
Transition-Metal-Catalyzed Transformations for the Synthesis of Marine Drugs


Abstract
1. Introduction
2. Results
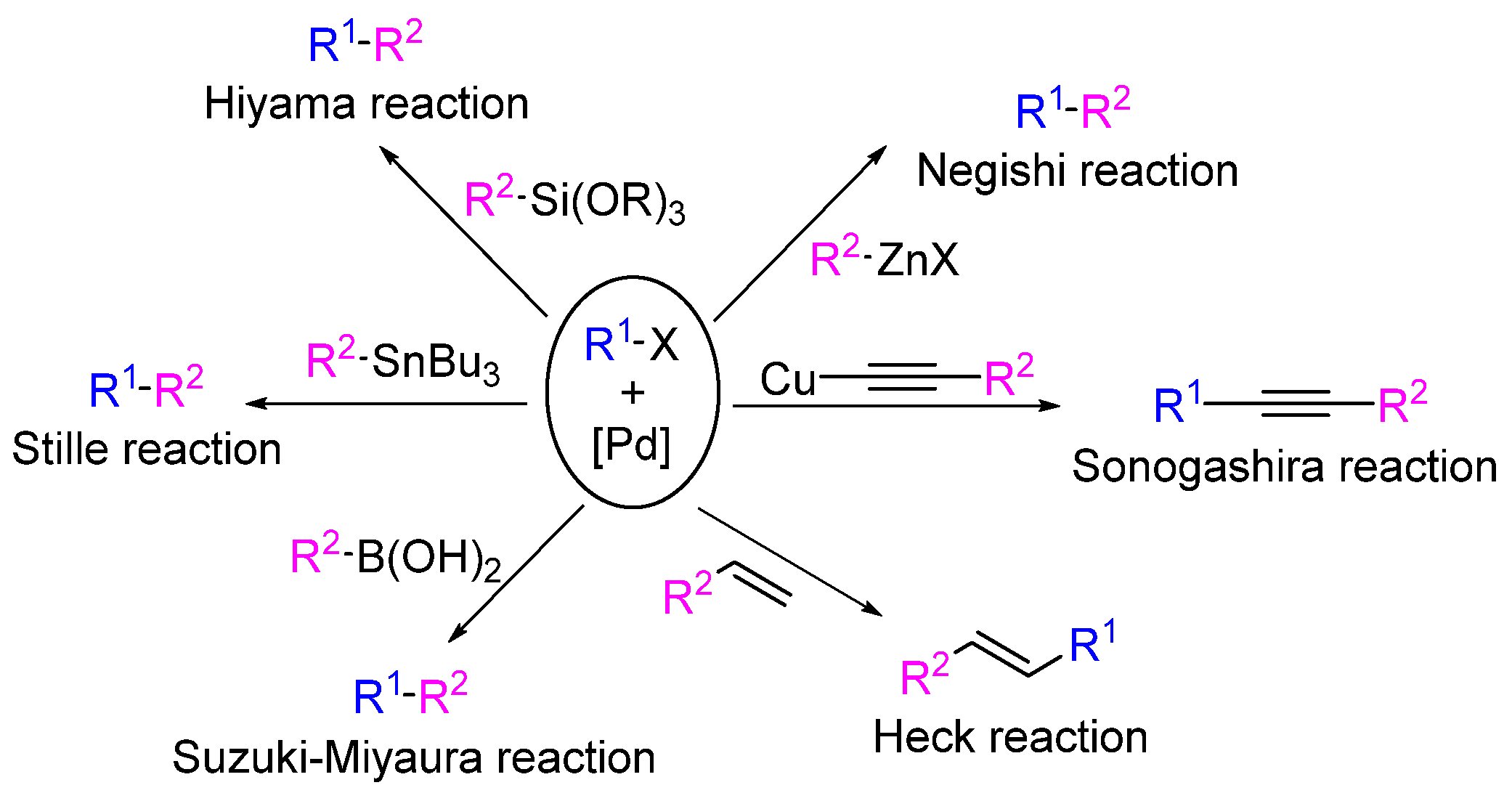
2.1. Cross-Coupling Reactions in the Synthesis of Marine Drugs
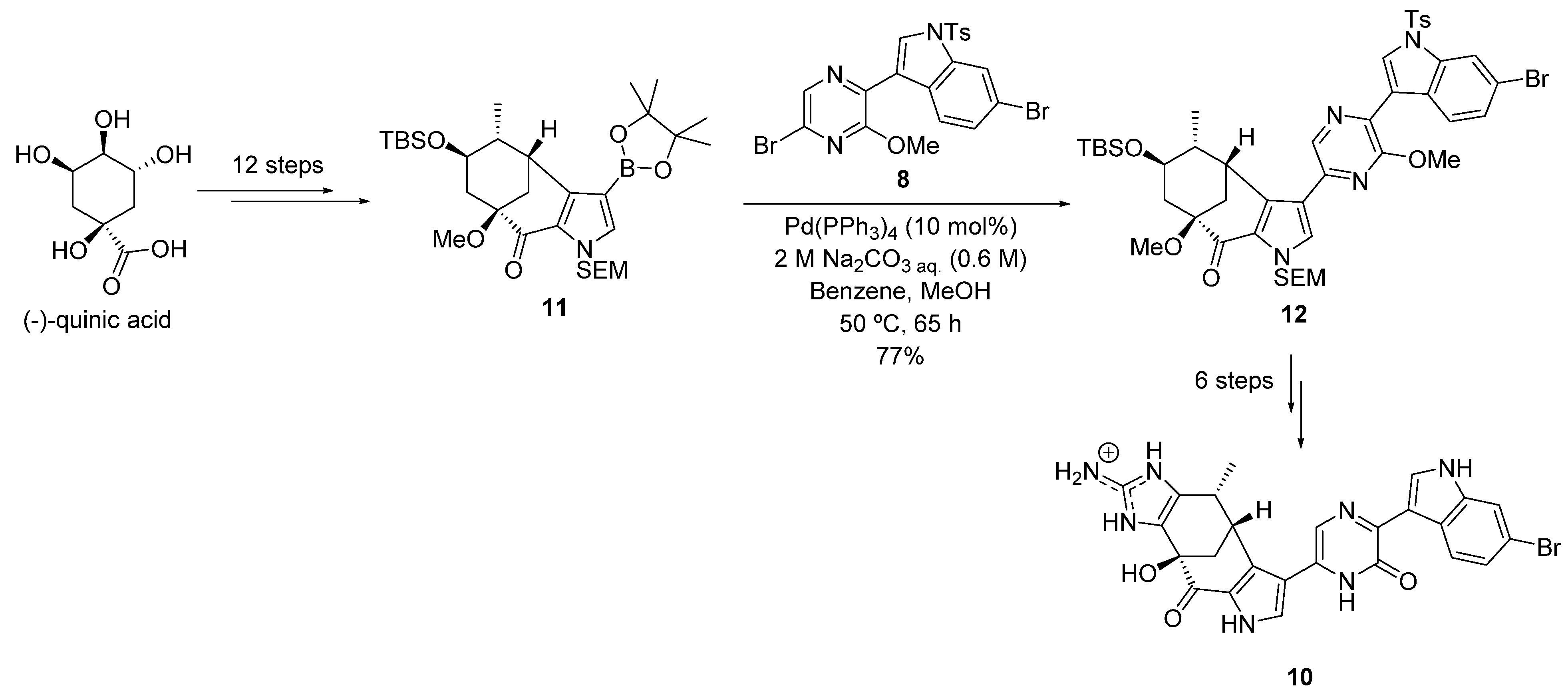
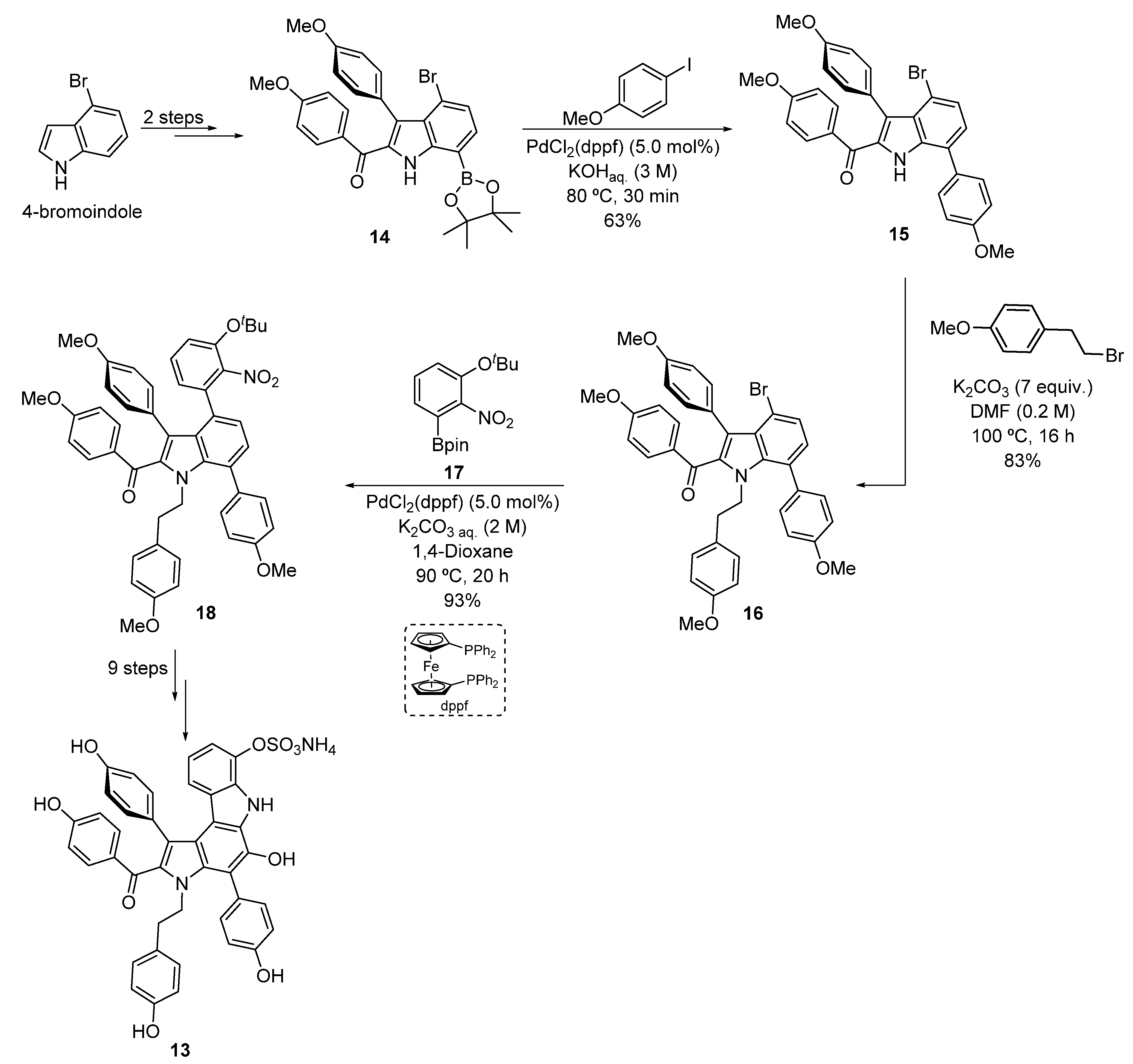
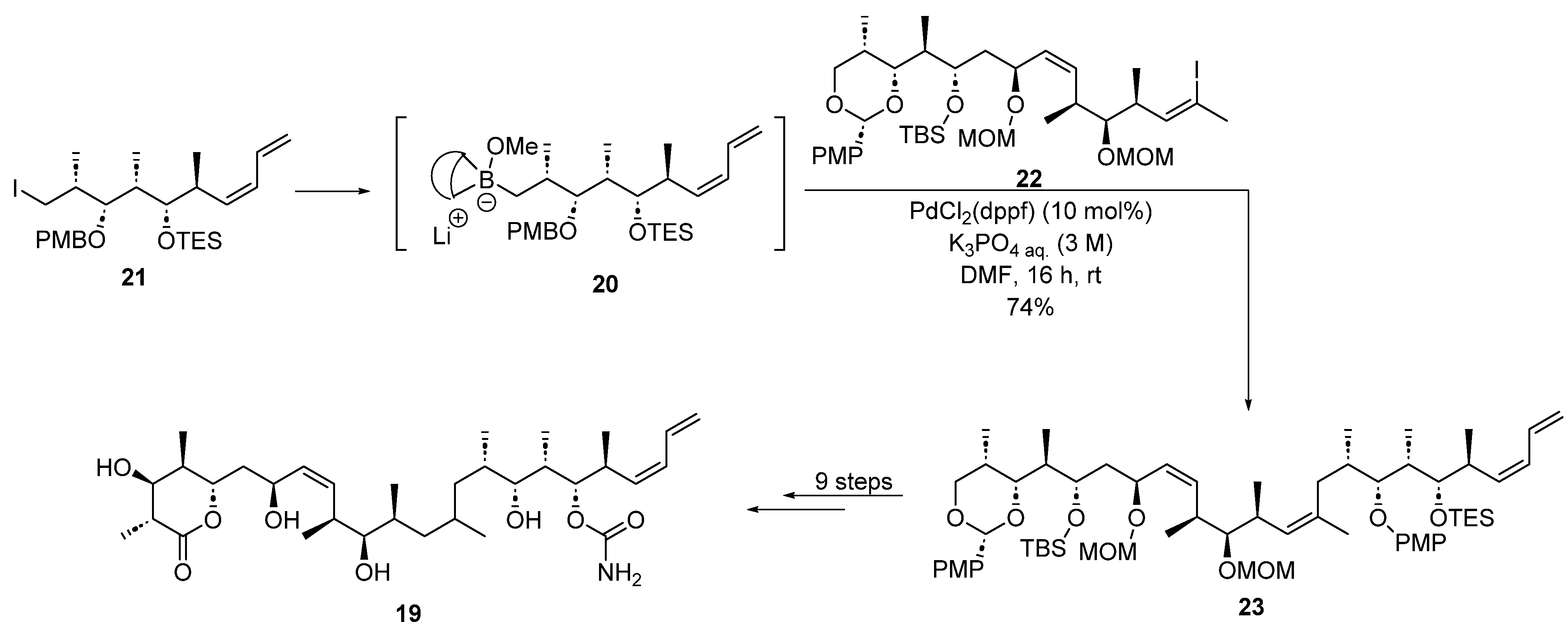
2.1.1. Suzuki–Miyaura Cross-Coupling Reaction
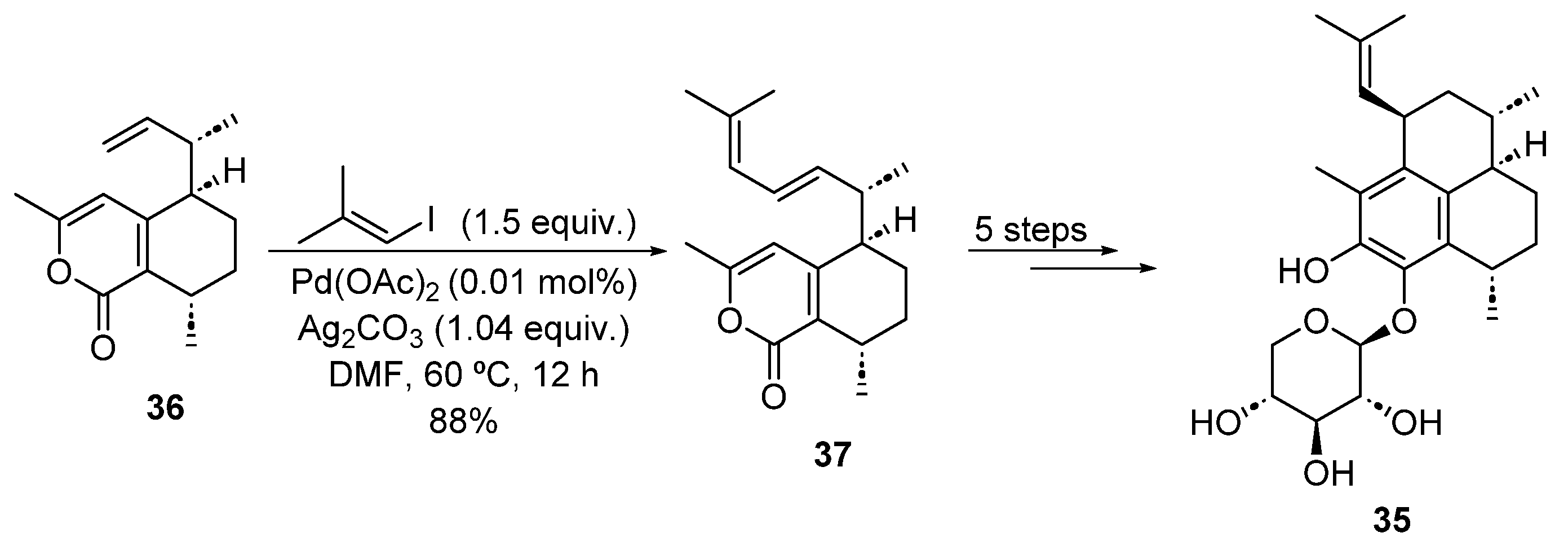
2.1.2. Heck Cross-Coupling Reaction
2.1.3. Negishi Cross-Coupling Reaction
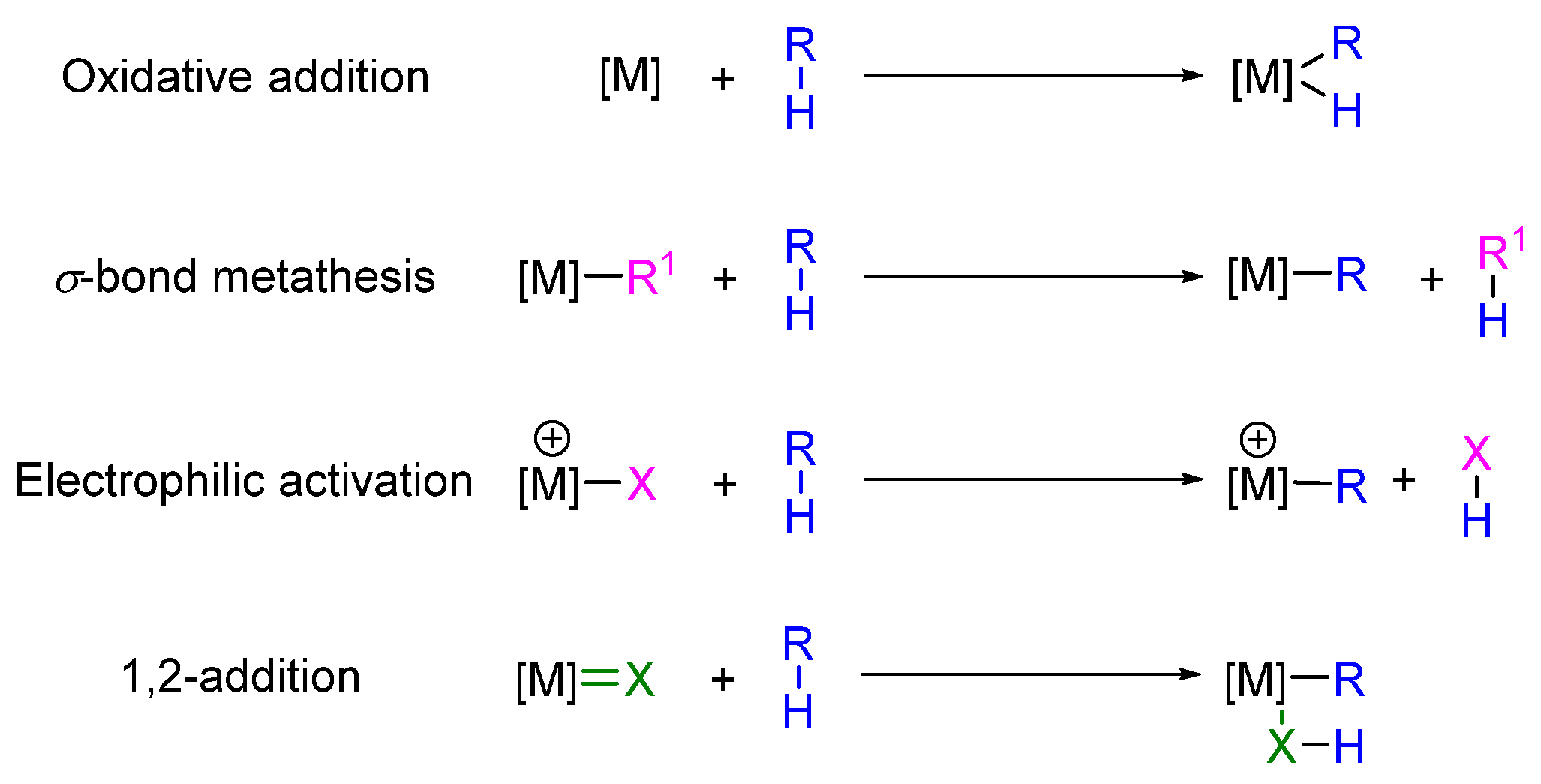
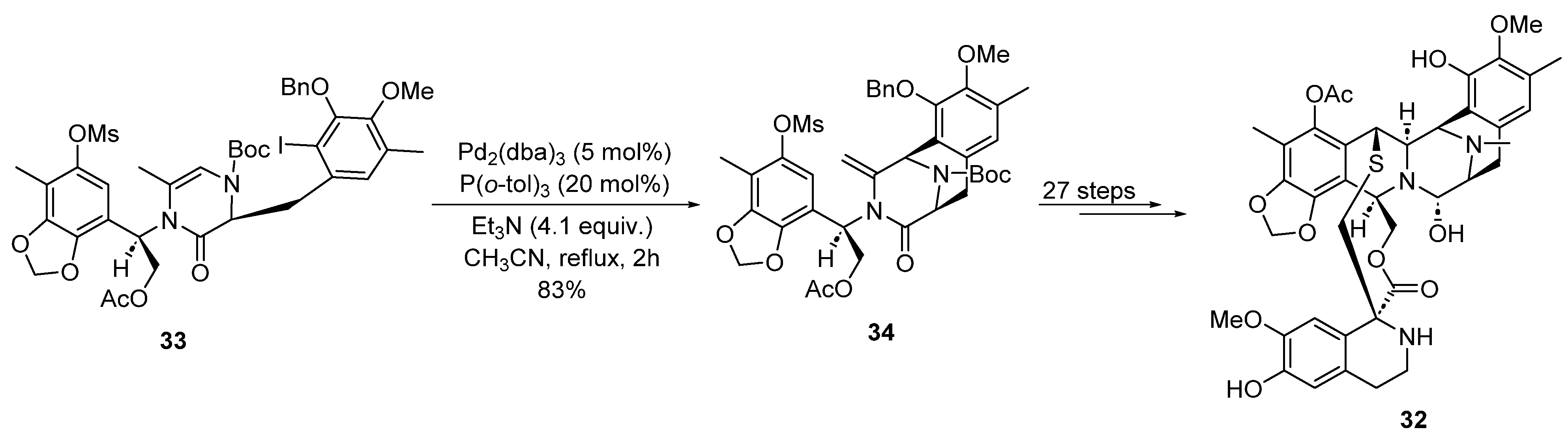
2.1.4. C-H Activation
2.1.5. Tsuji–Trost Allylation
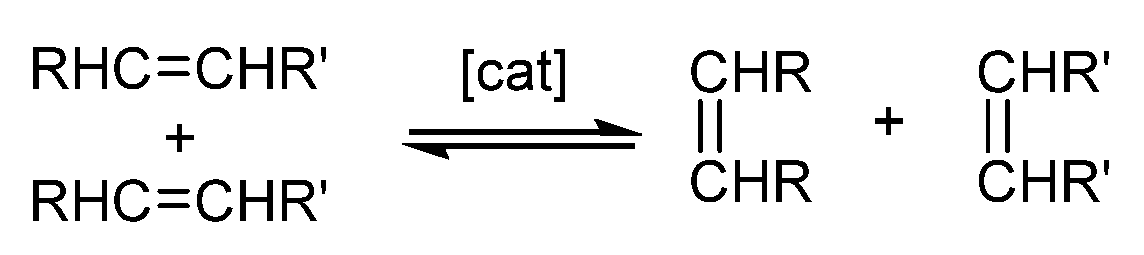
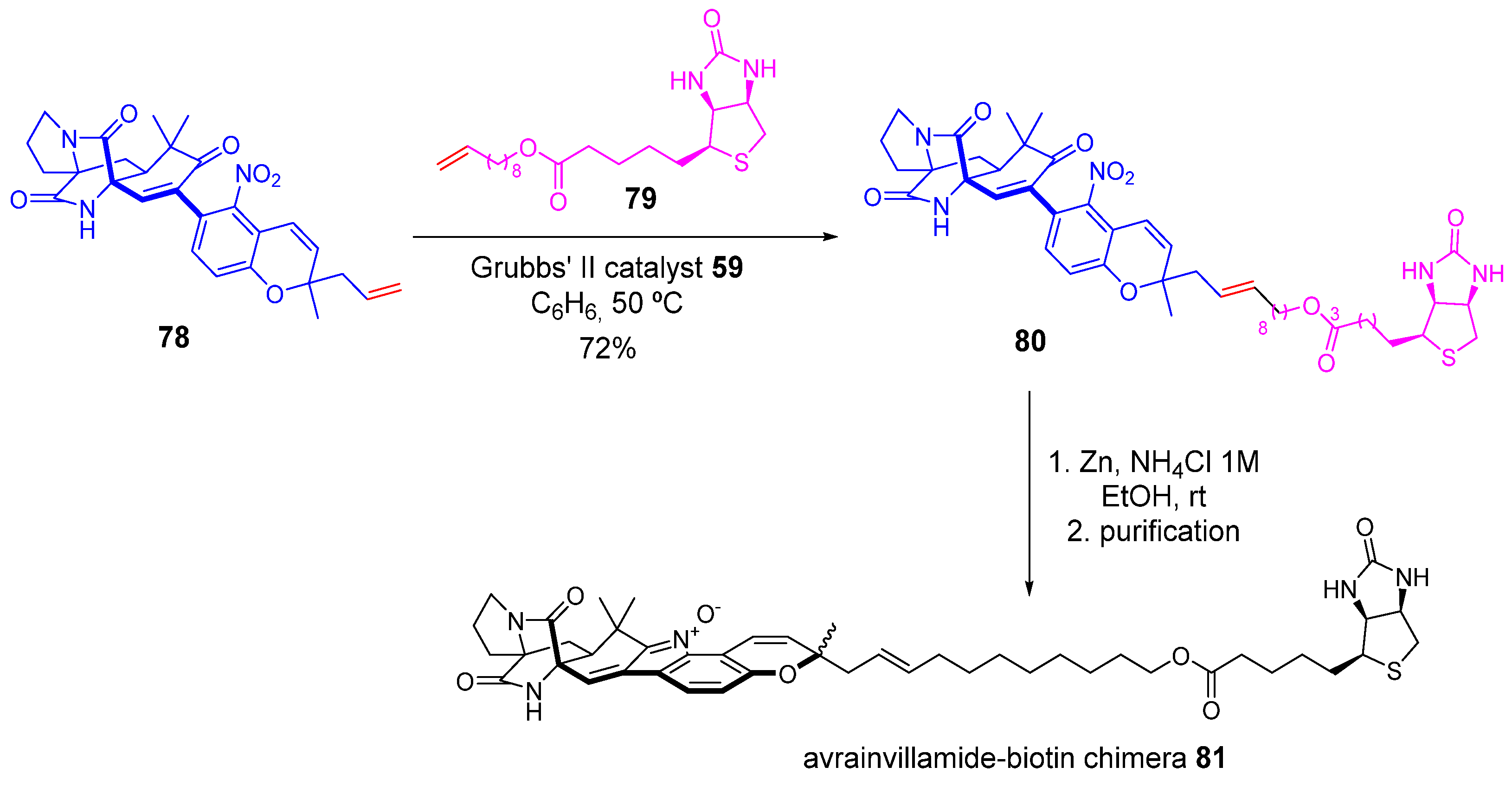
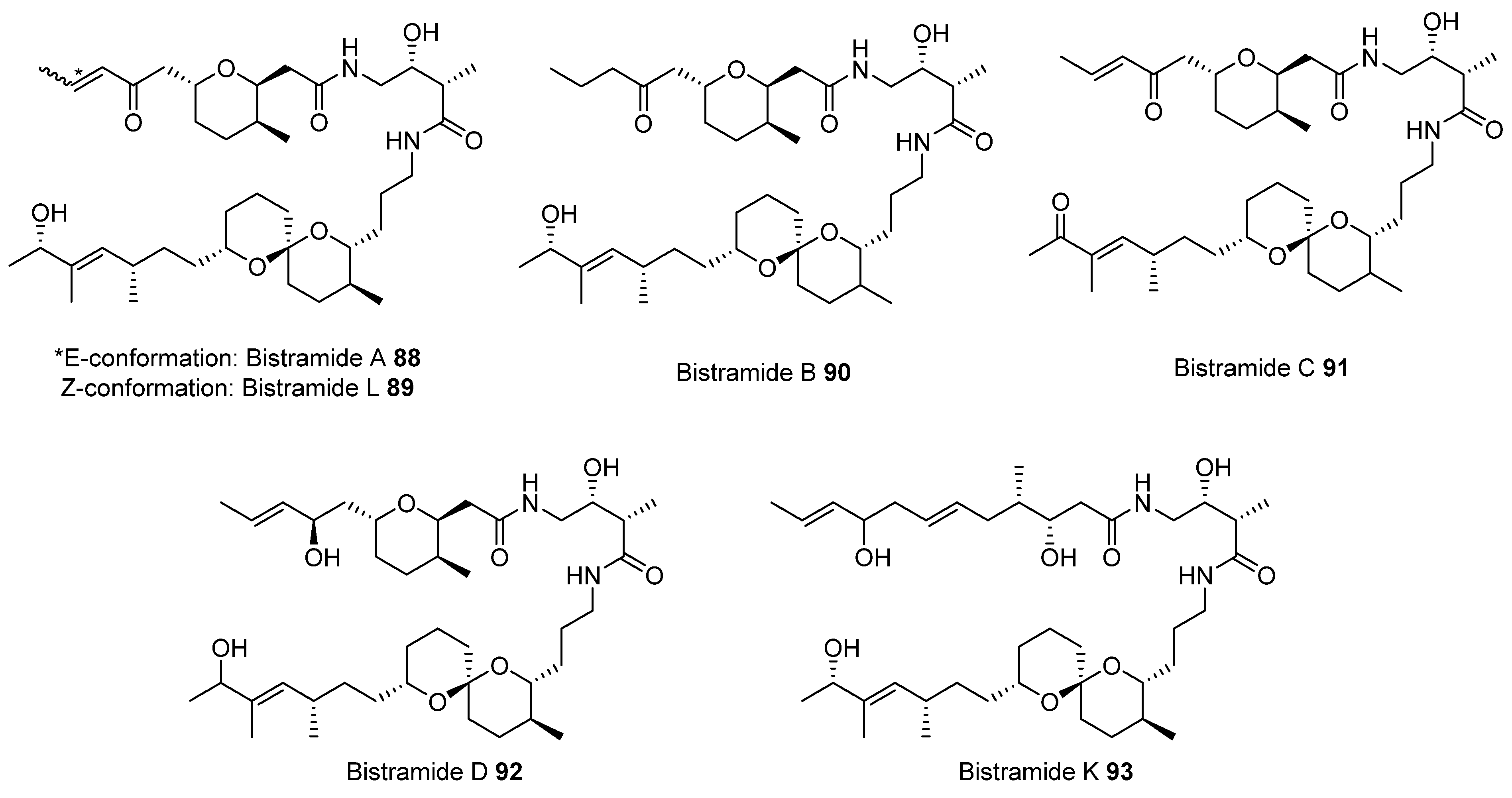
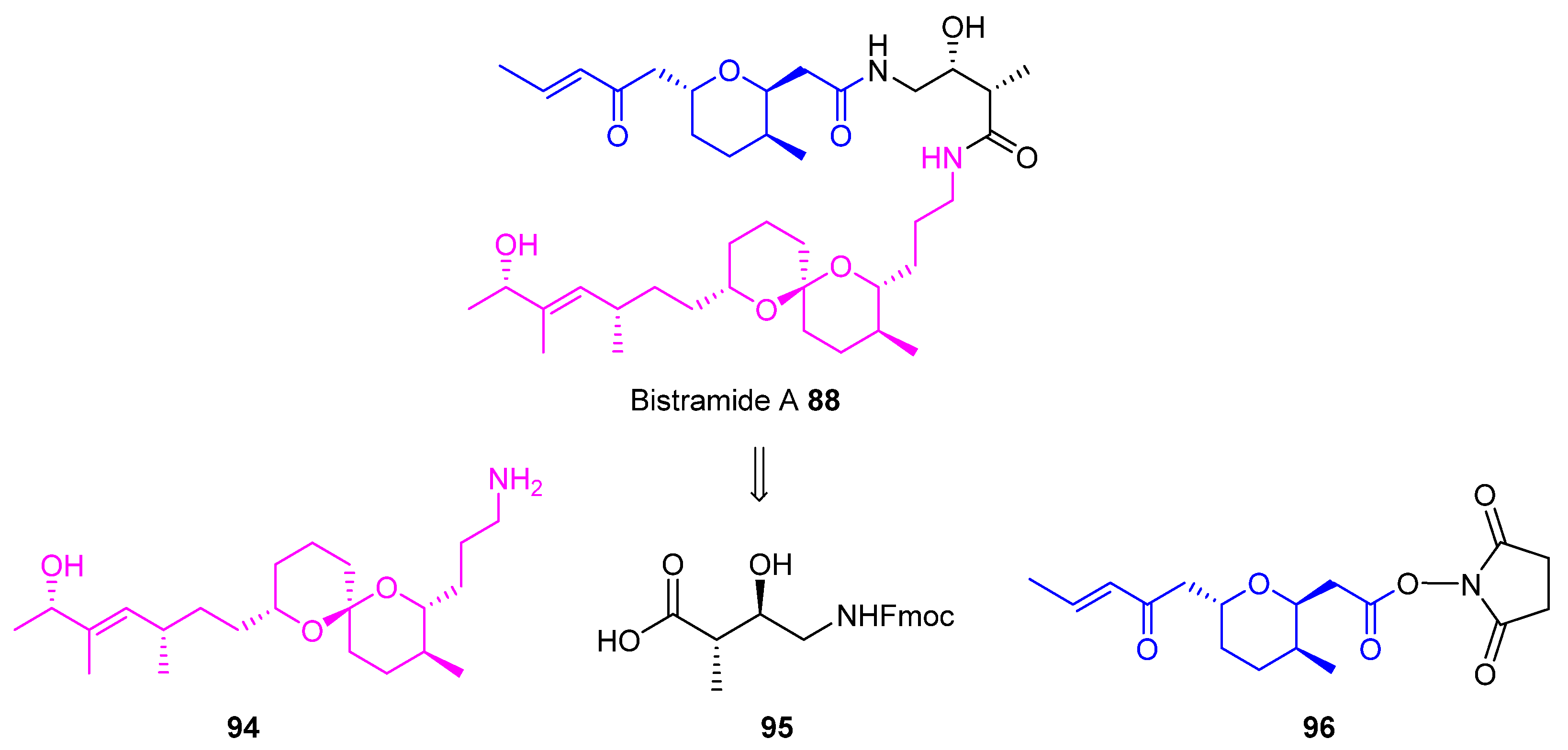
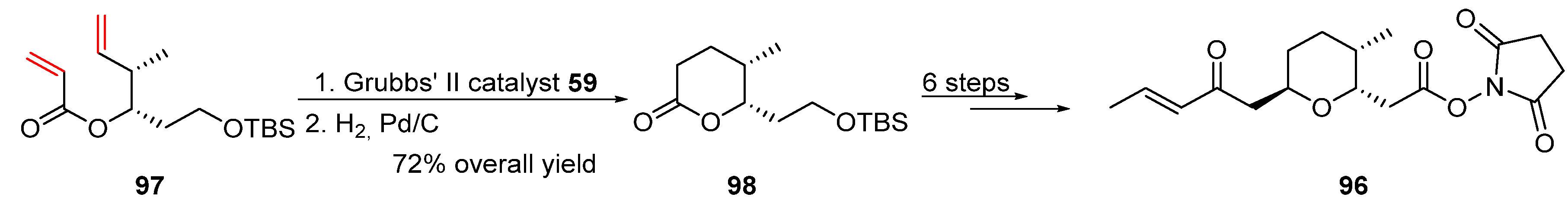
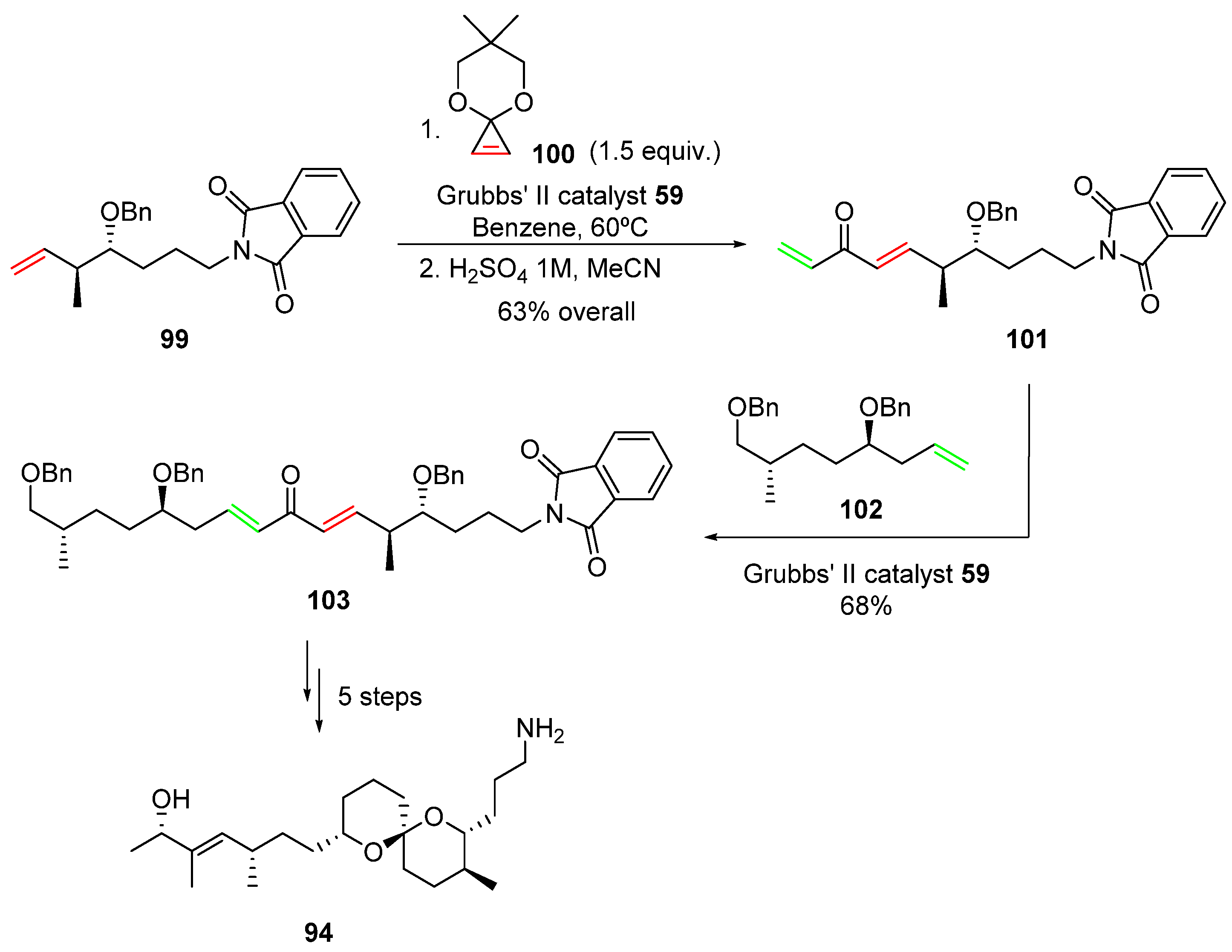
2.2. Olefin Metathesis
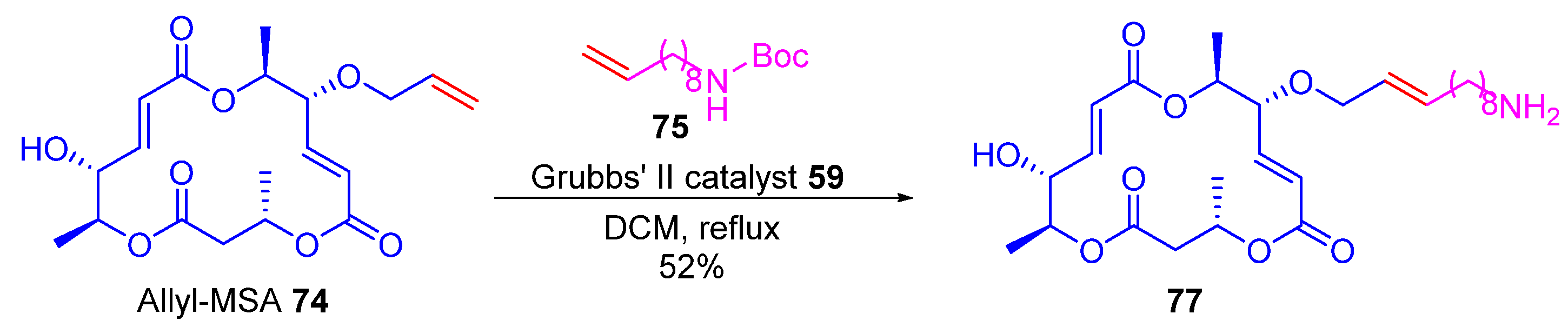
2.2.1. Cross-Metathesis (CM): Type 1 Reaction
2.2.2. Ring-Closing Metathesis (RCM): Type 2 Reaction
2.2.3. Ring-Opening Cross-Metathesis (ROCM): Type 3 Reaction
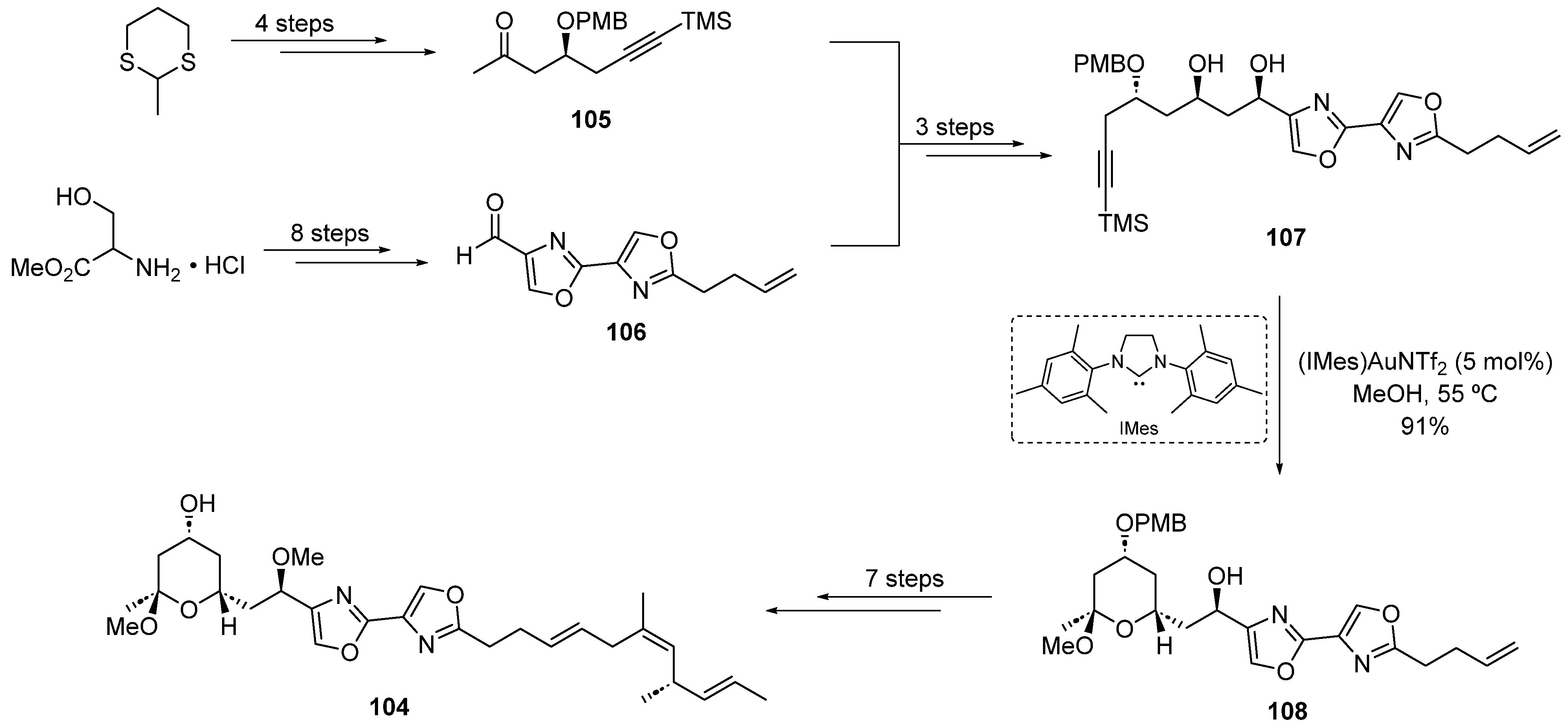
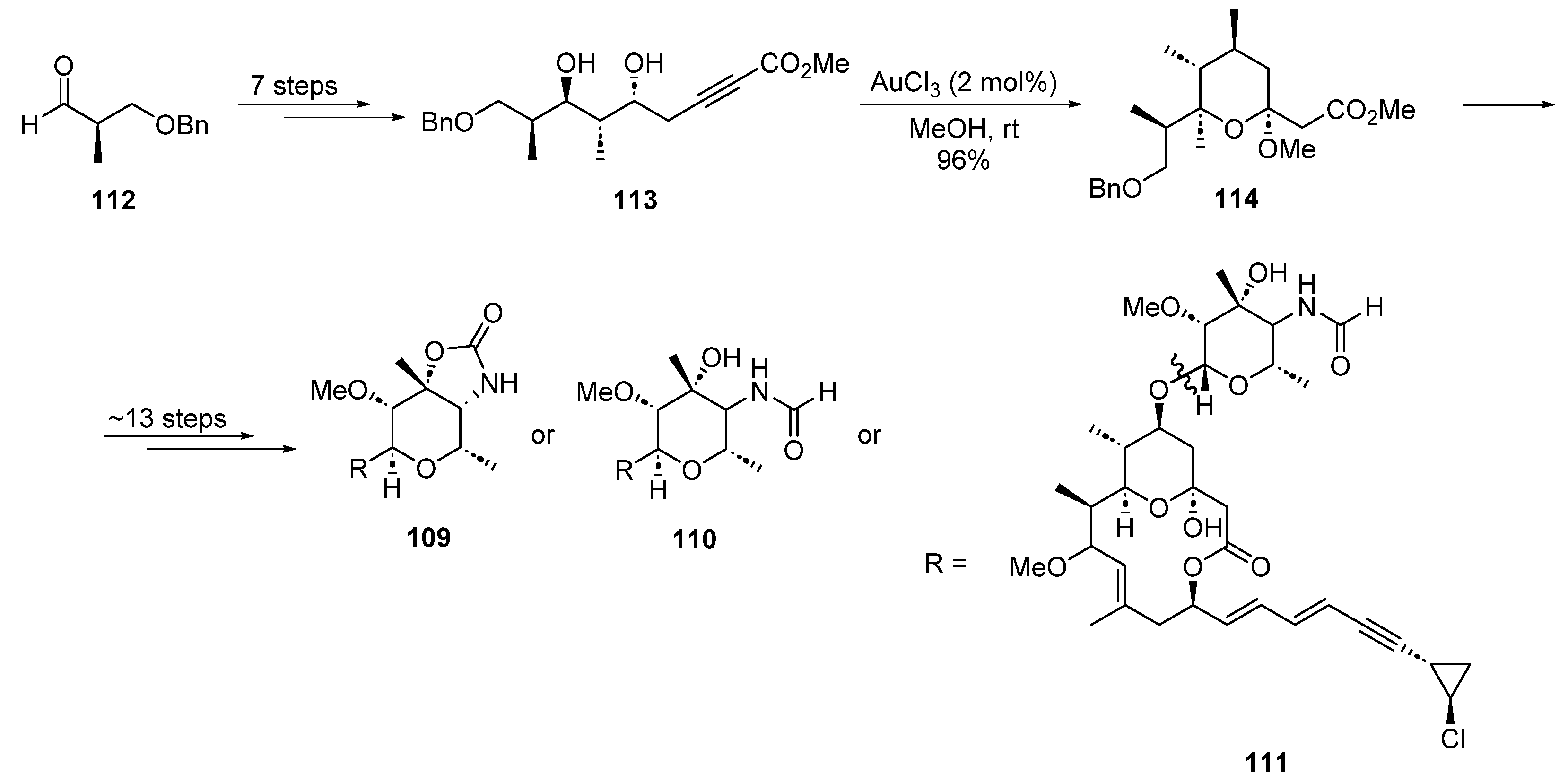
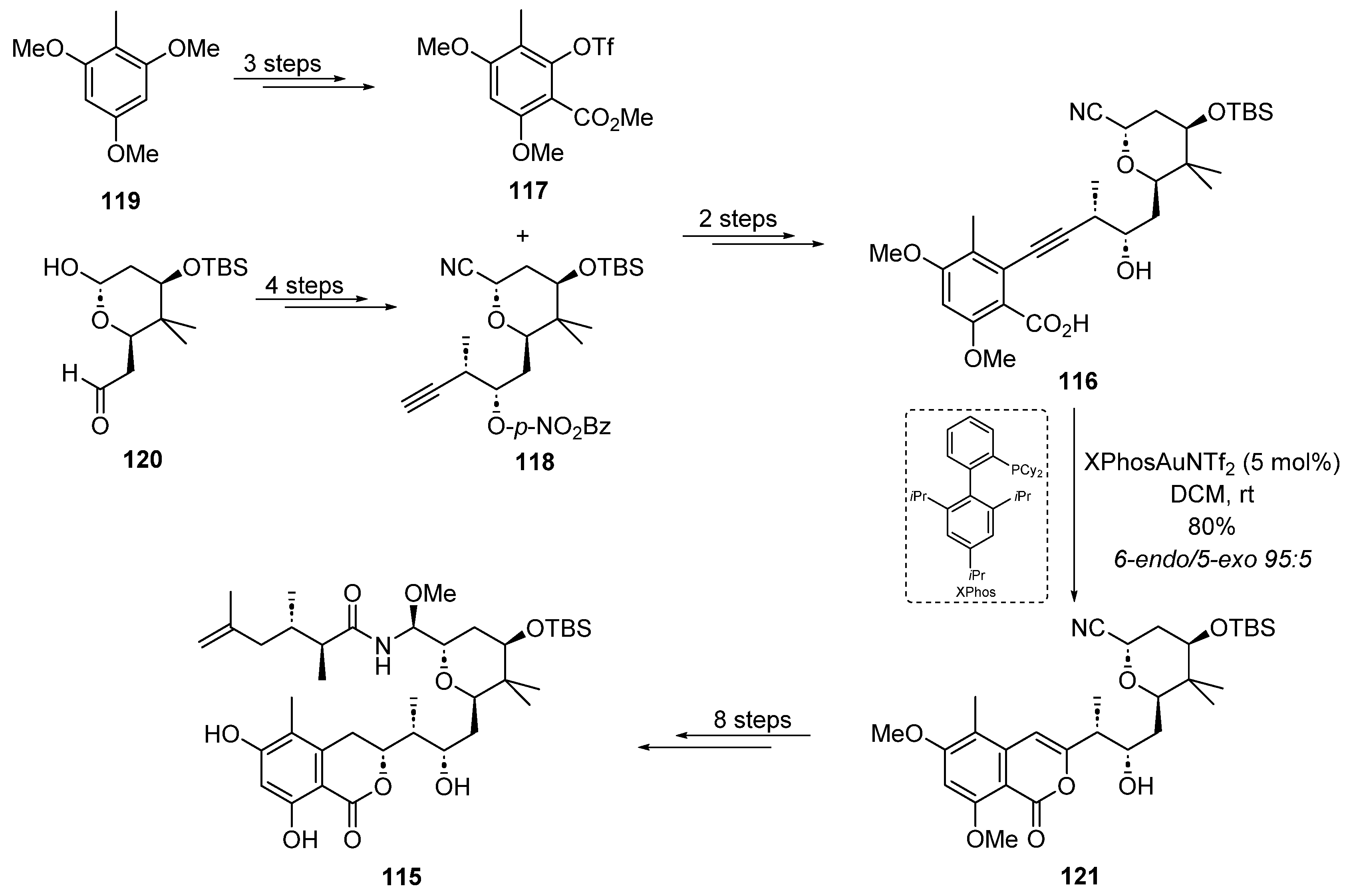
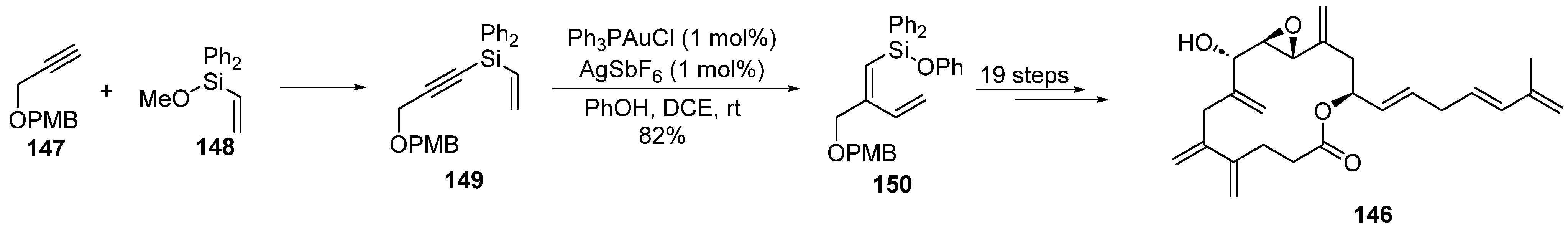
2.3. Gold Catalysis in the Total Synthesis of Marine Drugs
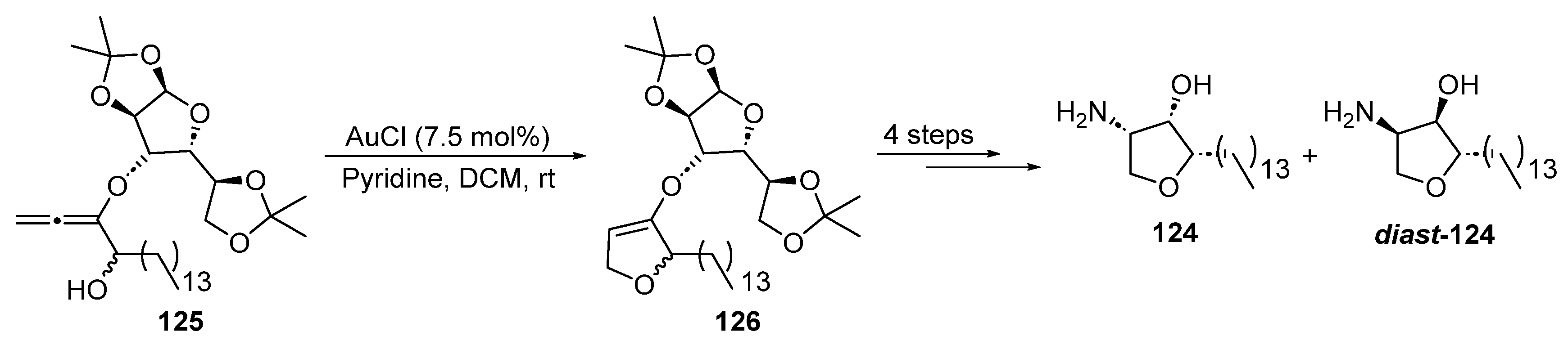
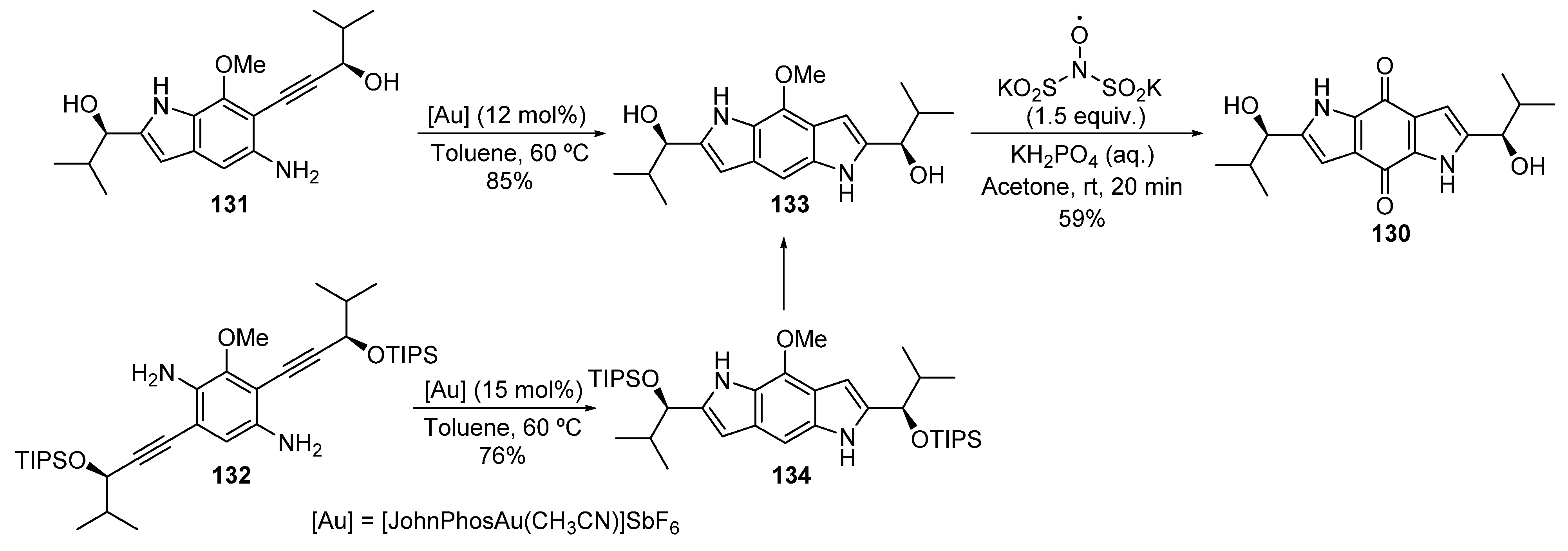
2.3.1. Addition of O-/N-Nucleophiles to Alkynes and Allenes
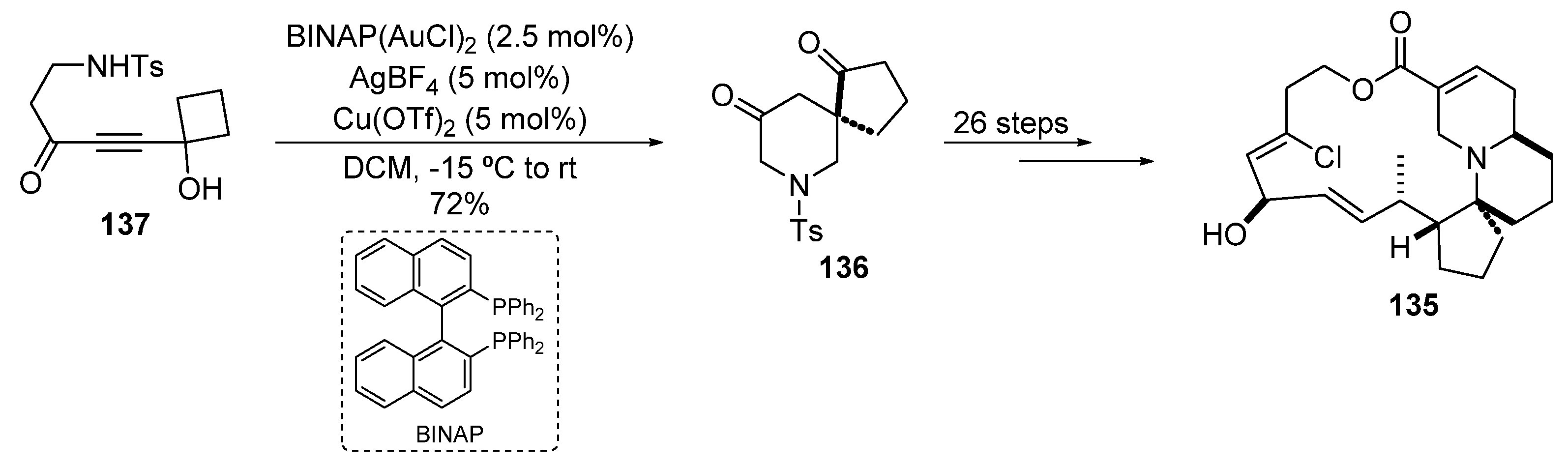
2.3.2. Gold-Catalyzed Cycloisomerization and Cycloadditions
2.3.3. Other Gold-Catalyzed Rearrangements
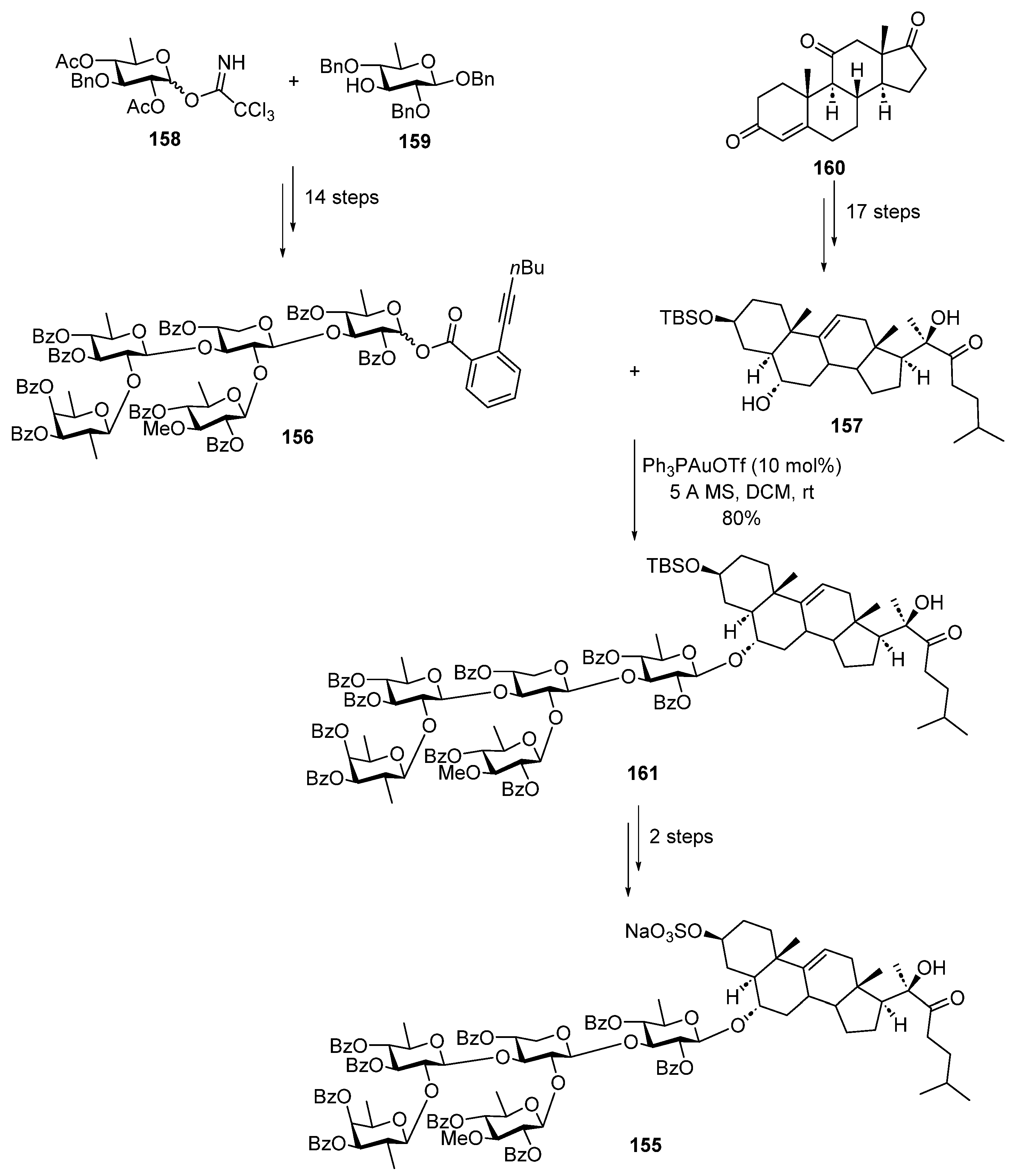
2.3.4. Gold-Catalyzed Glycosylation
2.4. Other Transition-Metal-Catalyzed Transformations in the Total Synthesis of Marine Drugs
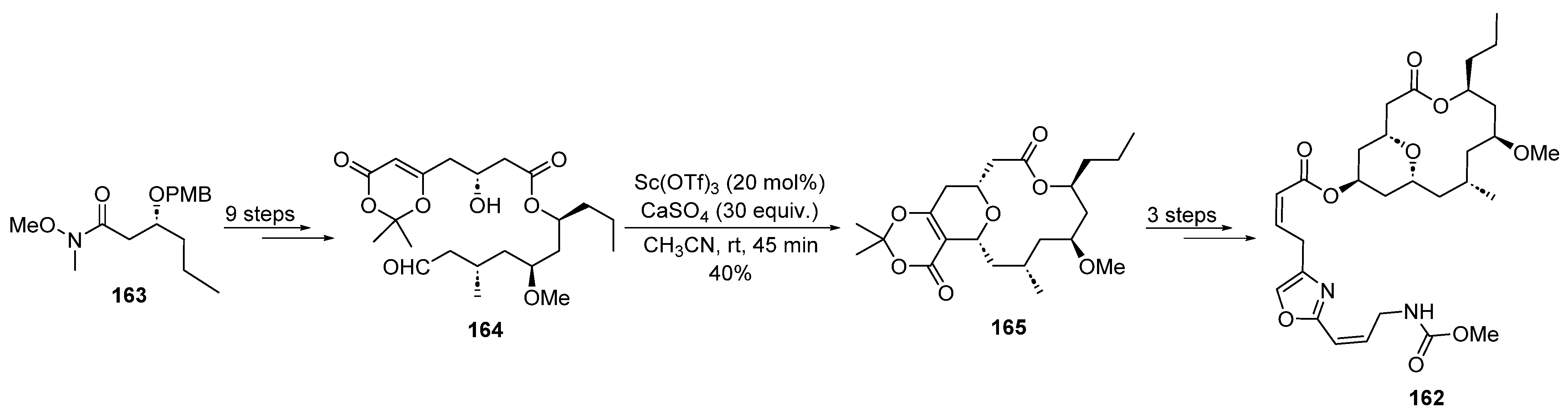
2.4.1. Scandium Catalysis
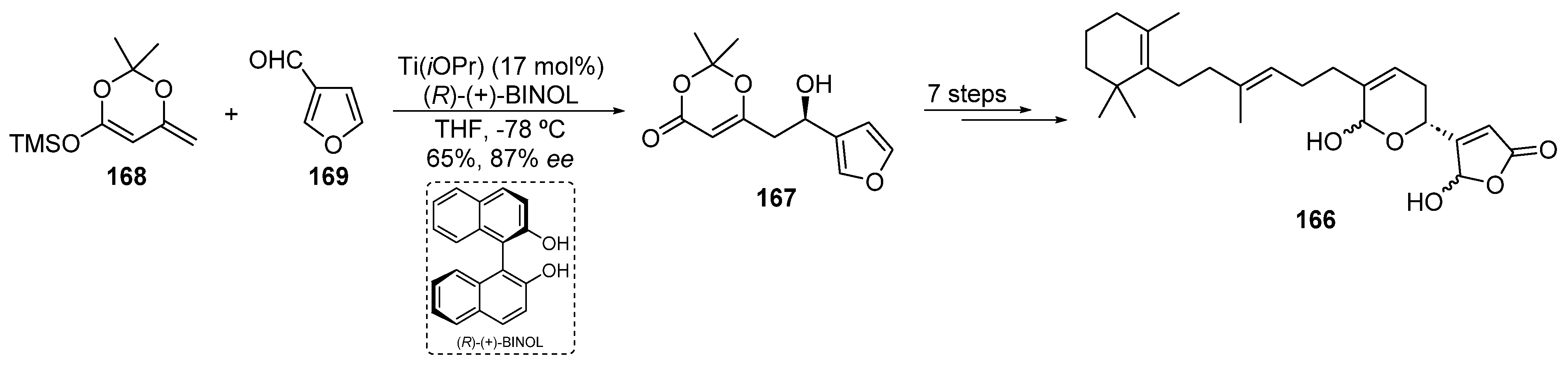
2.4.2. Titanium Catalysis
2.4.3. Iron Catalysis
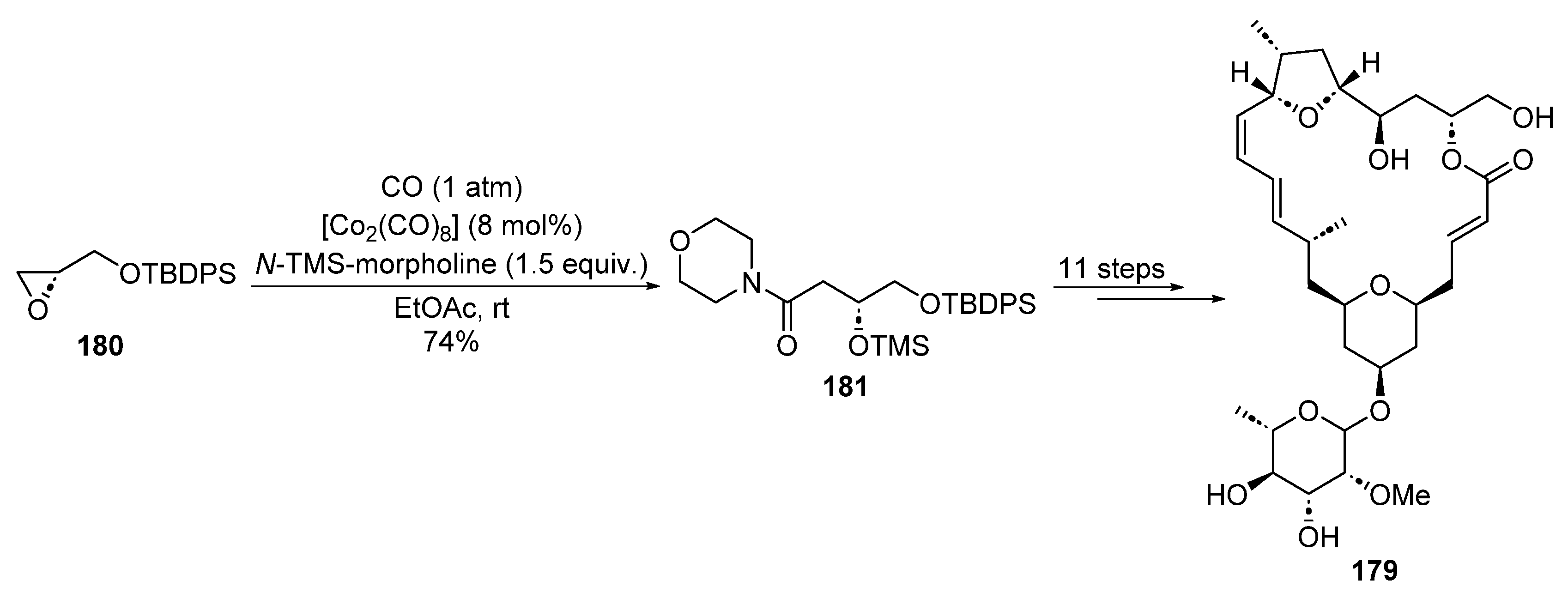
2.4.4. Cobalt Catalysis
2.4.5. Nickel Catalysis
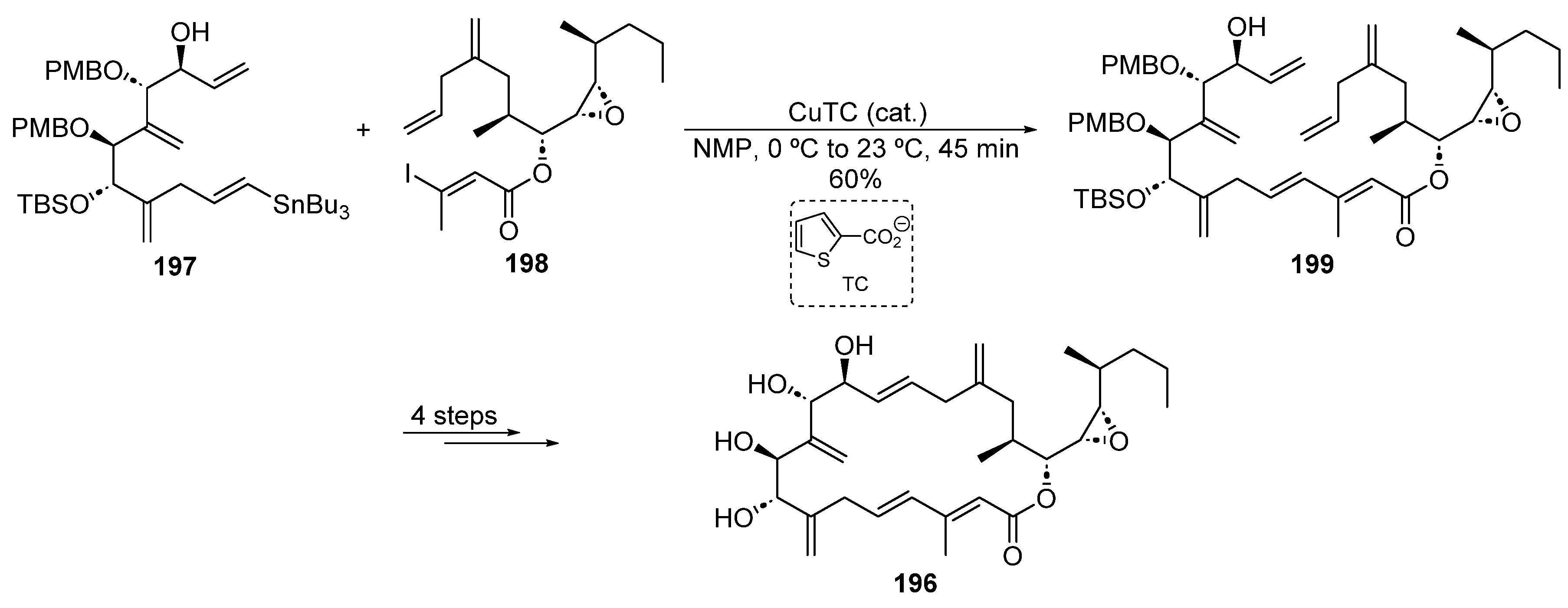
2.4.6. Copper Catalysis
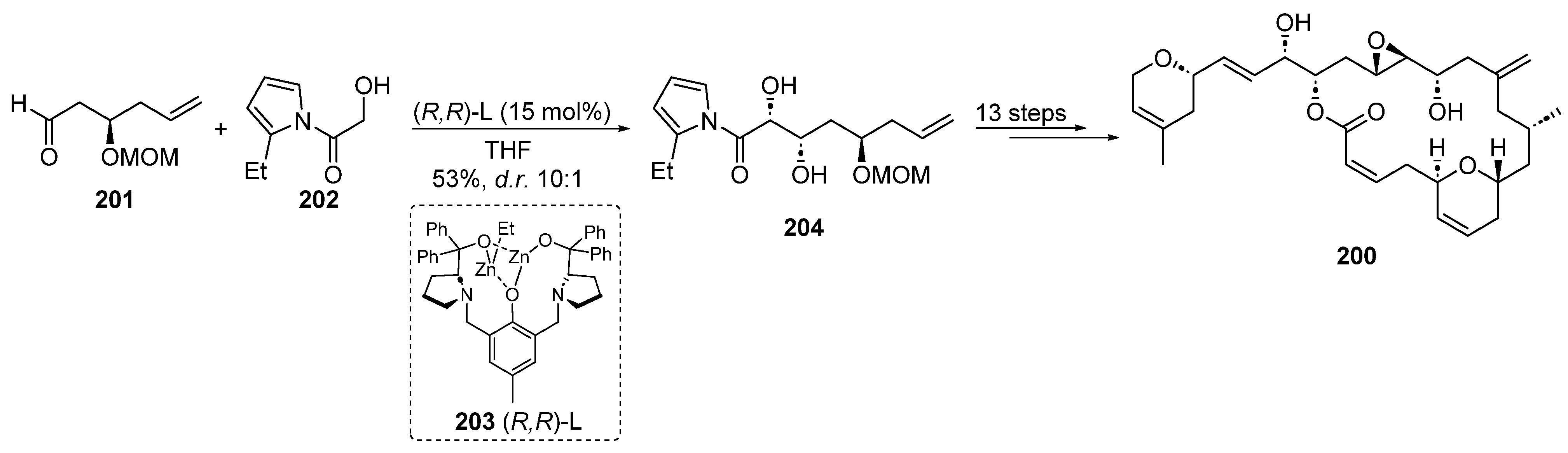
