3. Materials and Methods
3.1. General Experiment
All reactions were conducted in flame-dried or oven-dried glassware under an atmosphere of dry nitrogen or argon. Oxygen- and/or moisture-sensitive solids and liquids were transferred appropriately. The concentration of solutions in vacuo was accomplished using a rotary evaporator fitted with a diaphragm vacuum pump. Residual solvents were removed under an oil vacuum pump (0.1–0.2 mm Hg). All reaction solvents were purified before use: Tetrahydrofuran (THF) was distilled from Na/benzophenone. Toluene was distilled over molten sodium metal. Dichloromethane (DCM), 1,2-dichloroethane (DCE), N,N-dimethylformamide (DMF), acetonitrile (MeCN), N,N-diisopropylethylamine (DIPEA), 2,4,6-collidine, and trimethylamine (Et3N) were distilled from CaH2. The reagents were purchased at the highest commercial quality and used without further purification unless otherwise stated. Flash column chromatography was performed using the indicated solvents on silica gel (100–200 mesh, Xinnuo, Yantai, China). Reactions were monitored using thin-layer chromatography (TLC), which was carried out using pre-coated sheets (Xinnuo silica gel coated with fluorescent indicator F254, 0.25 mm). Compounds were visualized with UV light, iodine, and ceric ammonium molybdate stainer phosphomolybdic acid in EtOH. The 1H NMR spectra were recorded on 500 MHz or 600 MHz spectrometers (Bruker Avance, Karlsruhe, Germany). Chemical shifts are reported in parts per million (ppm), relative to either a tetramethylsilane (TMS) internal standard or the signals of the deuterated solvent. The following abbreviations are used to describe the spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, ddd = doublet of doublet of doublets; other combinations are derived from those listed above. Coupling constants (J) are reported in hertz (Hz) for the corresponding solutions, and chemical shifts are reported as parts per million (ppm) relative to residual CHCl3 δH (7.26 ppm). 13C-NMR nuclear magnetic resonance spectra were recorded at 126 MHz or 151 MHz for the corresponding solutions, and chemical shifts are reported as parts per million (ppm) relative to residual CDCl3 δC (77.16 ppm). All high-resolution mass spectra (HRMS) were obtained by Thermo Scientific’s UltiMate 3000 Series liquid system (Germering, Bavaria, Germany) and Thermo Scientific’s Q-Exactive combined quadrupole Orbitrap mass spectrometer (Germering, Bavaria, Germany). Optical rotations were recorded on a Rudolph AutoPol-I polarimeter (Shanghai, China) at 589 nm with a 50 mm cell. Data are reported as follows: specific rotation (c (g/100 mL), solvent).
3.2. Synthesis of Dipeptide 10
To a solution of Boc-N-Me-ᴅ-Phe-OH (1.00 g, 3.6 mmol) and N-Me-Gly-OMe⸱HCl (0.65 g, 4.7 mmol) in anhydrous DMF (5.0 mL), HOAt (0.10 g, 0.72 mmol), DIPEA (3.1 mL, 17.9 mmol), and HATU (2.72 g, 7.2 mmol) were added at room temperature, and the mixture was stirred overnight. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (20.0 mL × 3). The combined organic layers were then washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE(petroleum ether)/EA(Ethyl acetate) = 2:1), yielding dipeptide 10 (1.20 g, 93%) as a colorless oil. = +110.7 (c 0.7, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 7.31–7.11 (m, 5H), 5.33 (dd, J = 8.4, 6.7 Hz, 0.33H), 5.21 (dd, J = 9.1, 6.0 Hz, 0.17H), 5.02 (dd, J = 9.6, 5.1 Hz, 0.33H), 4.85 (dd, J = 9.2, 5.3 Hz, 0.17H), 4.39–4.14 (m, 1H), 4.04–3.90 (m, 0.67H), 3.78 (d, J = 17.2 Hz, 0.33H), 3.72 (s, 1H), 3.71 (s, 1H), 3.68 (s, 0.5H), 3.67 (s, 0.5H), 3.14 (dd, J = 13.9, 6.7 Hz, 0.5H), 3.07 (dd, J = 22.3, 5.7 Hz, 0.5H), 3.04–2.98 (m, 3H), 2.98–2.90 (m, 1H), 2.84 (s, 1H), 2.81 (s, 1H), 2.73 (s, 0.5H), 2.70 (s, 0.5H), 1.31 (s, 3H), 1.25 (s, 1.5H), 1.17 (s, 3H), 1.12 (s, 1.5H); 13C NMR (126 MHz, CDCl3) exists as rotational conformers: δ 170.9, 170.6, 170.5, 170.1, 169.5, 169.5, 169.4, 169.1, 155.2, 155.2, 154.3, 153.9, 138.1, 138.0, 137.6, 137.5, 129.5, 129.5, 129.5, 128.4, 128.4, 128.2, 126.4, 126.3, 80.4, 80.1, 80.1, 80.0, 58.1, 57.9, 55.8, 55.6, 52.3, 52.2, 52.1, 52.1, 50.7, 50.5, 50.0, 49.9, 36.0, 35.8, 35.1, 35.1, 29.6, 28.9, 28.9, 28.2, 28.1, 27.9, 27.7; HRMS (ESI-TOF) m/z: C19H28N2NaO5+ [M + Na]+: calcd: 387.1890; found: 387.1896.
