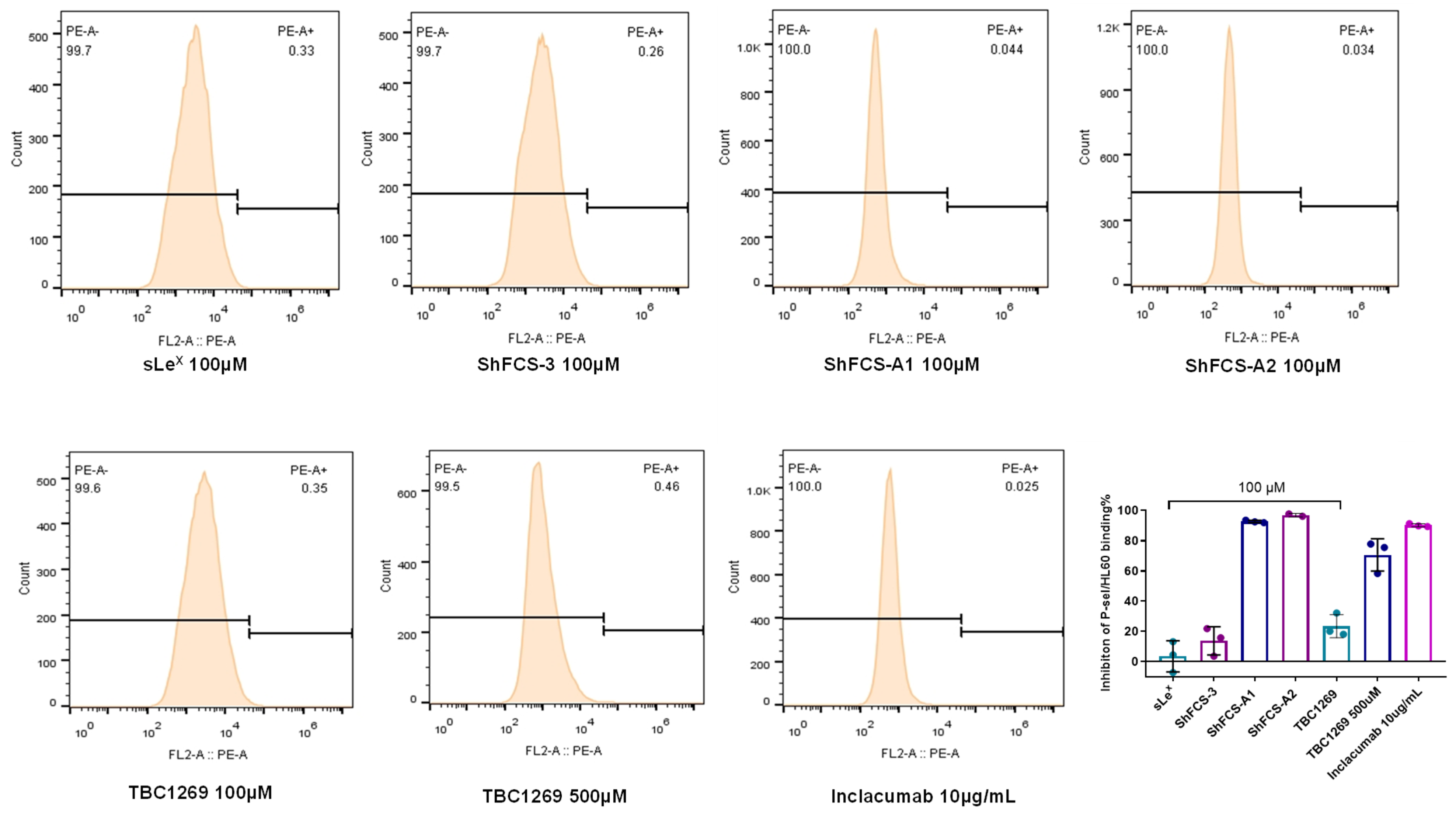
2.6. The Ability of ShFCS-A1 and ShFCS-A2 to Inhibit Binding of P-Selectin to PSGL-1
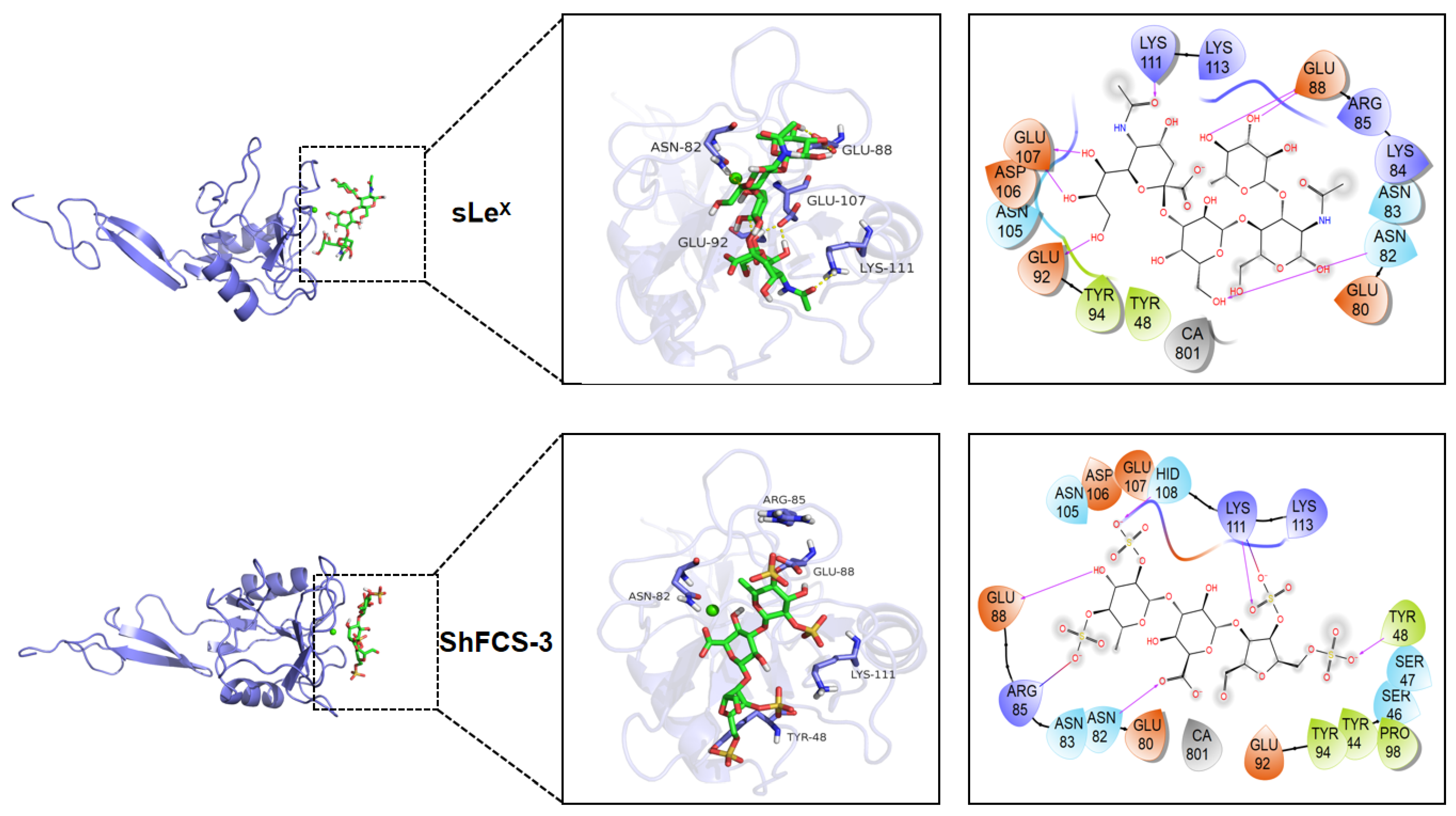
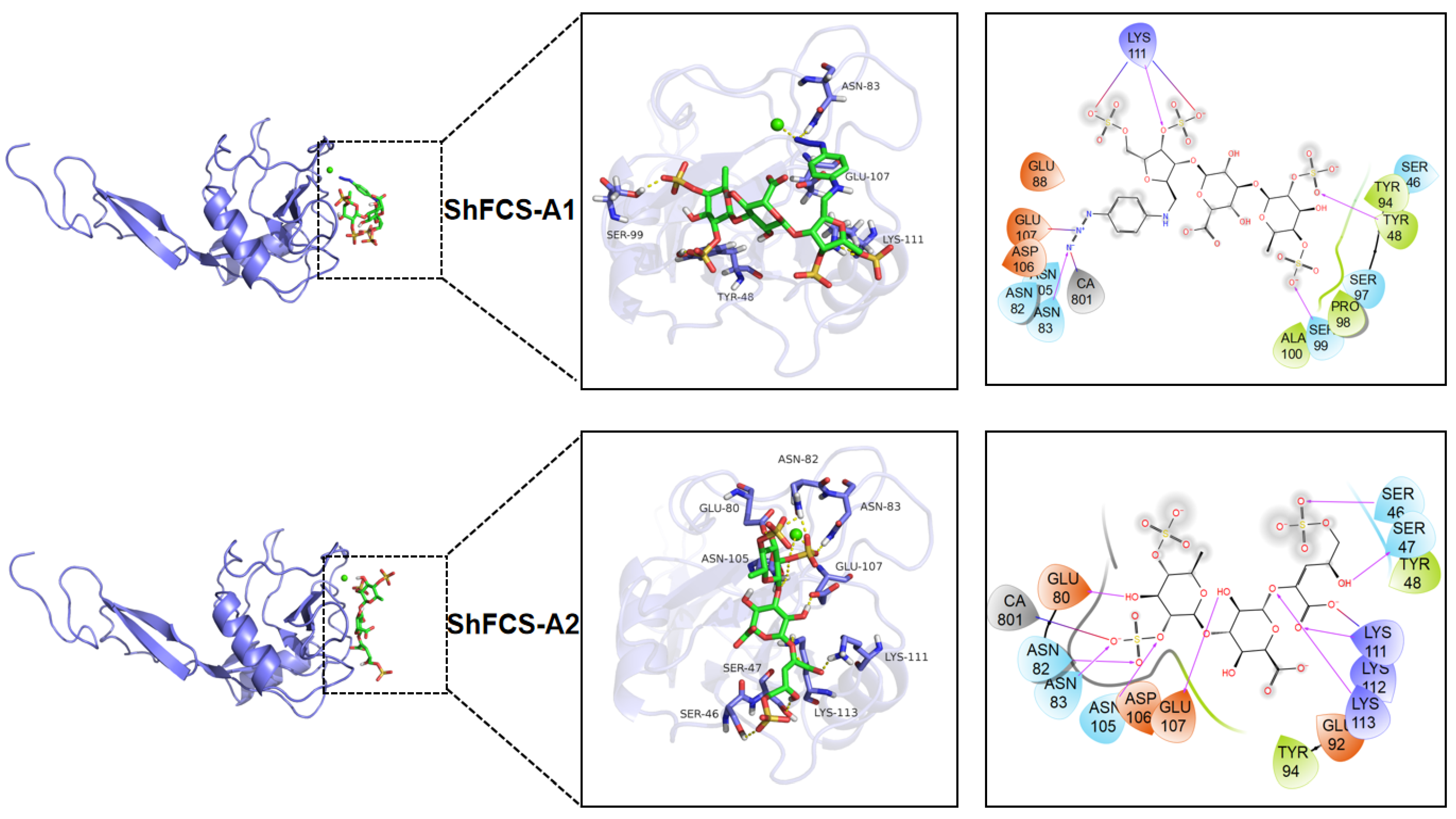
2.7. Molecular Docking
3. Materials and Methods
3.1. Materials and Reagents
3.2. Extraction, Isolation, and Purification of ShFCS-3
3.2.1. Extraction of ShFCS
3.2.2. Deacetylation and Deamination Depolymerization
3.2.3. Isolation and Purification of Oligosaccharide ShFCS-3
3.2.4. Nuclear Magnetic Resonance (NMR) Spectroscopy
3.3. Synthesis and Purification of ShFCS-A1 and ShFCS-A2
3.3.1. Synthesis
3.3.2. Purification
3.3.3. 1D/2D NMR Analysis
3.4. Inhibition of P-Selectin Binding to PSGL-1 by Compounds
3.5. Inhibition of P-Selectin Binding to HL-60 Cells by Compounds
3.6. Molecular Docking
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Myron, P.; Siddiquee, S.; Al Azad, S. Fucosylated Chondroitin Sulfate Diversity in Sea Cucumbers: A Review. Carbohydr. Polym. 2014, 112, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lai, S.; Huang, R.; Wu, M.; Gao, N.; Xu, L.; Qin, H.; Peng, W.; Zhao, J. Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan Degraded by Deaminative Cleavage. Carbohydr. Polym. 2013, 98, 1514–1523. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, M.; Xiao, C.; Yang, L.; Zhou, L.; Gao, N.; Li, Z.; Chen, J.; Chen, J.; Liu, J.; et al. Discovery of an Intrinsic Tenase Complex Inhibitor: Pure Nonasaccharide from Fucosylated Glycosaminoglycan. Proc. Natl. Acad. Sci. USA 2015, 112, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.H.; Bertozzi, C.R. The Clinical Impact of Glycobiology: Targeting Selectins, Siglecs and Mammalian Glycans. Nat. Rev. Drug Discov. 2021, 20, 217–243. [Google Scholar] [CrossRef]
- Kappelmayer, J.; Nagy, B. The Interaction of Selectins and PSGL-1 as a Key Component in Thrombus Formation and Cancer Progression. Biomed. Res. Int. 2017, 2017, 6138145. [Google Scholar] [CrossRef]
- Somers, W.S.; Tang, J.; Shaw, G.D.; Camphausen, R.T. Insights into the Molecular Basis of Leukocyte Tethering and Rolling Revealed by Structures of P- and E-Selectin Bound to SLe(X) and PSGL-1. Cell 2000, 103, 467–479. [Google Scholar] [CrossRef]
- Pouyani, T.; Seed, B. PSGL-1 Recognition of P-Selectin Is Controlled by a Tyrosine Sulfation Consensus at the PSGL-1 Amino Terminus. Cell 1995, 83, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.J.; Park, D.D.; Park, S.S.; Haller, C.A.; Chen, J.; Dai, E.; Liu, L.; Mandhapati, A.R.; Dhakal, B.; Wever, W.J.; et al. A PSGL-1 Glycomimetic Reduces Thrombus Burden without Affecting Hemostasis. Blood 2021, 138, 1182–1193. [Google Scholar] [CrossRef]
- Kerr, K.M.; Auger, W.R.; Marsh, J.J.; Comito, R.M.; Fedullo, R.L.; Smits, G.J.; Kapelanski, D.P.; Fedullo, P.F.; Channick, R.N.; Jamieson, S.W.; et al. The Use of Cylexin (CY-1503) in Prevention of Reperfusion Lung Injury in Patients Undergoing Pulmonary Thromboendarterectomy. Am. J. Respir. Crit. Care Med. 2000, 162, 14–20. [Google Scholar] [CrossRef]
- Hicks, A.E.R.; Abbitt, K.B.; Dodd, P.; Ridger, V.C.; Hellewell, P.G.; Norman, K.E. The Anti-Inflammatory Effects of a Selectin Ligand Mimetic, TBC-1269, Are Not a Result of Competitive Inhibition of Leukocyte Rolling In Vivo. J. Leukoc. Biol. 2005, 77, 59–66. [Google Scholar] [CrossRef]
- Kogan, T.P.; Dupré, B.; Bui, H.; McAbee, K.L.; Kassir, J.M.; Scott, I.L.; Hu, X.; Vanderslice, P.; Beck, P.J.; Dixon, R.A. Novel Synthetic Inhibitors of Selectin-Mediated Cell Adhesion: Synthesis of 1,6-Bis [3-(3-Carboxymethylphenyl)-4-(2-Alpha-D- Mannopyranosyloxy)Phenyl]Hexane (TBC1269). J. Med. Chem. 1998, 41, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Davenpeck, K.L.; Berens, K.L.; Dixon, R.A.; Dupre, B.; Bochner, B.S. Inhibition of Adhesion of Human Neutrophils and Eosinophils to P-Selectin by the Sialyl Lewis Antagonist TBC1269: Preferential Activity against Neutrophil Adhesion In Vitro. J. Allergy Clin. Immunol. 2000, 105, 769–775. [Google Scholar] [CrossRef]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, a Novel Pan-Selectin Antagonist, Reverses Acute Vascular Occlusions in Sickle Cell Mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef]
- Tammara, B.; Shafer, F.E.; Harnisch, L. Dose Selection Process for Younger Children Participating in a Phase 3 Study to Evaluate the Efficacy and Safety of Rivipansel (GMI-1070) in the Treatment of Vaso-Occlusive Crisis in Hospitalized Patients with Sickle Cell Disease. Blood 2018, 132, 3643. [Google Scholar] [CrossRef]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized Phase 2 Study of GMI-1070 in SCD: Reduction in Time to Resolution of Vaso-Occlusive Events and Decreased Opioid Use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B. Analogues of the Pan-Selectin Antagonist Rivipansel (GMI-1070). Eur. J. Med. Chem. 2024, 272, 116455. [Google Scholar] [CrossRef]
- Kaila, N.; Janz, K.; Huang, A.; Moretto, A.; DeBernardo, S.; Bedard, P.W.; Tam, S.; Clerin, V.; Keith, J.C.; Tsao, D.H.H.; et al. 2-(4-Chlorobenzyl)-3-Hydroxy-7,8,9,10-Tetrahydrobenzo[H]Quinoline-4-Carboxylic Acid (PSI-697): Identification of a Clinical Candidate from the Quinoline Salicylic Acid Series of P-Selectin Antagonists. J. Med. Chem. 2007, 50, 40–64. [Google Scholar] [CrossRef]
- Myers, D.D.; Henke, P.K.; Bedard, P.W.; Wrobleski, S.K.; Kaila, N.; Shaw, G.; Meier, T.R.; Hawley, A.E.; Schaub, R.G.; Wakefield, T.W. Treatment with an Oral Small Molecule Inhibitor of P Selectin (PSI-697) Decreases Vein Wall Injury in a Rat Stenosis Model of Venous Thrombosis. J. Vasc. Surg. 2006, 44, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Myers, D.; Wrobleski, S.; Longo, C.; Bedard, P.; Kaila, N.; Shaw, G.; Londy, F.; Rohrer, S.; Fex, B.; Zajkowski, P.; et al. Resolution of Venous Thrombosis Using a Novel Oral Small-Molecule Inhibitor of P-Selectin (PSI-697) without Anticoagulation. Thromb. Haemost. 2007, 97, 400–407. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. New Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Blair, H.A. Crizanlizumab: First Approval. Drugs 2020, 80, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, V.R.; Sardar, M.Y.R.; Ying, Y.; Song, X.; Haller, C.; Dai, E.; Wang, X.; Hanjaya-Putra, D.; Sun, L.; Morikis, V.; et al. Glycopeptide Analogues of PSGL-1 Inhibit P-Selectin In Vitro and In Vivo. Nat. Commun. 2015, 6, 6387. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.E.R.; Leppänen, A.; Cummings, R.D.; McEver, R.P.; Hellewell, P.G.; Norman, K.E. Glycosulfopeptides Modeled on P-selectin Glycoprotein Ligand-1 Inhibit P-selectin-dependent Leukocyte Rolling In Vivo. FASEB J. 2002, 16, 1461–1462. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wang, L.; Cavalcante, M.C.M.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.S.; Esko, J.D.; Pavão, M.S.G. Selectin Blocking Activity of a Fucosylated Chondroitin Sulfate Glycosaminoglycan from Sea Cucumber. Effect on Tumor Metastasis and Neutrophil Recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef]