2.4.7. Zinc Catalysis
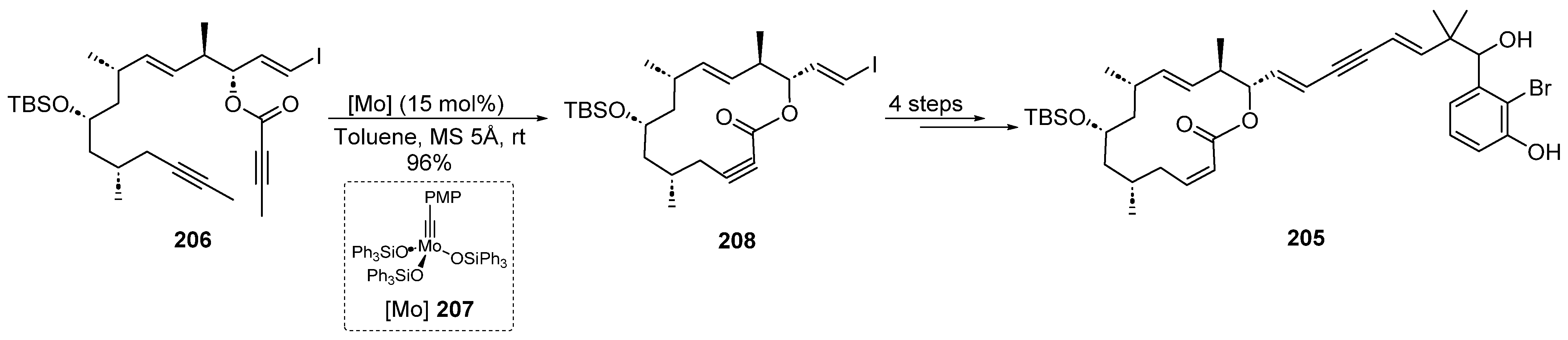
2.4.8. Molybdenum Catalysis
2.4.9. Rhodium Catalysis
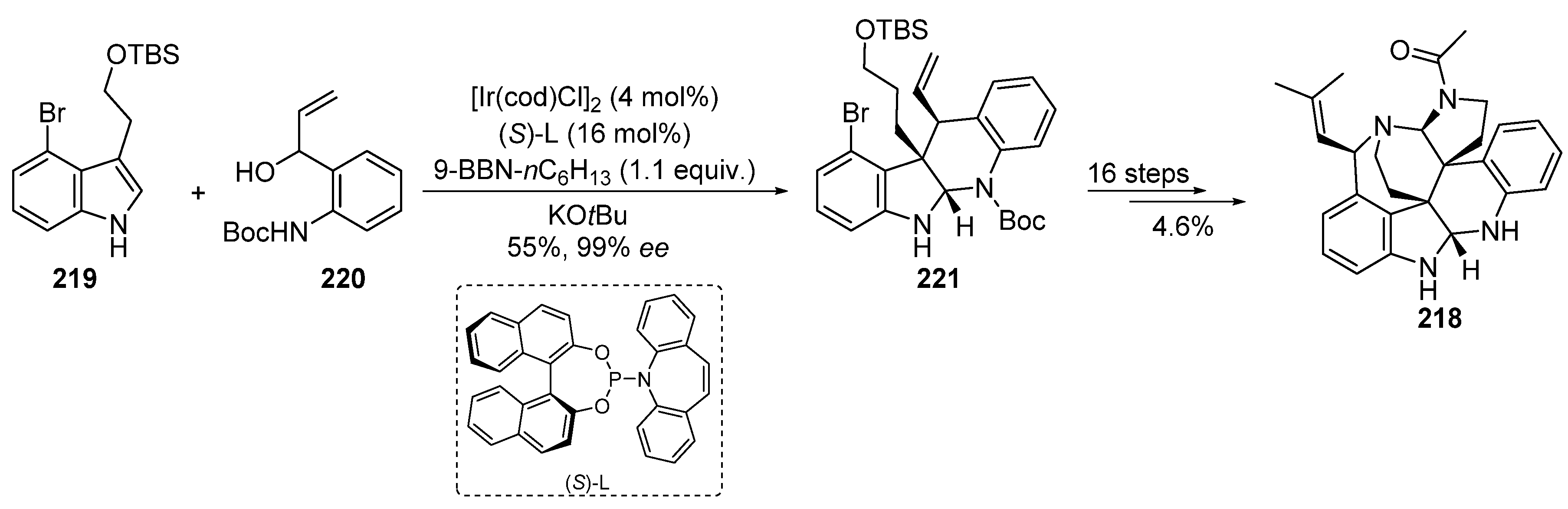
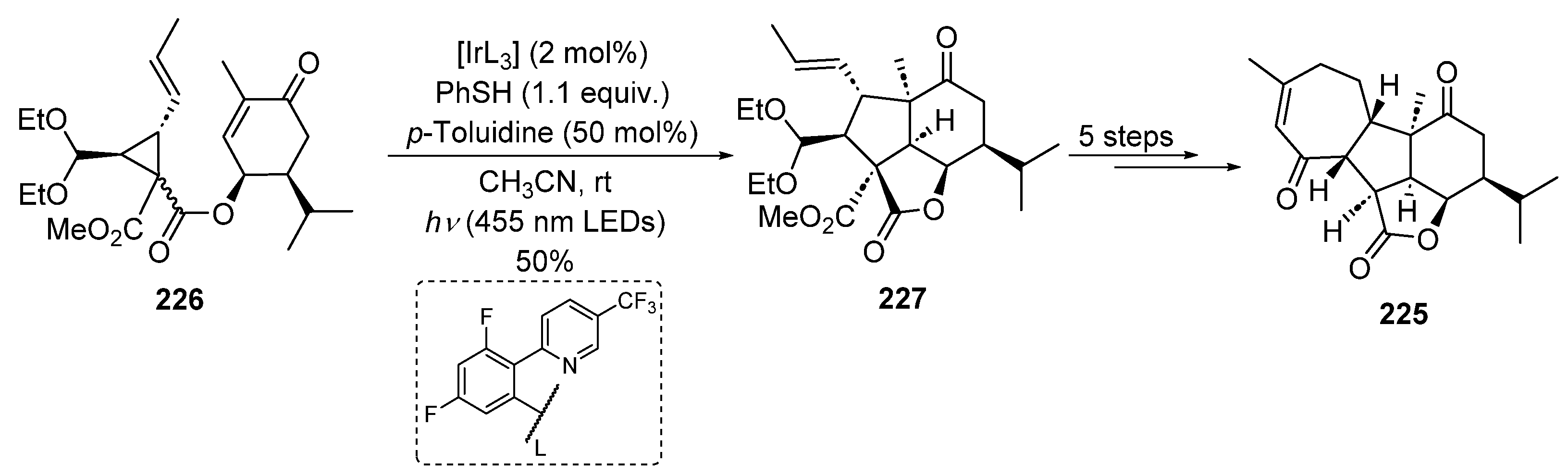
2.4.10. Iridium Catalysis
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cui, X.; Li, W.; Ryabchuk, P.; Junge, K.; Beller, M. Bridging Homogeneous and Heterogeneous Catalysis by Heterogeneous Single-Metal-Site Catalysts. Nat. Catal. 2018, 1, 385–397. [Google Scholar] [CrossRef]
- Shelke, Y.G.; Yashmeen, A.; Gholap, A.V.A.; Gharpure, S.J.; Kapdi, A.R. Homogeneous Catalysis: A Powerful Technology for the Modification of Important Biomolecules. Chem. Asian J. 2018, 13, 2991–3013. [Google Scholar] [CrossRef]
- Xiang, S.H.; Tan, B. Advances in Asymmetric Organocatalysis over the Last 10 Years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- Han, B.; He, X.H.; Liu, Y.Q.; He, G.; Peng, C.; Li, J.L. Asymmetric Organocatalysis: An Enabling Technology for Medicinal Chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Peña, L.F.; González-Andrés, P.; Parte, L.G.; Escribano, R.; Guerra, J.; Barbero, A.; López, E. Continuous Flow Chemistry: A Novel Technology for the Synthesis of Marine Drugs. Mar. Drugs 2023, 21, 402. [Google Scholar] [CrossRef]
- Magano, J.; Dunetz, J.R. Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C-H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef] [PubMed]
- Zweig, J.E.; Kim, D.E.; Newhouse, T.R. Methods Utilizing First-Row Transition Metals in Natural Product Total Synthesis. Chem. Rev. 2017, 117, 11680–11752. [Google Scholar] [CrossRef]
- Cheung, K.P.S.; Sarkar, S.; Gevorgyan, V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122, 1543–1625. [Google Scholar] [CrossRef]
- Jana, R.; Pathak, T.P.; Sigman, M.S. Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-Organometallics as Reaction Partners. Chem. Rev. 2011, 111, 1417–1492. [Google Scholar] [CrossRef]
- Choury, M.; Lopes, A.B.; Blond, G.; Gulea, M. Synthesis of Medium-Sized Heterocycles by Transition-Metal-Catalyzed Intramolecular Cyclization. Molecules 2020, 25, 3147. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Lan, Y. Recent Advances in Theoretical Studies on Transition-Metal-Catalyzed Carbene Transformations. Acc. Chem. Res. 2021, 54, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Fang, P.; Mei, T.S. Recent Advances in C-H Functionalization Using Electrochemical Transition Metal Catalysis. ACS Catal. 2018, 8, 7179–7189. [Google Scholar] [CrossRef]
- Busacca, C.A.; Fandrick, D.R.; Song, J.J.; Senanayake, C.H. Transition Metal Catalysis in the Pharmaceutical Industry. In Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective; Crawley, M.L., Trost, B.M., Eds.; John Wiley & Sons: New York, NY, USA, 2012; pp. 1–376. [Google Scholar]
- Firsan, S.J.; Sivakumar, V.; Colacot, T.J. Emerging Trends in Cross-Coupling: Twelve-Electron-Based L1Pd(0) Catalysts, Their Mechanism of Action, and Selected Applications. Chem. Rev. 2022, 122, 16983–17027. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Chen, Y.; Zhu, X.; Yu, L. Polyaniline-Supported Nano Metal-Catalyzed Coupling Reactions: Opportunities and Challenges. Chin. Chem. Lett. 2023, 34, 107728. [Google Scholar] [CrossRef]
- Papon, N.; Copp, B.R.; Courdavault, V. Marine Drugs: Biology, Pipelines, Current and Future Prospects for Production. Biotechnol. Adv. 2022, 54, 107871. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine Natural Products: A New Wave of Drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Manzo, E. Synthesis of Marine Natural Products and Molecules Inspired by Marine Substances. Mar. Drugs 2021, 19, 208. [Google Scholar] [CrossRef]
- Fernández-Peña, L.; Matos, M.J.; López, E. Recent Advances in Biologically Active Coumarins from Marine Sources: Synthesis and Evaluation. Mar. Drugs 2023, 21, 37. [Google Scholar] [CrossRef]
- Wu, X.; Lamb, K.J.; Lara-Sánchez, A.; Alonso-Moreno, C.; North, M.; Castro-Osma, J.A. Homogeneous Aluminum and Iron Catalysts for the Synthesis of Organic Molecules and Biodegradable Polymers. In Synthetic Inorganic Chemistry: New Perspectives; Elsevier: Amsterdam, The Netherlands, 2021; pp. 3–43. [Google Scholar]
- Miyaura, N.; Tamao, K. Introduction to Cross-Coupling Reactions. Top. Curr. Chem. 2002, 219, 1–10. [Google Scholar]
- Trzeciak, A.M.; Augustyniak, A.W. The Role of Palladium Nanoparticles in Catalytic C–C Cross-Coupling Reactions. Coord. Chem. Rev. 2019, 84, 1–20. [Google Scholar] [CrossRef]
- Wu, X.F.; Anbarasan, P.; Neumann, H.; Beller, M. From Noble Metal to Nobel Prize: Palladium-Catalyzed Coupling Reactions as Key Methods in Organic Synthesis. Angew. Chem. Int. Ed. 2010, 49, 9047–9050. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A. Organoborates in New Synthetic Reactions. Acc. Chem. Res. 1982, 15, 178–184. [Google Scholar] [CrossRef]
- Miyaura, N.; Yanagi, T.; Suzuki, A. The Palladium-Catalyzed Cross-Coupling Reaction of Phenylboronic Acid with Haloarenes in the Presence of Bases. Synth. Commun. 1981, 11, 513–519. [Google Scholar] [CrossRef]
- Negishi, E.; King, A.O.; Okukado, N. Selective Carbon-Carbon Bond Formation via Transition Metal Catalysis. 3. A Highly Selective Synthesis of Unsymmetrical Biaryls and Diarylmethanes by the Nickel- or Palladium-Catalyzed Reaction of Aryl- and Benzylzinc Derivatives with Aryl Halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- Heck, K.F.; Nolley, J.P. Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Stille, J.K. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles [New Synthetic Methods (58)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Hiyama, T. Cross-Coupling of Organosilanes with Organic Halides Mediated by Palladium Catalyst and Tris(Diethylamino)Sulfonium Difluorotrimethylsilicate. J. Org. Chem. 1988, 53, 918–920. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A Convenient Synthesis of Acetylenes: Catalytic Substitutions of Acetylenic Hydrogen with Bromoalkenes, Iodoarenes and Bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Basu, D.; Kumar, S.; SaiSudhir, V.; Bandichhor, R. Transition Metal Catalyzed C-H Activation for the Synthesis of Medicinally Relevant Molecules: A Review. J. Chem. Sci. 2018, 130, 71. [Google Scholar] [CrossRef]
- Kohmoto, S.; Kashman, Y.J.; McConnell, O.; Rinehart, K.L., Jr.; Wright, A.; Koehn, F. Dragmacidin, a New Cytotoxic Bis(indole) Alkaloid from a Deep Water Marine Sponge, Dragmacidon sp. J. Org. Chem. 1988, 53, 3116–3118. [Google Scholar] [CrossRef]
- Yamazaki, K.; Okuda, Y.; Takaya, A.; Nemoto, T. Total Synthesis of Dragmacidins G and H. Org. Lett. 2024, 26, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.A.; Peterson, T.A.; Wright, A.E. The Marine Natural Compound Dragmacidin D Selectively Induces Apoptosis in Triple-Negative Breast Cancer Spheroids. Mar. Drugs 2023, 21, 642. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.K.; Sarpong, R.; Stoltz, B.M. The First Total Synthesis of Dragmacidin D. J. Am. Chem. Soc. 2002, 124, 13179–13184. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Bifulco, G.; Bruno, I.; Casapullo, A.; Gomez-Paloma, L.; Riccio, R. Dragmacidin F: A New Antiviral Bromoindole Alkaloid from the Mediterranean Sponge Halicortex sp. Tetrahedron 2000, 56, 3743–3748. [Google Scholar] [CrossRef]
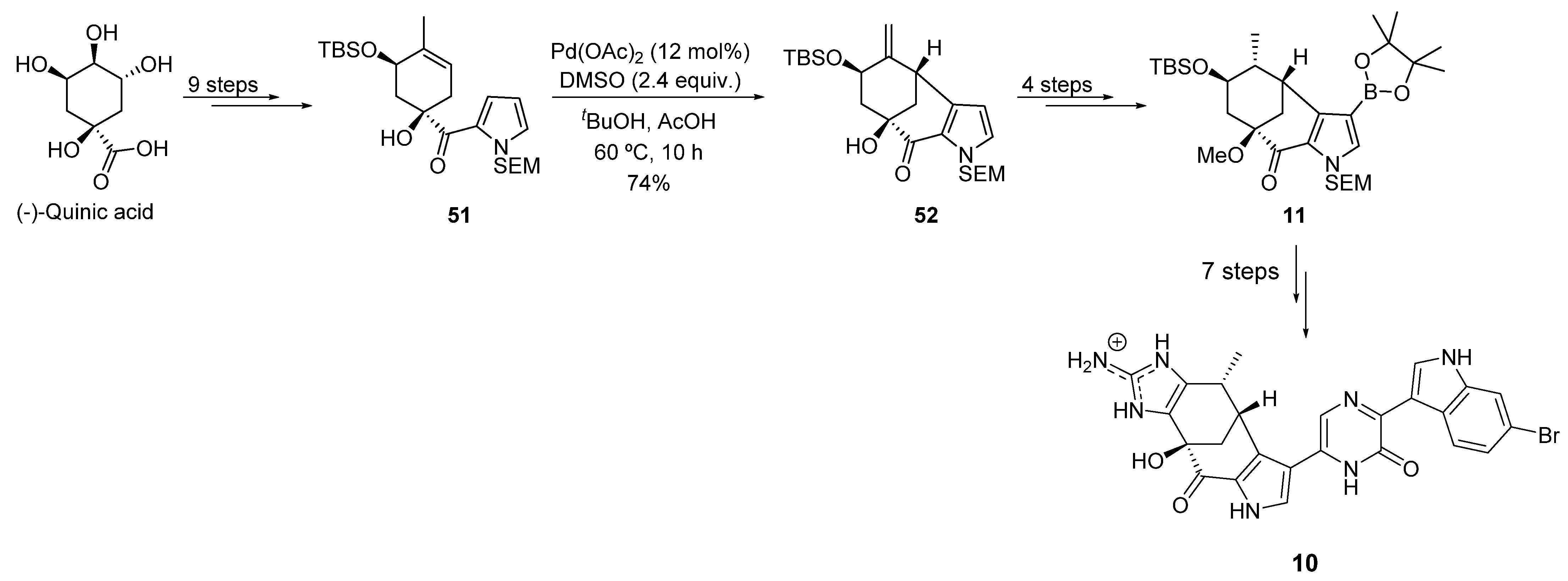
- Garg, N.K.; Caspi, D.D.; Stoltz, B.M. Development of an Enantiodivergent Strategy for the Total Synthesis of (+)- and (−)-Dragmacidin f from a Single Enantiomer of Quinic Acid. J. Am. Chem. Soc. 2005, 127, 5970–5978. [Google Scholar] [CrossRef] [PubMed]
- Warabi, K.; Matsunaga, S.; Van Soest, R.W.M.; Fusetani, N. Dictyodendrins A-E, the First Telomerase-Inhibitory Marine Natural Products from the Sponge Dictyodendrilla Verongiformis. J. Org. Chem. 2003, 68, 2765–2770. [Google Scholar] [CrossRef]
- Pitts, A.K.; O’Hara, F.; Snell, R.H.; Gaunt, M.J. A Concise and Scalable Strategy for the Total Synthesis of Dictyodendrin B Based on Sequential C-H Functionalization. Angew. Chem. Int. Ed. 2015, 127, 5541–5545. [Google Scholar] [CrossRef]
- Kato, Y.; Fusetani, N.; Matsunaga, S.; Hashimoto, K.; Koseki, K.; Konosu, S. Discodermolide: A New Bioactive Polyhydroxylated Lactone from the Marine Sponge Discodermia Dissoluta. Mod. J. Am. Chem. Soc. 1990, 55, 35. [Google Scholar]
- Florence, G.J.; Gardner, N.M.; Paterson, I. Development of practical syntheses of the marine anticancer agents discodermolide and dictyostatin. Nat. Prod. Rep. 2008, 25, 342–375. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Johns, B.A. Total Synthesis of (+)-Discodermolide. J. Org. Chem. 1998, 63, 7885–7892. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Gao, F.; Boyd, M.R.; Schmidt, J.M. Isolation and Structure of the Cancer Cell Growth Inhibitor Dictyostatin 1. J. Chem. Soc. Chem. Commun. 1994, 9, 1111–1112. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Srivastava, A.; Hazra, D. Total Synthesis of Potential Antitumor Agent, (−)-Dictyostatin. Org. Lett. 2007, 9, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Cheng, J.; Ohta, T.; Nakamura, H.; Nozoe, S.; Hirata, Y.; Ohizumi, Y.; Sasaki, T. Iejimalides A and B, Novel 24-Membered Macrolides with Potent Antileukemic Activity from the Okinawan Tunicate Eudistoma cf. rigida. J. Org. Chem. 1988, 53, 6147–6150. [Google Scholar] [CrossRef]
- Nozawa, K.; Tsuda, M.; Ishiyama, H.; Sasaki, T.; Tsuruo, T.; Kobayashi, J. Absolute Stereochemistry and Antitumor Activity of Iejimalides. Bioorg. Med. Chem. 2006, 14, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Moulin, E.; Nevado, C.; Gagnepain, J.; Kelter, G.; Fiebig, H.H.; Fürstner, A. Synthesis and Evaluation of an Iejimalide-Archazolid Chimera. Tetrahedron 2010, 66, 6421–6428. [Google Scholar] [CrossRef]
- Sasse, F.; Steinmetza, H.; Hoflea, G.; Gbf, H.R. Archazolids, New Cytotoxic Macrolactones from Archangium Gephyra (Myxobacteria) Production, Isolation, Physico-Chemical and Biological Properties. J. Antibiot. 2003, 56, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Luong, B.; Schwenk, R.; Bräutigam, J.; Müller, R.; Menche, D.; Bischoff, I.; Fürst, R. The Vacuolar-Type ATPase Inhibitor Archazolid Increases Tumor Cell Adhesion to Endothelial Cells by Accumulating Extracellular Collagen. PLoS ONE 2018, 13, e0203053. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent Antitumor Agents from the Caribbean Tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Lu, W.Y.; Li, H.J.; Li, Q.Y.; Wu, Y.C. Application of Marine Natural Products in Drug Research. Bioorg. Med. Chem. 2021, 35, 116058. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Nakata, H.; Yokoshima, S.; Fukuyama, T. Synthetic Studies toward Ecteinascidin 743 (Trabectedin). Synthesis 2012, 44, 2743–2753. [Google Scholar]
- Endo, A.; Yanagisawa, A.; Abe, M.; Tohma, S.; Kan, T.; Fukuyama, T. Total Synthesis of Ecteinascidin 743. J. Am. Chem. Soc. 2002, 124, 6552–6554. [Google Scholar] [CrossRef] [PubMed]
- Look, S.A.; Fenical, W.; Jacobs, R.S.; Clardy, J. The Pseudopterosins: Anti-Inflammatory and Analgesic Natural Products from the Sea Whip Pseudopterogorgia Elisabethae. Proc. Natl. Acad. Sci. USA 1986, 83, 6238–6240. [Google Scholar] [CrossRef] [PubMed]
- Caplan, S.L.; Zheng, B.; Dawson-Scully, K.; White, C.A.; West, L.M. Pseudopterosin a: Protection of Synaptic Function and Potential as a Neuromodulatory Agent. Mar. Drugs 2016, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Flachsmann, F.; Schellhaas, K.; Moya, C.E.; Jacobs, R.S.; Fenical, W. Synthetic Pseudopterosin Analogues: A Novel Class of Antiinflammatory Drug Candidates. Bioorg. Med. Chem. 2010, 18, 8324–8333. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Su, F.; Liu, C.; Yuan, H.; Zhao, S.; Zhou, Z.; Quan, T.; Luo, T. Enantioselective Total Syntheses of Various Amphilectane and Serrulatane Diterpenoids via Cope Rearrangements. J. Am. Chem. Soc. 2016, 138, 6261–6270. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.W.; Schreiber, S.L.; Sigal, N.H.; Dumont, F.J.; Hung, D.T.; Jamison, T.F.; Gunasekera, S.P.; Gunasekera, M.; Longley, R.E.; Golec, J.M.C. Syntheses of Discodermolides Useful for Investigating Microtubule Binding and Stabilization. J. Am. Chem. Soc. 1996, 118, 11054–11080. [Google Scholar]
- Smith, A.B.; Scott Freeze, B.; Brouard, I.; Hirose, T. A Practical Improvement, Enhancing the Large-Scale Synthesis of (+)-Discodermolide: A Third-Generation Approach. Org. Lett. 2003, 5, 4405–4408. [Google Scholar] [CrossRef]
- Mandal, D.; Yamaguchi, A.D.; Yamaguchi, J.; Itami, K. Synthesis of Dragmacidin D via Direct C-H Couplings. J. Am. Chem. Soc. 2011, 133, 19660–19663. [Google Scholar] [CrossRef]
- Tahara, Y.; Hirata, Y. Studies on the puffer fish toxin. J. Pharm. Soc. Jpn. 1990, 29, 587–625. [Google Scholar]
- Manabe, A.; Ohfune, Y.; Shinada, T. Toward the Total Synthesis of Tetrodotoxin: Stereoselective Construction of the 7-Oxanorbornane Intermediate. Tetrahedron Lett. 2014, 55, 6077–6080. [Google Scholar] [CrossRef]
- Lázaro-Milla, C.; Quirós, M.T.; Cárdenas, D.J.; Almendros, P. Triflyl-Assisted Reductive Pd-Catalyzed Tsuji-Trost Type Reaction. Chem. Commun. 2020, 56, 6070–6073. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Metathesis Reactions in Total Synthesis. Angew. Chem. Int. Ed. 2005, 44, 4490–4527. [Google Scholar] [CrossRef]
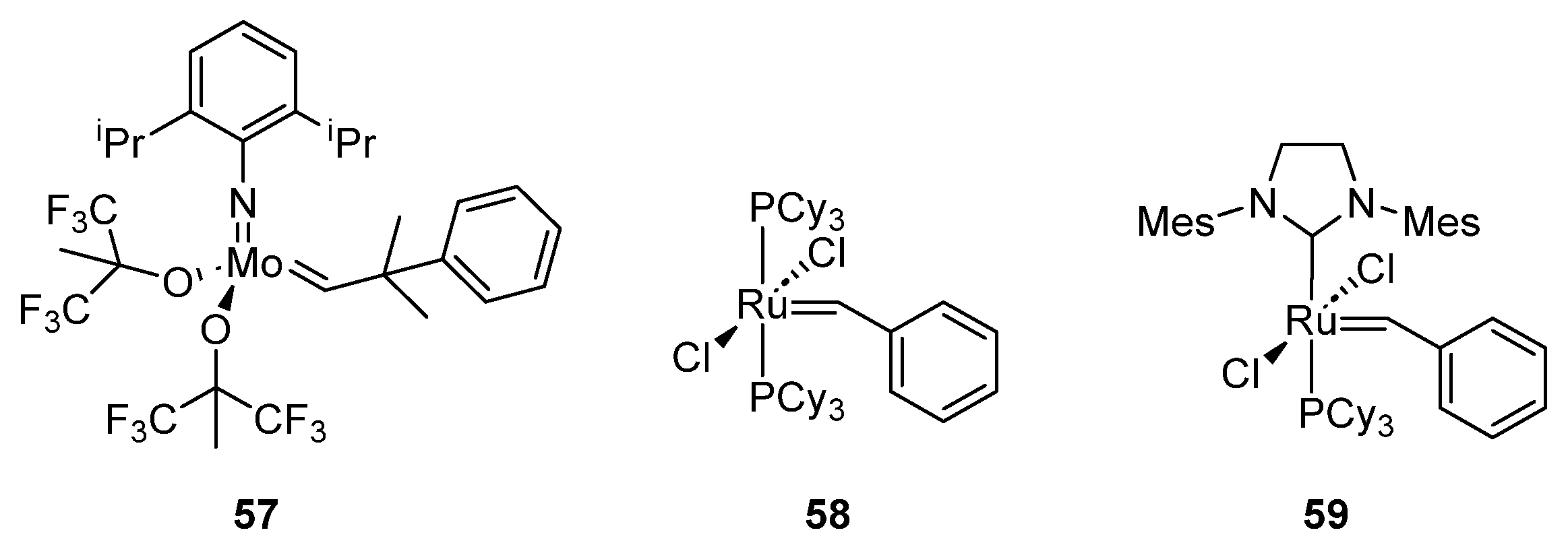
- Nguyen, S.T.; Grubbs, R.H.; Ziller, J.W. Syntheses and Activities of New Single-Component, Ruthenium-Based Olefin Metathesis Catalysts. J. Am. Chem. Soc. 1993, 115, 9858–9859. [Google Scholar] [CrossRef]
- Casey, C.P. 2005 Nobel Prize in Chemistry. Development of the Olefin Metathesis Method in Organic Synthesis. J. Chem. Educ. 2006, 83, 192. [Google Scholar] [CrossRef]
- Calderon, N.; Ofstead, E.A.; Ward, J.P.; Judy, W.A.; Scott, K.W. Olefin Metathesis. I. Acyclic Vinylenic Hydrocarbons. J. Am. Chem. Soc. 1968, 90, 4133–4140. [Google Scholar] [CrossRef]
- Ivin, K.J. Some Recent Applications of Olefin Metathesis in Organic Synthesis: A Review. J. Mol. Catal. A Chem. 1998, 133, 1–16. [Google Scholar] [CrossRef]
- Ha, M.W.; Song, B.R.; Chung, H.J.; Paek, S.-M. Design and Synthesis of Anti-Cancer Chimera Molecules Based on Marine Natural Products. Mar. Drugs 2019, 17, 500. [Google Scholar] [CrossRef]
- Paek, S.-M. Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules 2014, 19, 15982–16000. [Google Scholar] [CrossRef]
- Yun, H.; Sim, J.; An, H.; Lee, J.; Lee, H.S.; Shin, Y.K.; Paek, S.-M.; Suh, Y.-G. Design and Synthesis of a Macrosphelide A-Biotin Chimera. Org. Biomol. Chem. 2014, 12, 7127. [Google Scholar] [CrossRef] [PubMed]
- Wulff, J.E.; Siegrist, R.; Myers, A.G. The Natural Product Avrainvillamide Binds to the Oncoprotein Nucleophosmin. J. Am. Chem. Soc. 2007, 129, 14444–14451. [Google Scholar] [CrossRef] [PubMed]
- Eggen, M.; Mossman, C.J.; Buck, S.B.; Nair, S.K.; Bhat, L.; Ali, S.M.; Reiff, E.A.; Boge, T.C.; Georg, G.I. Total Synthesis of Cryptophycin-24 (Arenastatin A) Amenable to Structural Modifications in the C16 Side Chain. J. Am. