
Synthesis and Evaluation of Antitumor and Anti-Angiogenesis Activity of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A



Abstract
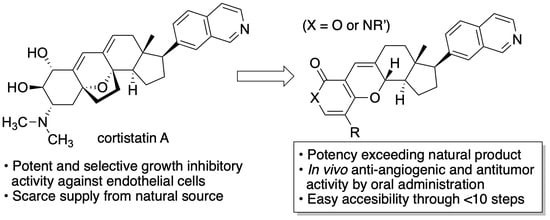
1. Introduction
2. Results and Discussions
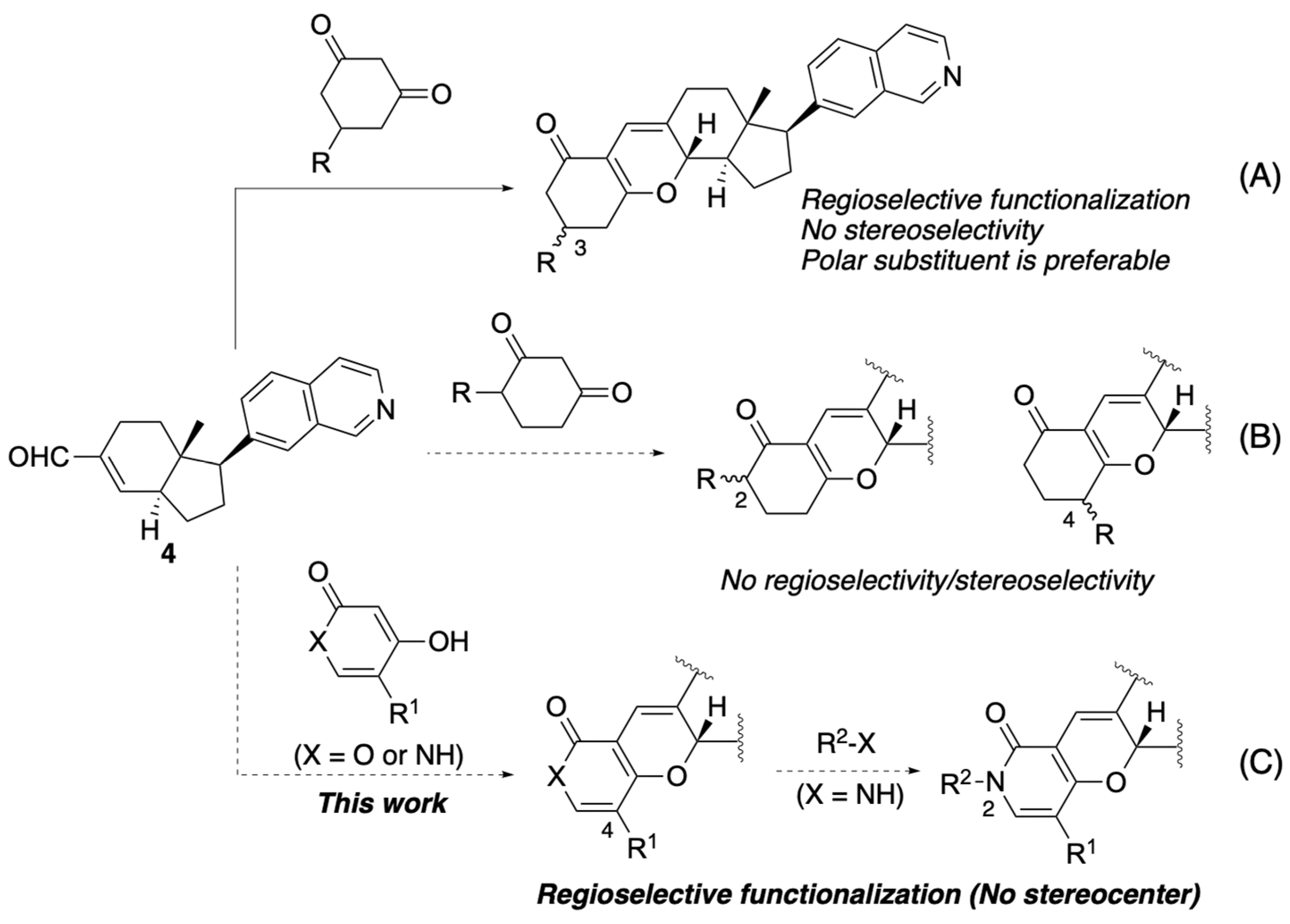
2.1. Design of Pyrone- or Pyridone-Embedded Analogs
2.2. Synthesis of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A
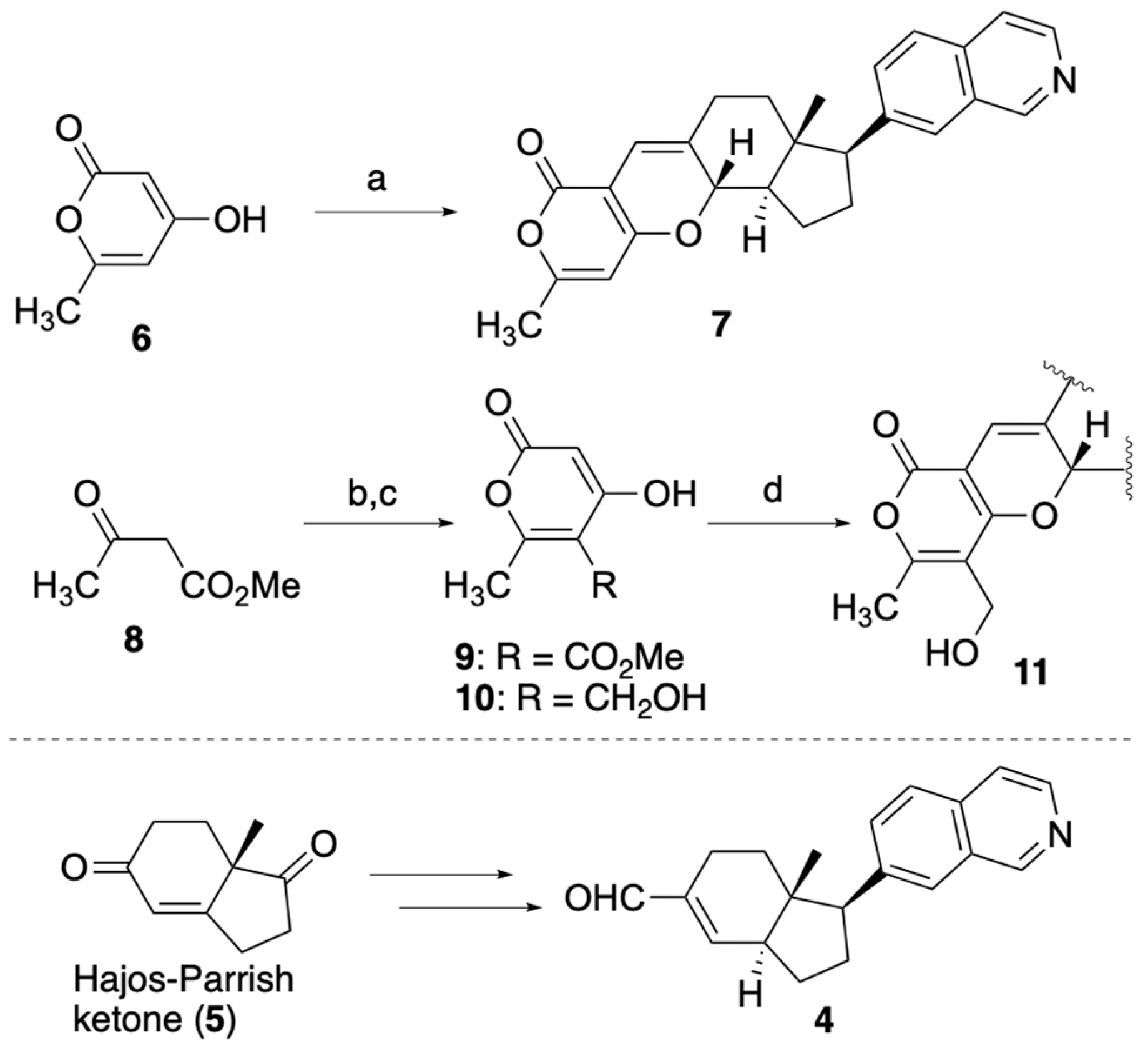
2.2.1. Synthesis of Pyrone-Embedded Analogs
2.2.2. Synthesis of Pyridone-Embedded Analogs
2.3. Biological Evaluation of Pyrone- or Pyridone-Embedded Analogs
2.3.1. Antiproliferative Activities of the Analogs Against Endothelial or Cancer Cells
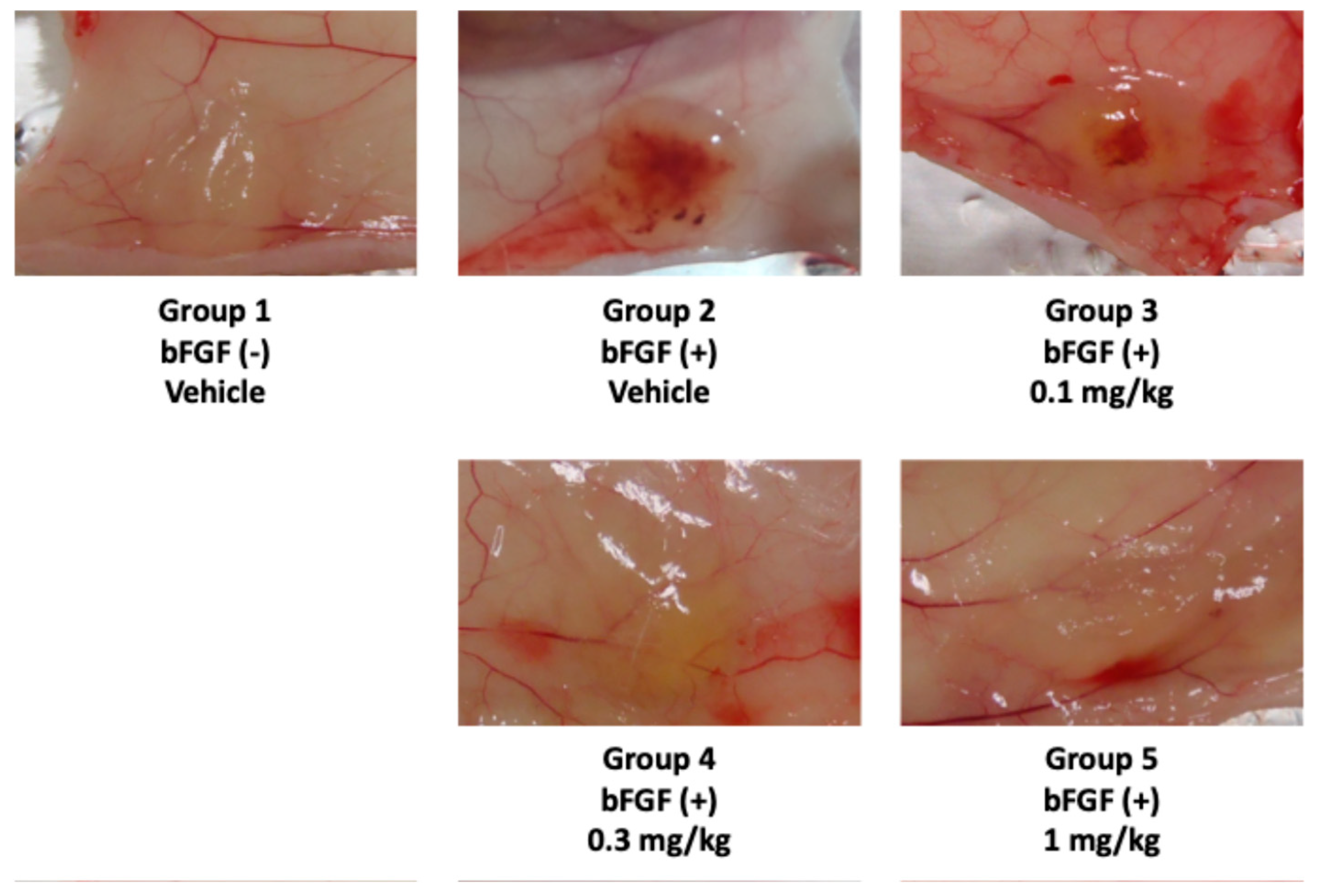
2.3.2. In Vivo Activity Evaluation of Analog 19
3. Materials and Methods
3.1. General
3.2. Assay for Cytotoxicity of Compounds Against HUVECs
3.3. Antiproliferative Activity of the Compounds Against Cancer Cells
3.4. In Vivo Matrigel Plug Assay and Toxicity Tests of Analog 19
3.5. In Vivo Antitumor Effect of Analog 19
3.6. Synthesis
3.6.1. (3S,3aR,11aS,11bR)-3-(Isoquinolin-7-yl)-3a,9-dimethyl-1,2,3,3a,4,5,11a,11b-octahydro-7H-cyclopenta[h]pyrano[4,3-b]chromen-7-one (7)
3.6.2. (3S,3aR,11aS,11bR)-10-(Hydroxymethyl)-3-(isoquinolin-7-yl)-3a,9-dimethyl-1,2,3,3a,4,5,11a,11b-octahydro-7H-cyclopenta[h]pyrano[4,3-b]chromen-7-one (11)
3.6.3. (3S,3aR,11aS,11bR)-3-(Isoquinolin-7-yl)-3a-methyl-2,3,3a,4,5,8,11a,11b-octahydrocyclopenta[7,8]chromeno[3,2-c]pyridin-7(1H)-one (13)
3.6.4. (3S,3aR,11aS,11bR)-3-(Isoquinolin-7-yl)-3a,8-dimethyl-2,3,3a,4,5,8,11a,11b-octahydrocyclopenta[7,8]chromeno[3,2-c]pyridin-7(1H)-one (15)
3.6.5. 4-Hydroxy-5-(hydroxymethyl)-1-methylpyridin-2(1H)-one (18)
3.6.6. (3S,3aR,11aS,11bR)-10-(Hydroxymethyl)-3-(isoquinolin-7-yl)-3a,8-dimethyl-2,3,3a,4,5,8,11a,11b-octahydrocyclopenta[7,8]chromeno[3,2-c]pyridin-7(1H)-one (19)
3.6.7. tert-Butyl (2-(2,2-Dimethyl-4-oxo-4H-1,3-dioxin-6-yl)-3-oxobutyl)carbamate (22)
3.6.8. tert-Butyl (Z)-(5-(methylamino)-2-(1-(methylamino)ethylidene)-3,5-dioxopentyl)carbamate (23)
3.6.9. N-((4-Hydroxy-1,2-dimethyl-6-oxo-1,6-dihydropyridin-3-yl)methyl)acetamide (26)
3.6.10. N-(((3S,3aR,11aS,11bR)-3-(Isoquinolin-7-yl)-3a,8,9-trimethyl-7-oxo-1,2,3,3a,4,5,7,8,11a,11b-decahydrocyclopenta[7,8]chromeno[3,2-c]pyridin-10-yl)methyl)acetamide (27)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine Natural Products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Wang, S.; Dong, G.; Sheng, C. Structural Simplification of Natural Products. Chem. Rev. 2019, 119, 4180–4220. [Google Scholar] [CrossRef]
- Wach, J.-Y.; Gademann, K. Reduce to the Maximum: Truncated Natural Products as Powerful Modulators of Biological Processes. Synlett 2012, 2012, 163–170. [Google Scholar] [CrossRef]
- Ueda, M. Chemical Biology of Natural Products on the Basis of Identification of Target Proteins. Chem. Lett. 2012, 41, 658–666. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins A, B, C, and D, Anti-angiogenic Steroidal Alkaloids, from the Marine Sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef]