- Panagos, C.G.; Thomson, D.S.; Moss, C.; Hughes, A.D.; Kelly, M.S.; Liu, Y.; Chai, W.; Venkatasamy, R.; Spina, D.; Page, C.P.; et al. Fucosylated Chondroitin Sulfates from the Body Wall of the Sea Cucumber Holothuria Forskali: Conformation, Selectin Binding, and Biological Activity. J. Biol. Chem. 2014, 289, 28284–28298. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.E.; Bilan, M.I.; Dmitrenok, A.S.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Two Structurally Similar Fucosylated Chondroitin Sulfates from the Holothurian Species Stichopus Chloronotus and Stichopus Horrens. Carbohydr. Polym. 2018, 189, 10–14. [Google Scholar] [CrossRef]
- Guan, R.; Peng, Y.; Zhou, L.; Zheng, W.; Liu, X.; Wang, P.; Yuan, Q.; Gao, N.; Zhao, L.; Zhao, J. Precise Structure and Anticoagulant Activity of Fucosylated Glycosaminoglycan from Apostichopus Japonicus: Analysis of Its Depolymerized Fragments. Mar. Drugs 2019, 17, 195. [Google Scholar] [CrossRef]
- Zhang, J.; Park, S.; Chang, S. Selective C−O Bond Cleavage of Sugars with Hydrosilanes Catalyzed by Piers’ Borane Generated In Situ. Angew. Chem. Int. Ed. 2017, 56, 13757–13761. [Google Scholar] [CrossRef]
- Pezzotti, F.; Therisod, M. Enzymatic Synthesis of Aldonic Acids. Carbohydr. Res. 2006, 341, 2290–2292. [Google Scholar] [CrossRef]
- Niu, W.; Molefe, M.N.; Frost, J.W. Microbial Synthesis of the Energetic Material Precursor 1,2,4-Butanetriol. J. Am. Chem. Soc. 2003, 125, 12998–12999. [Google Scholar] [CrossRef]
- Overend, W.G.; Stacey, M.; Wiggins, L.F. 288. Deoxy-Sugars. Part IV. A Synthesis of 2-Deoxy-D-Ribose from D-Erythrose. J. Chem. Soc. 1949, 1358. [Google Scholar] [CrossRef]
- Bienkowski, M.J.; Conrad, H.E. Structural Characterization of the Oligosaccharides Formed by Depolymerization of Heparin with Nitrous Acid. J. Biol. Chem. 1985, 260, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Moussa, A.; Crépet, A.; Ladavière, C.; Trombotto, S. Reducing-End “Clickable” Functionalizations of Chitosan Oligomers for the Synthesis of Chitosan-Based Diblock Copolymers. Carbohydr. Polym. 2019, 219, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Kenney, I.M.; Beckstein, O.; Iorga, B.I. Prediction of Cyclohexane-Water Distribution Coefficients for the SAMPL5 Data Set Using Molecular Dynamics Simulations with the OPLS-AA Force Field. J. Comput. Aided Mol. Des. 2016, 30, 1045–1058. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | δ | Couplings | COSY | TOCSY | ROESY | 13C | δ | HSQC | HMBC | |
|---|---|---|---|---|---|---|---|---|---|---|
| F | H1 | 5.458 | J(1,2) = 3.90 | H2 | H2,3,4 | H2; U2,3 | C1 | 99.65 | H1 | H5; U3 |
| H2 | 4.330 | J(2,3) = 10.50 | H1,3 | H1,3,4 | H1,3 | C2 | 77.86 | H2 | H1,2,3,4 | |
| H3 | 4.041 | J(3,4) = 3.18 | H2,4 | H1,2,4 | H2,4,5 | C3 | 69.13 | H3 | H1,2,3,4 | |
| H4 | 4.566 | H3 | H1,2,3 | H3,5,6 | C4 | 83.58 | H4 | H3,4,5,6 | ||
| H5 | 4.435 | J(5,6) = 6.60 | H6 | H5 | H3,4,6 | C5 | 68.82 | H5 | H1,4,6 | |
| H6 | 1.143 | H5 | H6 | H4,5 | C6 | 18.32 | H6 | H5 | ||
| U | H1 | 4.241 | J(1,2) = 7.62 | H2 | H2,3,4,5 | H3,5; T3 | C1 | 103.81 | H1 | H1,2,5; T3 |
| H2 | 3.503 | J(2,3) = 8.88 | H1,3 | H1,3,4,5 | H1,3; F1 | C2 | 75.53 | H2 | H2,3,5 | |
| H3 | 3.401 | J(3,4) = 9.00 | H2,4 | H1,2,4,5 | H1,2; F1 | C3 | 84.11 | H3 | H2,3,4; F1 | |
| H4 | 3.502 | J(4,5) = 10.02 | H3,5 | H1,2,3,5 | H5; F1 | C4 | 72.78 | H4 | H3,4,5 | |
| H5 | 3.413 | H4 | H1,2,3,4 | H1,3 | C5 | 79.39 | H5 | H4,5 | ||
| C6 | 178.15 | H4,5 | ||||||||
| T | H1 | 3.319 | J(1,2) = 4.68 | H2 | H2,3,4 | H2,3; A2/6 | C1 | 48.27 | H1/1′ | H1/1′,3 |
| H1′ | 3.263 | J(1,1′) = 14.16 | H2 | H2,3,4 | H2; A2/6 | |||||
| H2 | 4.186 | J(2,3) = 11.46 | H1,3 | H1,3,4 | H1,3,4 | C2 | 81.74 | H2 | H1/1′,2,4 | |
| H3 | 4.455 | J(3,4) = 5.52 | H2,4 | H1,2,4,6 | H1/1′,2,4,6 | C3 | 79.71 | H3 | H1,2,4; U1 | |
| H4 | 4.959 | J(4,5) = 4.80 | H3,5 | H1,2,3,5,6 | H3,5,6 | C4 | 80.05 | H4 | H3,4,5 | |
| H5 | 4.428 | J(5,6) = 2.64 | H4,6′ | H4,6/6′ | H4,6,6′ | C5 | 79.86 | H5 | H4,5,6,6′ | |
| H6 | 4.243 | J(6,6′) = 11.58 | H6′ | H4,5,6′ | H3,5,6′ | C6 | 70.65 | H6,6′ | H6,6′ | |
| H6′ | 4.072 | J(6′,5) = 9.36 | H5,6 | H4,5,6 | H3,5,6 | |||||
| A | -- | C1 | 147.65 | H3/5; T1/1′ | ||||||
| H2/6 | 6.780 | J(2,3) = 8.76 | H3/5 | H3/5 | H3/5 | C2/6 | 118.40 | H2/6 | H3/5,2/6 | |
| H3/5 | 6.900 | J(5,6) = 8.76 | H2/6 | H2/6 | H2/6 | C3/5 | 122.81 | H3/5 | H3/5,2/6 | |
| -- | -- | C4 | 132.91 | H3/5,2/6 |
| 1H | δ | Couplings | COSY | TOCSY | ROESY | 13C | δ | HSQC | HMBC | |
|---|---|---|---|---|---|---|---|---|---|---|
| F | H1 | 5.488 | J(1,2) = 3.96 | H2 | H2,3,4 | H2; U3 | C1 | 99.80 | H1 | H1,5; U3 |
| H2 | 4.346 | J(2,3) = 10.50 | H1,3 | H1,3,4 | H1,3 | C2 | 77.87 | H2 | H1,2,3,4 | |
| H3 | 4.048 | J(3,4) = 3.36 | H2,4 | H1,2,4 | H2,4,5 | C3 | 69.17 | H3 | H1,2,3,4 | |
| H4 | 4.575 | H3 | H1,2,3,5 | H3,5,6 | C4 | 83.51 | H4 | H3,4,5,6 | ||
| H5 | 4.448 | J(5,6) = 6.66 | H6 | H4,6 | H3,4,6 | C5 | 68.91 | H5 | H1,4,5,6 | |
| H6 | 1.144 | H5 | H5 | H4,5 | C6 | 18.31 | H6 | H5,6 | ||
| U | H1 | 4.979 | J(1,2) = 7.92 | H2 | H2,3,4,5 | H5; A3 | C1 | 103.54 | H1 | H1,2 |
| H2 | 3.737 | J(2,3) = 8.28 | H1,3 | H1,3,4,5 | -- | C2 | 75.44 | H2 | H3 | |
| H3 | 3.642 | J(3,4) = 8.82 | H2,4 | H1,2,4,5 | F1 | C3 | 83.75 | H3 | H2,4; F1 | |
| H4 | 3.608 | J(4,5) = 9.36 | H3,5 | H1,2,3,5 | -- | C4 | 72.51 | H4 | H3,4,5 | |
| H5 | 3.851 | H4 | H1,2,3,4 | H1 | C5 | 79.67 | H5 | -- | ||
| C6 | 178.80 | -- | ||||||||
| A | -- | C1 | 172.32 | -- | H3,5 | |||||
| -- | C2 | 146.77 | -- | U1 | ||||||
| H3 | 6.636 | J(3,4) = 1.98 | H4 | H4,5,5′ | U1 | C3 | 123.21 | H3 | H3,5 | |
| H4 | 5.269 | J(4,5) = 3.06 | H3,5,5′ | H3,5,5′ | H3,5 | C4 | 80.93 | H4 | H3,5′ | |
| H5 | 4.316 | J(6,6′) = 11.82 | H4,6′ | H3,4,5′ | H4,5′ | C6 | 70.25 | H5,5′ | H5,5′ | |
| H5′ | 3.989 | J(6′,5) = 6.36 | H4,6 | H3,4,5 | H5 |
| Compounds | Docking Score (kcal/mol) |
|---|---|
| sLeX | −5.620 |
| ShFCS-3 | −5.066 |
| ShFCS-A1 | −5.954 |
| ShFCS-A2 | −6.140 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Sun, H.; Gu, X.; Long, W.; Zhu, G.; Wu, X.; Wang, Y.; Li, P.; Sha, L.; Zhang, J.; et al. Exploring the Inhibitory Effects of Fucosylated Chondroitin Sulfate (FCS) Oligosaccharide Isolated from Stichopus horrens and the Derivatives on P-Selectin. Mar. Drugs 2025, 23, 236. https://doi.org/10.3390/md23060236
Li C, Sun H, Gu X, Long W, Zhu G, Wu X, Wang Y, Li P, Sha L, Zhang J, et al. Exploring the Inhibitory Effects of Fucosylated Chondroitin Sulfate (FCS) Oligosaccharide Isolated from Stichopus horrens and the Derivatives on P-Selectin. Marine Drugs. 2025; 23(6):236. https://doi.org/10.3390/md23060236
Chicago/Turabian StyleLi, Caiyi, Huifang Sun, Xi Gu, Wen Long, Guangyu Zhu, Xiaolu Wu, Yu Wang, Pengfei Li, Le Sha, Jiali Zhang, and et al. 2025. "Exploring the Inhibitory Effects of Fucosylated Chondroitin Sulfate (FCS) Oligosaccharide Isolated from Stichopus horrens and the Derivatives on P-Selectin" Marine Drugs 23, no. 6: 236. https://doi.org/10.3390/md23060236
APA StyleLi, C., Sun, H., Gu, X., Long, W., Zhu, G., Wu, X., Wang, Y., Li, P., Sha, L., Zhang, J., Sun, W., Gao, N., Zuo, Z., & Zhao, J. (2025). Exploring the Inhibitory Effects of Fucosylated Chondroitin Sulfate (FCS) Oligosaccharide Isolated from Stichopus horrens and the Derivatives on P-Selectin. Marine Drugs, 23(6), 236. https://doi.org/10.3390/md23060236