3.3. Synthesis of Tripeptide 12
Dipeptide 10 (2.97 g, 8.2 mmol) was dissolved in a mixture of THF-MeOH-H2O (10.0 mL/10.0 mL/5.0 mL), and LiOH (1 M in H2O, 16.5 mL, 16.5 mmol) was added at room temperature. The mixture was stirred for 2 h and then acidified to pH = 5 with 1 M HCl (aq.). The aqueous phase was extracted with ethyl acetate (30.0 mL × 3), and the combined organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated to yield the crude acid, which was not further purified. To a solution of the residue acid and amine 11 (3.62 g, 7.5 mmol) in anhydrous DMF (25.0 mL), HOAt (1.03 g, 7.5 mmol), DIPEA (9.2 mL, 52.7 mmol), and HATU (35.7 g, 15.1 mmol) were added at room temperature, and the mixture was stirred overnight. The reaction mixture was quenched with saturated NH4Cl (aq.) and extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 2:1), yielding tripeptide 12 (3.60 g, 68%) as a colorless oil. = +35.8 (c 1.5, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 7.85–7.74 (m, 2H), 7.66–7.55 (m, 2H), 7.46–7.38 (m, 2H), 7.38–7.31 (m, 2H), 7.30–7.14 (m, 5H), 6.82 (d, J = 8.1 Hz, 0.15H), 6.63 (d, J = 8.6 Hz, 0.3H), 6.56 (d, J = 8.6 Hz, 0.45H), 6.21 (d, J = 8.6 Hz, 0.1H), 5.37 (t, J = 7.6 Hz, 0.45H), 5.15 (t, J = 7.7 Hz, 0.15H), 5.06 (dd, J = 9.2, 5.4 Hz, 0.3H), 5.00 (dd, J = 10.4, 4.2 Hz, 0.1H), 4.64–4.51 (m, 3H), 4.28–4.17 (m, 1.5H), 4.09–3.95 (m, 0.8H), 3.95–3.80 (m, 0.3H), 3.75 (d, J = 15.3 Hz, 0.4H), 3.17 (dd, J = 14.0, 7.1 Hz, 0.45H), 3.12–3.06 (m, 0.45H), 3.07 (s, 1H), 3.06–3.02 (m, 1.55H), 3.02–2.93 (m, 1.55H), 2.89–2.81 (m, 3H), 1.88–1.75 (m, 1H), 1.42–1.32 (m, 1H), 1.35 (s, 3H), 1.31–1.26 (m, 1H), 1.25–1.13 (m, 1H), 1.20 (s, 3H), 1.11 (s, 1H), 1.03–0.91 (m, 1H), 0.88–0.75 (m, 6H); 13C NMR (126 MHz, CDCl3) exists as rotational conformers: δ 171.6, 171.5, 171.5, 171.2, 168.5, 168.3, 155.4, 154.3, 143.6, 143.4, 143.4, 141.4, 141.4, 137.9, 137.4, 129.5, 129.4, 128.5, 128.3, 128.3, 127.9, 127.2, 126.5, 126.5, 124.9, 124.8, 120.1, 80.3, 80.1, 66.7, 66.6, 57.8, 56.5, 56.5, 55.6, 53.5, 52.7, 52.5, 46.8, 37.6, 37.5, 36.4, 36.1, 35.3, 29.7, 29.1, 28.2, 28.1, 28.0, 27.7, 24.9, 24.9, 24.8, 15.3, 15.3, 11.6; HRMS (ESI-TOF) m/z: C38H48N3O6+ [M + H]+: calcd: 642.3538; found: 642.3542.
3.4. Synthesis of Tetrapeptide 14
To a solution of tripeptide 12 (3.58 g, 5.6 mmol) in anhydrous CH2Cl2 (25.0 mL), TFA (5.0 mL) was added at room temperature. The mixture was stirred for 0.5 h and then concentrated under reduced pressure to yield the crude amine, which was not further purified. To a solution of the obtained amine and acid 13 in anhydrous DMF (25.0 mL), HOAt (0.68 g, 5.0 mmol), DIPEA (6.1 mL, 34.9 mmol), and HATU (3.79 g, 10.0 mmol) were added, and the mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (50.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.), water, and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 3:2), yielding tetrapeptide 14 (2.40 g, 61%) as a colorless oil. = +7.7 (c 1.0, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 7.85–7.75 (m, 2H), 7.65–7.58 (m, 2H), 7.45–7.38 (m, 2H), 7.33 (q, J = 7.8 Hz, 2H), 7.30–7.12 (m, 5H), 6.83 (d, J = 8.3 Hz, 0.3H), 6.64 (d, J = 8.7 Hz, 0.7H), 5.78 (dd, J = 9.2, 6.7 Hz, 0.7H), 5.62 (dd, J = 9.8, 6.2 Hz, 0.3H), 5.38 (d, J = 8.2 Hz, 0.7H), 5.30 (d, J = 7.8 Hz, 0.3H), 4.67–4.50 (m, 3H), 4.49–4.36 (m, 1H), 4.22 (t, J = 6.1 Hz, 1H), 4.17 (d, J = 17.0 Hz, 0.3H), 4.13 (d, J = 15.5 Hz, 0.7H), 3.87 (d, J = 15.5 Hz, 0.7H), 3.72 (d, J = 17.0 Hz, 0.3H), 3.23–3.14 (m, 1H), 3.14–3.06 (m, 1H), 3.04 (s, 3H), 3.00 (s, 3H), 1.90–1.77 (m, 1H), 1.44–1.34 (m, 9H), 1.22–1.13 (m, 1H), 1.04–0.95 (m, 1H), 0.88 (d, J = 6.8 Hz, 3H), 0.85–0.75 (m, 6H); 13C NMR (126 MHz, CDCl3) exists as rotational conformers: δ 173.9, 173.3, 171.7, 171.5, 171.1, 170.2, 168.2, 155.3, 143.5, 143.3, 141.4, 141.3, 136.4, 129.3, 129.2, 128.4, 127.9, 127.9, 127.2, 127.2, 126.8, 124.9, 124.8, 120.1, 79.6, 79.5, 66.7, 66.7, 56.7, 56.4, 53.9, 53.4, 52.5, 52.4, 46.8, 46.8, 46.6, 37.6, 37.6, 36.6, 35.5, 35.4, 35.2, 30.4, 29.7, 28.3, 28.3, 25.0, 24.8, 17.7, 17.5, 15.3, 15.3, 11.7, 11.6; HRMS (ESI-TOF) m/z: C41H52N4NaO7+ [M + Na]+: calcd: 735.3728; found: 735.3726.
3.5. Synthesis of Pentapeptide 2
To a solution of tripeptide 14 (2.45 g, 3.4 mmol) in anhydrous CH2Cl2 (15.0 mL), TFA (5.0 mL) was added at room temperature. The mixture was stirred for 30 min and then concentrated under reduced pressure to yield the crude amine, which was not further purified. To a solution of the obtained amine and compound 16 in anhydrous DMF (15.0 mL), HOAt (0.46 g, 3.4 mmol), DIPEA (4.1 mL, 23.7 mmol), and HATU (2.57 g, 6.8 mmol) were added, and the mixture was stirred at room temperature overnight. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (50.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.), water, and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 1:5), yielding tetrapeptide 2 (1.58 g, 63%) as a colorless oil. = +12.9 (c 0.9, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 7.86–7.72 (m, 2H), 7.65–7.54 (m, 2H), 7.47–7.38 (m, 2H), 7.37–7.30 (m, 2H), 7.30–7.05 (m, 6H), 6.87 (d, J = 8.5 Hz, 0.3H), 6.63 (d, J = 8.7 Hz, 0.7H), 5.74 (t, J = 7.9 Hz, 0.7H), 5.61 (t, J = 8.0 Hz, 0.3H), 4.82 (q, J = 7.0 Hz, 0.7H), 4.72 (t, J = 7.0 Hz, 0.3H), 4.65–4.48 (m, 3H), 4.22 (t, J = 6.2 Hz, 1H), 4.20–4.09 (m, 1H), 4.09–4.02 (m, 1H), 3.88–3.76 (m, 1H), 3.23–3.13 (m, 1.3H), 3.12–3.02 (m, 4H), 3.01–2.92 (m, 3.7H), 1.91–1.74 (m, 2H), 1.53–1.42 (m, 1H), 1.35–1.24 (m, 1H), 1.24–1.13 (m, 1H), 1.09–0.96 (m, 1H), 0.96–0.88 (m, 6H), 0.88–0.71 (m, 9H); 13C NMR (126 MHz, CDCl3) δ exists as rotational conformers: 173.2, 173.1, 172.8, 171.8, 171.7, 170.9, 170.2, 168.1, 168.0, 143.5, 143.3, 141.4, 141.3, 136.3, 129.3, 129.1, 128.5, 127.9, 127.9, 127.2, 127.2, 126.9, 124.9, 124.8, 120.1, 73.9, 73.9, 66.8, 56.7, 56.4, 54.0, 53.4, 52.5, 52.3, 46.7, 45.5, 45.2, 38.6, 37.8, 37.6, 36.5, 35.4, 35.1, 30.6, 30.5, 29.7, 26.2, 25.0, 24.9, 17.6, 17.4, 15.3, 12.7, 12.6, 11.9, 11.7, 11.6; HRMS (ESI-TOF) m/z: C42H54N4NaO7+ ccc+: calcd: 749.3885; found: 749.3895.
3.6. Synthesis of Ketone 18
To a stirred solution of compound 17 (2.32 g, 4.4 mmol) and N,O-dimethylhydroxylamine hydrochloride (3.03 g, 31.1 mmol) in anhydrous THF (15.0 mL), i-PrMgCl (26.6 mL, 53.3 mmol) was added dropwise at −30 °C. After stirring for 1 h, the reaction mixture was warmed to −20 °C. After stirring for 1 h, the reaction mixture was warmed to 0 °C. After stirring for an additional hour, the reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (50.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 4:1), yielding Weinreb amide S1 (0.95 g, 78%) as a colorless oil. = −8.0 (c 1.0, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.43–7.25 (m, 5H), 4.54 (s, 2H), 3.75 (ddd, J = 8.7, 6.8, 3.1 Hz, 1H), 3.65 (s, 3H), 3.29–3.17 (m, 1H), 3.22 (s, 3H), 1.69–1.58 (m, 1H), 1.58–1.40 (m, 3H), 1.10 (d, J = 6.9 Hz, 3H), 0.95 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 176.4, 139.0, 128.2, 127.8, 127.3, 81.0, 72.8, 61.4, 39.1, 33.5, 32.0, 18.0, 14.4, 13.5; HRMS (ESI-TOF) m/z: C16H25NNaO3+ [M + Na]+: calcd: 302.1727; found: 302.1737.
To a stirred solution of compound S1 (1.50 g, 5.4 mmol) in anhydrous THF (20.0 mL), allylmagnesium bromide (13.6 mL, 13.6 mmol, 1.0 M in THF) was added at 0 °C, and the mixture was stirred for 0.5 h at room temperature. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 30:1), yielding ketone 18 (1.23 g, 86%) as a colorless oil. = −57.8 (c 0.9, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 5.95 (ddt, J = 17.1, 10.2, 6.9 Hz, 1H), 5.19 (dd, J = 10.2, 1.5 Hz, 1H), 5.10 (dd, J = 17.2, 1.6 Hz, 1H), 4.52 (d, J = 11.1 Hz, 1H), 4.43 (d, J = 11.0 Hz, 1H), 3.71 (ddd, J = 8.2, 6.0, 3.6 Hz, 1H), 3.28 (dd, J = 6.9, 1.7 Hz, 2H), 2.93 (dq, J = 8.3, 7.0 Hz, 1H), 1.58–1.39 (m, 4H), 1.05 (d, J = 7.0 Hz, 3H), 0.95 (t, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 211.8, 138.4, 130.7, 128.3, 127.9, 127.6, 118.6, 81.0, 72.3, 49.0, 48.3, 33.1, 17.7, 14.3, 12.6; HRMS (ESI-TOF) m/z: C17H24NaO2+ [M + Na]+: calcd: 283.1669; found: 283.1677.
3.7. Synthesis of Compound 20
To a solution of ketone 18 (0.40 g, 1.5 mmol) in anhydrous THF (20.0 mL), (R)-Me-CBS (2.3 mL, 2.3 mmol, 1.0 M in THF) was added under an argon atmosphere, and the mixture was stirred for 10 min at room temperature. After cooling the reaction mixture to −78 °C, BH3⸱Me2S (0.20 mL, 2.0 mmol, 10.0 M in THF) was added, and the mixture was stirred for 3 h. The reaction mixture was then warmed to −20 °C and stirred for 0.5 h. The reaction mixture was quenched with MeOH (0.10 mL) and saturated NH4Cl (aq.) (5.0 mL) at 0 °C, and the aqueous phase was extracted with ethyl acetate (20.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 30:1), yielding alcohol S2 (0.40 g, 89%) as a colorless oil. = +4.1 (c 1.0, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.42–7.28 (m, 5H), 6.00–5.86 (m, 1H), 5.25–5.12 (m, 2H), 4.62–4.46 (m, 2H), 3.68–3.57 (m, 2H), 2.49–2.38 (m, 1H), 2.22–2.08 (m, 1H), 2.00–1.89 (m, 1H), 1.68–1.57 (m, 1H), 1.57–1.49 (m, 2H), 1.48–1.40 (m, 1H), 0.95 (t, J = 7.2 Hz, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 138.5, 135.2, 128.4, 127.9, 127.6, 117.8, 82.2, 73.17, 7.35, 4.55, 39.2, 32.3 18.0, 14.4, 11.9; HRMS (ESI-TOF) m/z: C17H26NaO2+ [M + Na]+: calcd: 285.1825; found: 285.1832.
To a solution of alcohol S2 (2.22 g, 8.5 mmol) in anhydrous CH2Cl2 (20.0 mL), Et3N (4.7 mL, 33.8 mmol) and TBSOTf (3.9 mL, 16.9 mmol) were added at room temperature under an argon atmosphere, and the mixture was stirred for 1 h. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were then washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 30:1), yielding compound 20 (2.80 g, 89%) as a colorless oil. = +4.5 (c 0.9, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.45–7.24 (m, 5H), 5.95–5.83 (m, 1H), 5.11–5.01 (m, 2H), 4.56–4.44 (m, 2H), 3.82 (td, J = 6.1, 4.5 Hz, 1H), 3.53 (ddd, J = 8.6, 5.7, 3.2 Hz, 1H), 2.39–2.26 (m, 1H), 2.26–2.18 (m, 1H), 2.11–1.99 (m, 1H), 1.63–1.54 (m, 1H), 1.54–1.42 (m, 2H), 1.42–1.28 (m, 1H), 0.94 (t, J = 6.9 Hz, 3H), 0.92 (s, 9H), 0.86 (d, J = 7.0 Hz, 3H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 139.2, 135.4, 128.3, 127.8, 127.3, 116.7, 79.8, 72.8, 71.0, 40.2, 38.1, 32.3, 25.9, 18.9, 18.1, 14.5, 10.1, −4.0, −4.7; HRMS (ESI-TOF) m/z: C23H40NaO2Si+ [M + Na]+: calcd: 399.2690; found: 399.2699.
3.8. Synthesis of Alcohol 4
To a solution of compound 20 (1.62 g, 4.3 mmol) in wet DCE (25.0 mL, analytically pure), 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (3.89 g, 17.2 mmol) was added at 50 °C, and the mixture was stirred for 1 h. The reaction mixture was cooled to room temperature and quenched with saturated Na2S2O3 (aq.) (20.0 mL), after which the aqueous phase was extracted with DCM (50.0 mL × 3). The combined organic layers were washed with 1 M NaOH (aq.)-saturated Na2S2O3 (aq.) (VNaOH: VNa2S2O3 = 1:1) and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 20:1), yielding alcohol 4 (0.85 g, 69%) as a colorless oil. = +3.3 (c 1.2, MeOH); 1H NMR (500 MHz, CDCl3) δ 5.94–5.82 (m, 1H), 5.18–4.99 (m, 2H), 3.80 (q, J = 5.4 Hz, 1H), 3.61–3.54 (m, 1H), 2.74 (s, 1H), 2.41–2.33 (m, 1H), 2.32–2.23 (m, 1H), 1.76–1.68 (m, 1H), 1.58–1.49 (m, 2H), 1.43–1.33 (m, 2H), 0.95 (t, J = 6.6 Hz, 3H), 0.93 (s, 9H), 0.84 (d, J = 7.0 Hz, 3H), 0.11 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 135.0, 117.1, 75.9, 73.8, 43.1, 39.3, 36.5, 25.9, 18.6, 18.0, 14.2, 12.8, −4.2, −4.7; HRMS (ESI-TOF) m/z: C16H34NaO2Si+ [M + Na]+: calcd: 309.2220; found: 309.2226.
3.9. Synthesis of Acetonide 21
To a solution of compound 4 (53.0 mg, 0.18 mmol) in anhydrous THF (3.0 mL), TBAF (0.4 mL, 0.4 mmol) was added at room temperature under an argon atmosphere, and the mixture was stirred for 30 min. The reaction mixture was concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 1:1), yielding diol S4 as a colorless oil.
To a solution of the diol S4 obtained above in anhydrous CH2Cl2 (1.0 mL), 2,2-dimethoxypropane (3.0 mL, 25.1 mmol) and PTSA (2.0 mg, 11.6 mmol) were added at room temperature under an argon atmosphere, and the mixture was stirred for 5 min. The reaction mixture was quenched with saturated Et3N (0.5 mL, 3.6 mmol), and the solvent was concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 50:1), yielding acetonide 21 (36.7 mg, 93%) as a colorless oil. = +14.7 (c 0.8, MeOH); 1H NMR (500 MHz, CDCl3) δ 5.93 (ddt, J = 17.2, 10.3, 6.8 Hz, 1H), 5.11–5.02 (m, 2H), 3.54 (ddd, J = 10.2, 7.0, 3.1 Hz, 1H), 3.46 (ddd, J = 10.3, 8.0, 2.4 Hz, 1H), 2.39 (dddd, J = 12.8, 6.5, 3.2, 1.6 Hz, 1H), 2.20 (dt, J = 14.6, 7.1 Hz, 1H), 1.60–1.48 (m, 2H), 1.43 (s, 3H), 1.39 (s, 3H), 1.37–1.29 (m, 3H), 0.91 (t, J = 7.0 Hz, 3H), 0.79 (d, J = 6.6 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 135.2, 116.2, 97.8, 74.1, 73.7, 37.7, 37.5, 35.2, 30.2, 19.6, 18.2, 14.0, 12.2.
3.10. Synthesis of Ester 22
To a solution of Fmoc-N-Me-ʟ-Ala-OH (3.17 g, 9.8 mmol) in anhydrous CH2Cl2 (15.0 mL) at 0 °C, oxalyl chloride (2.8 mL, 32.5 mmol) and catalytic DMF (32 μL, 0.41 mmol) were added, and the mixture was warmed to room temperature. The reaction mixture was stirred for 1 h and then concentrated under reduced pressure to yield the crude acyl chloride, which was used directly in the subsequent step. To a solution of alcohol 4 (932 mg, 3.3 mmol) and DMAP (0.99 g, 8.1 mmol) in anhydrous CH2Cl2 (15.0 mL), Et3N (4.5 mL, 32.5 mmol) was added at room temperature under an argon atmosphere. The reaction mixture was cooled to 0 °C, and a solution of the crude acyl chloride obtained above in anhydrous CH2Cl2 (15.0 mL) was added. The mixture was then warmed to room temperature and stirred for 1 h. The reaction was quenched with methanol (1.0 mL) and saturated NH4Cl (aq.) (5.0 mL), and the aqueous phase was extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were washed with 1 M HCl (aq.), saturated NH4Cl (aq.), and brine and then dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 20:1), yielding ester 22 (1.26 g, 66%) as a colorless oil. = −6.0 (c 0.7, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 7.85–7.77 (m, 2H), 7.70–7.57 (m, 2H), 7.46–7.37 (m, 2H), 7.37–7.30 (m, 2H), 5.91–5.78 (m, 1H), 5.20–4.97 (m, 3H), 4.97–4.91 (m, 0.7H), 4.85–4.79 (m, 0.3H), 4.49–4.35 (m, 2H), 4.33–4.23 (m, 1H), 3.72–3.63 (m, 1H), 2.97–2.91 (m, 3H), 2.34–2.24 (m, 1H), 2.22–2.13 (m, 1H), 1.99–1.90 (m, 1H), 1.64–1.48 (m, 2H), 1.45 (d, J = 7.3 Hz, 3H), 1.40–1.20 (m, 2H), 1.00–0.82 (m, 15H), 0.12–0.01 (m, 6H); 13C NMR (126 MHz, CDCl3) exists as rotational conformers: δ 171.5, 171.3, 156.5, 155.9, 144.1, 144.9, 141.3, 135.4, 135.0, 127.7, 127.1, 125.1, 125.1, 120.0, 119.8, 116.9, 76.1, 76.0, 72.9, 67.9, 67.8, 54.2, 47.3, 41.6, 41.5, 37.9, 37.7, 32.8, 32.5, 30.4, 30.2, 25.9, 18.8, 18.7, 18.1, 15.4, 14.9, 14.1, 10.4, −4.1, −4.7; HRMS (ESI-TOF) m/z: C35H51NNaO5Si+ [M + Na]+: calcd: 616.3429; found: 616.3436.
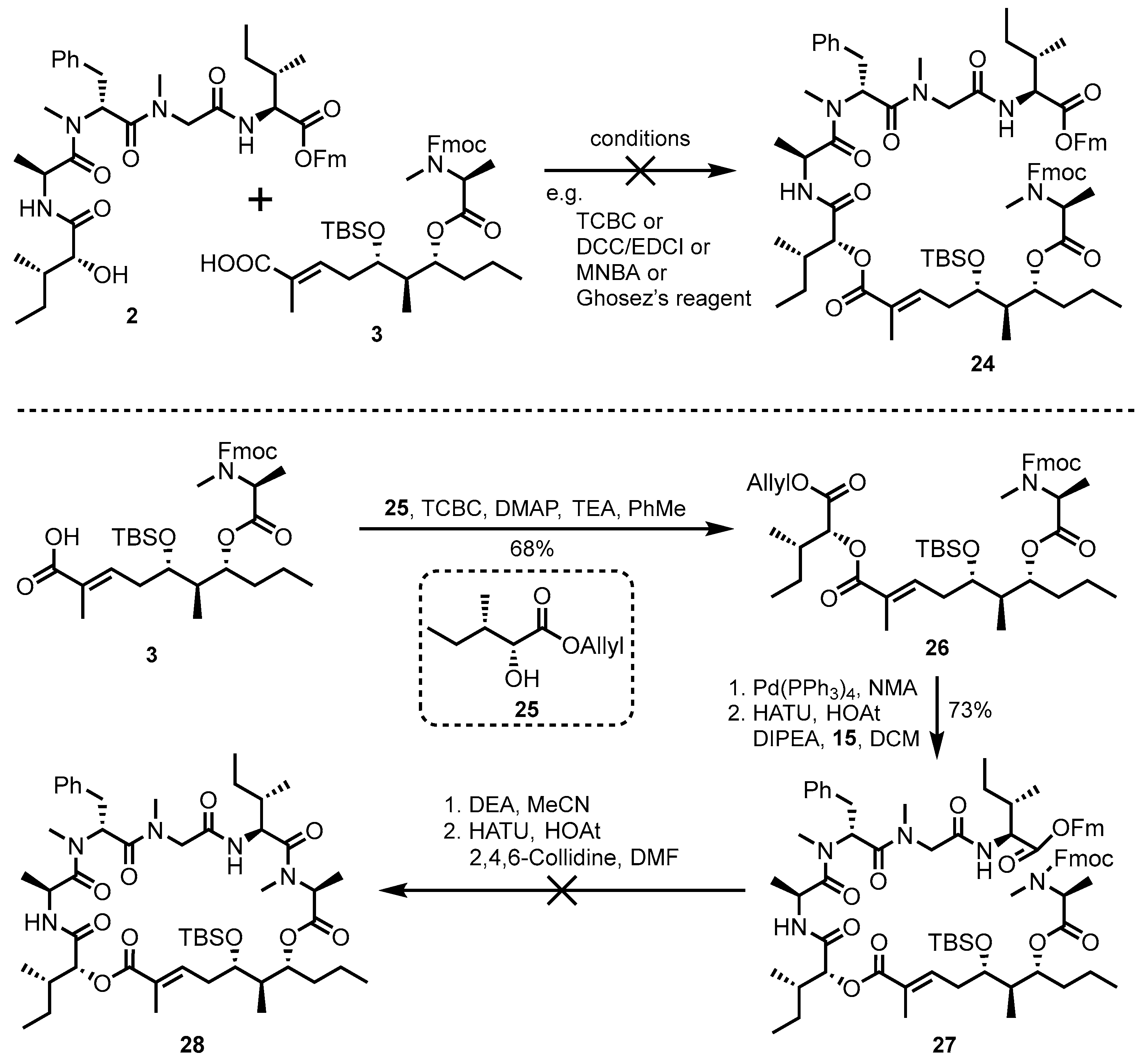
3.11. Synthesis of Ester 26
In a sealed tube, α-methylacrolein (11.1 mL, 3.5 mmol) and Grubbs II catalyst (8.9 mg, 10.0 μmol) were added to a solution of ester 22 (1.22 g, 2.1 mmol) in anhydrous CH2Cl2 (10.0 mL) at room temperature. After purging with nitrogen gas for 30 min, the reaction mixture was warmed to 50 °C and stirred overnight. The mixture was then concentrated under reduced pressure. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 10:1), yielding aldehyde S3 (1.06 g, 81%) as a colorless oil. = −21.3 (c 0.8, MeOH); 1H NMR (500 MHz, CDCl3) exists as rotational conformers: δ 9.38 (s, 0.73H), 9.35 (s, 0.27H), 7.87–7.74 (m, 2H), 7.68–7.55 (m, 2H), 7.52–7.41 (m, 2H), 7.41–7.30 (m, 2H), 6.67–6.48 (m, 1H), 5.14–5.00 (m, 1H), 4.94–4.81 (m, 1H), 4.52–4.33 (m, 2H), 4.33–4.20 (m, 1H), 3.95–3.79 (m, 1H), 2.96 (s, 2.2H), 2.92 (s, 0.8H), 2.59–2.42 (m, 2H), 2.02–1.93 (m, 1H), 1.74 (s, 3H), 1.65–1.51 (m, 2H), 1.49–1.42 (m, 3H), 1.41–1.22 (m, 2H), 0.99–0.84 (m, 15H), 0.13–−0.02 (m, 6H); 13C NMR (126 MHz, CDCl3) δ exists as rotational conformers: 195.3, 195.0, 171.9, 156.5, 152.0, 151.0, 144.0, 143.9, 141.3, 140.3, 127.7, 127.1, 127.1, 125.0, 120.0, 120.0, 76.0, 75.8, 71.6, 71.4, 68.0, 67.8, 54.2, 47.2, 42.6, 42.3, 33.4, 32.2, 30.2, 25.8, 18.6, 18.5, 18.0, 15.3, 14.9, 14.2, 14.1, 10.3, 10.2, 9.4. −4.4, −4.7; HRMS (ESI-TOF) m/z: C37H53NNaO6Si+ [M + Na]+: calcd: 658.3534; found: 658.3541.
To a stirred solution of aldehyde S3 (1.05 g, 1.7 mmol) and 2-methyl-2-butene (16 mL) in t-BuOH/pH = 7 phosphate buffer (2:1, 24.0 mL), NaClO2 (4.33 g, 49.5 mmol) and NaH2PO4 (9.53 g, 61.1 mmol) were added at room temperature, and the mixture was stirred for 1 h. The reaction mixture was acidified to pH = 3 with 1 M HCl (aq.), and the aqueous phase was then extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were washed with saturated NH4Cl (aq.) and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 1:1), yielding acid 3 (1.05 g, 98%) as a colorless oil.
To a solution of acid 3 (353 mg, 0.58 mmol), alcohol 25 (300 mg, 1.70 mmol), and DMAP (0.35 g, 2.90 mmol) in anhydrous PhMe (10.0 mL) at −20 °C, 2,4,6-trichlorobenzoyl chloride (0.30 mL, 1.70 mmol) was added, and the mixture was stirred for 0.5 h. The reaction mixture was then warmed to room temperature and stirred overnight. The mixture was acidified to pH = 3 with 1 M HCl (aq.), and the organic phase was extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were washed with 1 M HCl (aq.), saturated NaHCO3 (aq.), and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 1:3), yielding ester 26 (318 mg, 68%) as a colorless oil. = −25.0 (c 0.8, MeOH); 1H NMR (600 MHz, CDCl3) exists as rotational conformers: δ 7.84–7.76 (m, 2H), 7.72–7.56 (m, 2H), 7.48–7.39 (m, 2H), 7.39–7.31 (m, 2H), 7.00–6.88 (m, 1H), 5.91 (ddt, J = 16.5, 10.9, 5.6 Hz, 1H), 5.33 (d, J = 17.4 Hz, 1H), 5.24 (d, J = 10.3 Hz, 1H), 5.08 (d, J = 3.5 Hz, 1H), 5.07–5.03 (m, 1H), 4.95–4.86 (m, 0.66H), 4.83–4.79 (m, 0.34H), 4.75–4.60 (m, 2H), 4.49–4.36 (m, 2H), 4.32–4.23 (m, 1H), 3.86–3.79 (m, 1H), 2.93 (s, 2H), 2.91 (s, 1H), 2.42–2.30 (m, 2H), 2.10–2.00 (m, 1H), 2.00–1.91 (m, 1H), 1.88 (s, 2.5H), 1.85 (s, 0.5H), 1.63–1.51 (m, 3H), 1.51–1.39 (m, 4H), 1.37–1.23 (m, 2H), 0.99 (d, J = 6.9 Hz, 3H), 0.98–0.81 (m, 18H), 0.10–0.00 (m, 6H);13C NMR (151 MHz, CDCl3) exists as rotational conformers: δ 171.6, 171.2, 169.9, 167.4, 156.4, 155.8, 144.1, 143.9, 141.3, 141.3, 140.6, 140.2, 131.8, 127.7, 127.1, 125.1, 125.1, 120.0, 118.5, 76.2, 76.0, 74.8, 71.7, 71.6, 67.9, 67.8, 65.5, 54.3, 54.1, 47.2, 42.5, 42.4, 36.8, 33.4, 33.1, 32.4, 32.3, 30.2, 29.7, 26.1, 25.8, 18.5, 17.9, 15.3, 14.9, 14.5, 14.1, 12.7, 11.7, 10.4. −4.3, −4.7; HRMS (ESI-TOF) m/z: C46H67NNaO9Si+ [M + Na]+: calcd: 828.4477; found: 828.4487.
3.12. Synthesis of Compound 27
To a solution of compound 26 (0.14 g, 0.17 mmol) and Pd(PPh3)4 (19.6 mg, 17 μmol) in anhydrous THF (2.0 mL), PhNHMe (37.7 μL, 0.34 mmol) was added. The resulting mixture was stirred at room temperature for 1 h. The reaction was quenched with TFA (75.6 μL, 1.02 mmol), diluted with ethyl acetate (30.0 mL), and washed sequentially with 1 M HCl (aq.) (3.0 mL × 2). The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude acid, which was used in the next step without further purification. The crude acid was dissolved in anhydrous DMF (5.0 mL); HOAt (22.74 mg, 0.17 mmol), DIPEA (0.29 mL, 1.7 mmol), amine 15 (0.11 g, 0.15 mmol), and HATU (0.12 g, 0.34 mmol) were added at room temperature; and the mixture was stirred overnight. The reaction mixture was quenched with saturated NH4Cl (aq.), and the aqueous phase was extracted with ethyl acetate (20.0 mL × 3). The combined organic layers were then washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 1:1), yielding compound 27 (0.17 g, 73%) as a colorless oil. = −12.9 (c 0.6, MeOH); 1H NMR (600 MHz, Chloroform-d) exists as rotational conformers: δ 7.83–7.74 (m, 4H), 7.66–7.53 (m, 4H), 7.49–7.38 (m, 4H), 7.37–7.30 (m, 4H), 7.27–7.15 (m, 5H), 6.95–6.89 (m, 1H), 6.65 (d, J = 7.4 Hz, 1H), 6.52 (d, J = 8.6 Hz, 1H), 5.84 (dd, J = 9.5, 6.5 Hz, 0.7H), 5.76 (t, J = 8.2 Hz, 0.3H), 5.23–5.18 (m, 1H), 5.07–4.98 (m, 1H), 4.93–4.87 (m, 1H), 4.75–4.68 (m, 1H), 4.59–4.51 (m, 3H), 4.45–4.37 (m, 2H), 4.30–4.20 (m, 2H), 4.06–3.94 (m, 2H), 3.90–3.82 (m, 1H), 3.22–3.12 (m, 2H), 3.12–2.89 (m, 9H), 2.83 (s, 1H), 2.39–2.33 (m, 2H), 2.00–1.93 (m, 2H), 1.90–1.87 (m, 3H), 1.85–1.78 (m, 1H), 1.61–1.52 (m, 2H), 1.48–1.21 (m, 7H), 1.21–0.55 (m, 31H), 0.11–−0.02 (m, 6H); 13C NMR (151 MHz, CDCl3) exists as rotational conformers: 172.4, 171.7, 171.6, 170.8, 170.8, 169.5, 168.1, 166.7, 156.4, 144.1, 143.9, 143.6, 143.4, 141.4, 141.3, 136.5, 129.3, 128.3, 127.9, 127.9, 127.7, 127.2, 127.1, 124.8, 120.1, 76.1, 76.1, 75.9, 71.7, 67.8, 66.7, 56.6, 56.4, 56.3, 54.3, 54.1, 53.8, 53.7, 52.7, 52.2, 47.2, 46.8, 45.0, 42.6, 38.6, 37.8, 37.7, 37.6, 37.5, 36.7, 36.5, 35.2, 35.1, 33.4, 32.5, 31.9, 30.6, 30.4, 30.2, 29.7, 29.7, 29.4, 26.1, 25.8, 24.9, 18.5, 18.0, 17.3, 17.2, 15.3, 14.9, 14.1, 14.1, 13.9, 12.8, 11.7, 11.6, 11.5, 10.4, −4.2, −4.7. HRMS (ESI-TOF) m/z: C79H105N5NaO13Si+ [M + Na]+: calcd: 1382.7370; found: 1382.7390.
3.13. Synthesis of Compound 31
To a solution of compound 14 (28 mg, 36 μmol) in anhydrous CH3CN (2.0 mL), diethylamine (Et2NH) (1.0 mL) was added, and the mixture was stirred for 2 h at room temperature. The reaction mixture was then concentrated under reduced pressure, and the residue was dissolved in ethyl acetate (50.0 mL) and washed with 1 M HCl (aq.) (3.0 mL × 2) and brine. The organic phase was then dried over anhydrous Na2SO4, filtered, and concentrated to afford crude 29, which was used in the next step without further purification. To a solution of compound 26 (16 mg, 20 μmol) in anhydrous CH3CN (2.0 mL), diethylamine (Et2NH) (1.0 mL) was added, and the mixture was stirred for 2 h at room temperature. The mixture was then concentrated under reduced pressure to afford crude 30, which was used in the next step without further purification. The above crude acid 29 and amine 30 were dissolved in anhydrous THF (3.0 mL), and DEPBT (13 mg, 45 μmol) and DIPEA (7.8 μL, 45 μmol) were added successively at room temperature. The mixture was then stirred overnight. The reaction mixture was quenched with saturated NH4Cl (5.0 mL), and the organic phase was extracted with ethyl acetate (30.0 mL × 3). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 2:3), yielding compound 31 (11.5 mg, 53%) as a colorless oil. = −16.4 (c 0.5, MeOH); 1H NMR (600 MHz, CDCl3) exists as rotational conformers: δ 7.39–7.13 (m, 5H), 7.13–7.03 (m, 1H), 7.02–6.91 (m, 1H), 6.81–6.66 (m, 1H), 5.98–5.87 (m, 1H), 5.78–5.68 (m, 1H), 5.67–5.59 (m, 1H), 5.38–5.30 (m, 1H), 5.30–5.22 (m, 1H), 5.13–5.07 (m, 1H), 5.07–4.96 (m, 1H), 4.96–4.82 (m, 1H), 4.71–4.61 (m, 2H), 4.59–4.36 (m, 2H), 4.34–4.26 (m, 1H), 3.94–3.84 (m, 1H), 3.84–3.72 (m, 1H), 3.32–3.11 (m, 2H), 3.11–2.80 (m, 8H), 2.42–2.27 (m, 2H), 2.10–2.01 (m, 1H), 1.97–1.80 (m, 4H), 1.79–1.38 (m, 18H), 1.38–1.06 (m, 15H), 1.04–0.82 (m, 18H), 0.09–−0.02 (m, 6H); 13C NMR (151 MHz, CDCl3) exists as rotational conformers: δ 173.1, 172.0, 171.3, 170.9, 170.7, 169.8, 167.9, 167.5, 155.3, 140.8, 136.5, 131.8, 129.9, 129.4, 128.4, 128.3, 126.8, 118.6, 79.4, 76.1, 74.9, 71.8, 65.5, 54.2, 53.2, 52.2, 51.9, 46.6, 42.8, 37.6, 36.8, 36.4, 35.3, 33.4, 32.4, 31.9, 31.3, 30.4, 29.7, 29.7, 29.4, 28.3, 28.3, 26.1, 25.8, 24.0, 22.7, 18.7, 18.0, 17.7, 15.5, 14.5, 14.5, 14.1, 14.1, 12.6, 11.7, 11.2, 10.4, −4.3, −4.7, −4.7; HRMS (ESI-TOF) m/z: C58H97N5NaO13Si+ [M + Na]+: calcd: 1122.6744; found: 1122.6726.
3.14. Synthesis of Lagunamide D (1)
To a solution of compound 31 (26 mg, 24 μmol) and Pd(PPh3)4 (2.7 mg, 2.4 μmol) in anhydrous THF (2.0 mL), PhNHMe (5.2 μL, 48 μmol) was added. The resulting mixture was stirred at room temperature for 1 h. The reaction was quenched with TFA (10 μL, 144 μmol), diluted with ethyl acetate (30.0 mL), and washed sequentially with 1 M HCl (aq.) (3.0 mL × 2). The organic phase was washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated to afford the crude acid 32, which was used in the next step without further purification. To a solution of the crude acid in anhydrous CH2Cl2 (2.5 mL), TFA (0.50 mL) was added, and the mixture was stirred at room temperature for 2 h. The mixture was concentrated under reduced pressure, the residue was dissolved in DMF (12.0 mL), and HOAt (16 mg, 0.12 mmol), 2,4,6-collidine (94 μL, 0.72 mmol), and HATU (90 mg, 0.24 mmol) were added successively. The mixture was stirred for 48 h and then concentrated under reduced pressure. The residue was dissolved in ethyl acetate and washed with 1 M HCl (aq.), water, and brine. The organic phase was then dried over anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash chromatography on silica gel (PE/EA = 2:3 to pure EA), yielding lagunamide D (13 mg, 67%) as a colorless foam. = −57.1 (c 0.07, MeOH); 1H NMR (600 MHz, DMSO-d6) exists as rotational conformers: δ 8.49 (d, J = 6.4 Hz, 0.3H), 8.34 (d, J = 8.3 Hz, 0.1H), 7.23–7.05 (m, 5.1H), 6.92–6.74 (m, 0.2H), 6.74–6.54 (m, 0.2H), 5.81–5.67 (m, 0.2H), 5.28 (dd, J = 10.2, 5.3 Hz, 0.6H), 5.23–4.99 (m, 0.6H), 4.92 (d, J = 7.6 Hz, 0.6H), 4.90–4.81 (m, 1.0H), 4.81–4.72 (m, 0.7H), 4.72–4.44 (m, 0.9H), 4.31 (dq, J = 6.3, 6.3 Hz, 0.6H), 4.25–4.07 (m, 0.8H), 3.98 (d, J = −18.4 Hz, 0.5H), 3.92 (q, J = 6.6 Hz, 0.6H), 3.87–3.72 (m, 0.3H), 3.72–3.38 (m, 0.7H), 3.21 (s, 2.4H), 3.16–3.06 (m, 0.6H), 3.06–2.88 (m, 1.9H), 2.85 (s, 2.7H), 2.80–2.76 (m, 0.8H), 2.73 (s, 2.2H), 2.64 (s, 1.2H), 2.45–2.18 (m, 1.2H), 2.10 (ddd, J = −14.0, 9.8, 9.8 Hz, 0.9H), 1.95–1.85 (m, 1.2H), 1.85–1.64 (m, 4.9H), 1.62–1.08 (m, 11.0H), 1.06–0.41 (m, 21.0H); 13C NMR (151 MHz, DMSO-d6) δ 172.45, 170.80, 170.14, 170.11, 169.52, 168.23, 168.01, 144.45, 137.17, 129.18, 127.48, 126.83, 125.84, 75.36, 74.62, 69.30, 58.07, 52.45, 51.1, 50.40, 44.65, 40.89, 37.29, 37.10, 36.27, 35.79, 34.42, 33.86, 29.60, 29.36, 25.83, 23.19, 17.05, 14.89, 14.69, 14.06, 13.92, 12.93, 11.98, 11.40, 10.27, 9.60; HRMS (ESI-TOF) m/z: C44H69N5NaO10+ [M + Na]+: calcd: 850.4937; found: 850.4909.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}