- Watanabe, Y.; Aoki, S.; Tanabe, D.; Setiawan, A.; Arai, M.; Kobayashi, M. Cortistatins E, F, G, and H, four novel steroidal alkaloids from marine sponge Corticium simplex. Tetrahedron 2007, 63, 4074–4079. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Tanabe, D.; Setiawan, A.; Arai, M.; Kobayashi, M. Cortistatins J, K, L, novel abeo-9(10-19)-androstane-type steroidal alkaloids with isoquinoline unit, from marine sponge Corticium simplex. Tetrahedron Lett. 2007, 48, 4485–4488. [Google Scholar] [CrossRef]
- Aoki, S.; Watanabe, Y.; Tanabe, D.; Arai, M.; Suna, H.; Miyamoto, K.; Tsujibo, H.; Tsujikawa, K.; Yamamoto, H.; Kobayashi, M. Structure–activity relationship and biological property of cortistatins, anti-angiogenic spongean steroidal alkaloids. Bioorganic Med. Chem. 2007, 15, 6758–6762. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.R.H.; Simmons, E.M.; Sarpong, R. Synthetic Strategies Directed Towards the Cortistatin Family of Natural Products. Eur. J. Org. Chem. 2010, 2010, 3553–3567. [Google Scholar] [CrossRef]
- Chen, D.Y.-K.; Tseng, C.-C. Chemistry of the cortistatins–a novel class of anti-angiogenic agents. Org. Biomol. Chem. 2010, 8, 2900–2911. [Google Scholar] [CrossRef] [PubMed]
- Nising, C.F.; Bräse, S. Highlights in Steroid Chemistry: Total Synthesis versus Semisynthesis. Angew. Chem. Int. Ed. 2008, 47, 9389–9391. [Google Scholar] [CrossRef]
- Shi, J.; Manolikakes, G.; Yeh, C.-H.; Guerrero, C.A.; Shenvi, R.A.; Shigehisa, H.; Baran, P.S. Scalable Synthesis of Cortistatin A and Related Structures. J. Am. Chem. Soc. 2011, 133, 8014–8027. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Peng, X.-S.; Sun, Y.-P.; Polet, D.; Zou, B.; Lim, C.S.; Chen, D.Y.-K. Total Synthesis and Biological Evaluation of Cortistatins A and J and Analogues Thereof. J. Am. Chem. Soc. 2009, 131, 10587–10597. [Google Scholar] [CrossRef]
- Lee, H.M.; Nieto-Oberhuber, C.; Shair, M.D. Enantioselective Synthesis of (+)-Cortistatin A, a Potent and Selective Inhibitor of Endothelial Cell Proliferation. J. Am. Chem. Soc. 2008, 130, 16864–16866. [Google Scholar] [CrossRef]
- Flyer, A.N.; Si, C.; Myers, A.G. Synthesis of cortistatins A, J, K and L. Nat. Chem. 2010, 2, 886–892. [Google Scholar] [CrossRef]
- Yamashita, S.; Iso, K.; Kitajima, K.; Himuro, M.; Hirama, M. Total Synthesis of Cortistatins A and J. J. Org. Chem. 2011, 76, 2408–2425. [Google Scholar] [CrossRef]
- Nilson, M.G.; Funk, R.L. Total Synthesis of (±)-Cortistatin J from Furan. J. Am. Chem. Soc. 2011, 133, 12451–12453. [Google Scholar] [CrossRef]
- Kotoku, N.; Sumii, Y.; Hayashi, T.; Tamura, S.; Kawachi, T.; Shiomura, S.; Arai, M.; Kobayashi, M. Creation of Readily Accessible and Orally Active Analogue of Cortistatin, A. ACS Med. Chem. Lett. 2012, 3, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Kotoku, N.; Ito, A.; Shibuya, S.; Mizuno, K.; Takeshima, A.; Nogata, M.; Kobayashi, M. Short-step Synthesis and Structure-activity Relationship of Cortistatin A Analogs. Tetrahedron 2017, 73, 1342–1349. [Google Scholar] [CrossRef]
- Hsung, P.R.; Cole, P.K.; Zehnder, R.L.; Wang, J.; Wei, L.; Yang, X.; Coverdale, A.H. The total synthesis of (±)-arisugacin A. Tetrahedron 2003, 59, 311–324. [Google Scholar] [CrossRef]
- Odani, A.; Ishihara, K.; Ohtawa, M.; Tomoda, H.; Omura, S. Total synthesis of pyripyropene A. Tetrahedron 2011, 67, 8195–8203. [Google Scholar] [CrossRef]
- Fotiadou, A.D.; Zografos, A.L. Electrocyclization of Oxatrienes in the Construction of Structurally Complex Pyranopyridones. Org. Lett. 2012, 14, 5664–5667. [Google Scholar] [CrossRef]
- Mihara, H.; Shibuguchi, T.; Kuramochi, A.; Ohshima, T.; Shibasaki, M. Short Synthesis of (+)-Cylindricine C and Formal Total Synthesis of (−)-Lepadiformine. Heterocycles 2007, 72, 421–438. [Google Scholar] [CrossRef]
- Riveira, M.J.; Mischne, M.P. One-Pot Organocatalytic Tandem Aldol/Polycyclization Reactions between 1,3-Dicarbonyl Compounds and α,β,γ,δ-Unsaturated Aldehydes for the Straightforward Assembly of Cyclopenta[b]furan-Type Derivatives: New Insight into the Knoevenagel Reaction. Chem. Eur. J. 2012, 18, 2382–2388. [Google Scholar] [CrossRef]
- Aabid, H.B.; Quiroga, G.N.; La-Venia, A.; Grover, H.K.; Riveira, M.J. Biomimetic Domino Knoevenagel/Cycloisomerization Strategy for the Synthesis of Citridone A and Derivatives. J. Org. Chem. 2021, 86, 12226–12236. [Google Scholar] [CrossRef]
- Jacinto Demuner, A.; Moreira Valente, V.M.; Almeida Barbosa, L.C.; Rathi, A.; Donohoe, T.J.; Thompson, A.L. Synthesis and Phytotoxic Activity of New Pyridones Derived from 4-Hydroxy-6-Methylpyridin-2(1H)-one. Molecules 2009, 14, 4973–4986. [Google Scholar] [CrossRef]
- Kurdyumov, A.V.; Muñoz, R.L.P.; Hsung, R.P. Total Syntheses of Daldiniapyrone, Annularin B, and (±)-Annularin F. Synthesis 2006, 11, 1787–1790. [Google Scholar] [CrossRef]
- Shimizu, T.; Hiranuma, S.; Watanabe, T. Reduction of α-Pyrone Derivatives with Borane-Methyl Sulfide Complex. Heterocycles 1993, 36, 2445–2550. [Google Scholar] [CrossRef]
- Robins, J.M.; Yang, H.; Miranda, K.; Peterson, A.M.; Clercq, D.E.; Balzarini, J. Synthesis and Biological Evaluation of 3,3-Difluoropyridine-2,4(1H,3H)-dione and 3-Deaza-3-fluorouracil Base and Nucleoside Derivatives. J. Med. Chem. 2009, 52, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Baškovč, J.; Dahmann, G.; Golobič, A.; Grošelj, U.; Kočar, D.; Stanovnik, B.; Svete, J. Diversity-Oriented Synthesis of 1-Substituted 4-Aryl-6-oxo-1,6-dihydropyridine-3-carboxamides. ACS Comb. Sci. 2012, 14, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.B.; Mason, M.A.; Barret, M.G.A. Synthesis of 6-Substituted-4-Hydroxy-2-pyridinones via Intramolecular Ketene Trapping of Functionalized Enamine-Dioxinones. Org. Lett. 2011, 13, 5156–5159. [Google Scholar] [CrossRef]
- Barrett, N.T.; Patel, H.B.; Barrett, M.G.A. Synthesis of C-5-substituted resorcylates and resorcinamides via formylation–aromatization of functionalized keto-dioxinones. Tetrahedron 2014, 70, 6894–6901. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wang, E.S.; Johannessen, L.; Kwiatkowski, N.; Sim, T.; Gray, N.S. Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS. Med. Chem. Lett. 2018, 9, 540–545. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | S.I. a | |
|---|---|---|---|
| HUVECs | KB3-1 | ||
| 1 | 0.0018 b | 7.0 b | 3900 b |
| 2 | 0.035 c | 10.5 c | 300 c |
| 3 | 0.0026 c | 8.2 c | 3150 c |
| 7 | 0.09 | 62.8 | 700 |
| 11 | 0.02 | 10 | 500 |
| 13 | 0.01 | 11 | 1100 |
| 15 | 0.003 | 8.6 | 2870 |
| 19 | 0.001 | 6.4 | 6400 |
| 27 | 0.01 | 13.4 | 1340 |
| 28 | 0.56 | 4.8 | 8.6 |
| 19 | Body Weight (g) a | Hb Content | AST | ALT | |||
|---|---|---|---|---|---|---|---|
| bFGF | (mg/kg) | Day 1 | Day 9 | (µg/mg) b | (IU/L) | (IU/L) | |
| 1 | – | – | 26.03 ± 0.30 | 26.33 ± 0.33 | 0.65 ± 0.10 | – | – |
| 2 | + | – | 25.62 ± 0.52 | 25.98 ± 0.48 | 8.12 ± 1.25 | 59.6 ± 3.1 | 43.6 ± 4.7 |
| 3 | + | 0.1 | 25.70 ± 0.58 | 26.12 ± 0.58 | 3.27 ± 0.83 | – | – |
| 4 | + | 0.3 | 25.60 ± 0.64 | 26.14 ± 0.62 | 2.42 ± 0.79 | – | – |
| 5 | + | 1.0 | 26.30 ± 0.41 | 26.64 ± 0.39 | 0.79 ± 0.24 | 57.8 ± 4.5 | 48.4 ± 3.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimoto, Y.; Mizuno, K.; Nakamura, Y.; Arai, M.; Kotoku, N. Synthesis and Evaluation of Antitumor and Anti-Angiogenesis Activity of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A. Mar. Drugs 2025, 23, 179. https://doi.org/10.3390/md23040179
Fujimoto Y, Mizuno K, Nakamura Y, Arai M, Kotoku N. Synthesis and Evaluation of Antitumor and Anti-Angiogenesis Activity of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A. Marine Drugs. 2025; 23(4):179. https://doi.org/10.3390/md23040179
Chicago/Turabian StyleFujimoto, Yuri, Kanako Mizuno, Yuta Nakamura, Masayoshi Arai, and Naoyuki Kotoku. 2025. "Synthesis and Evaluation of Antitumor and Anti-Angiogenesis Activity of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A" Marine Drugs 23, no. 4: 179. https://doi.org/10.3390/md23040179
APA StyleFujimoto, Y., Mizuno, K., Nakamura, Y., Arai, M., & Kotoku, N. (2025). Synthesis and Evaluation of Antitumor and Anti-Angiogenesis Activity of Pyrone- or Pyridone-Embedded Analogs of Cortistatin A. Marine Drugs, 23(4), 179. https://doi.org/10.3390/md23040179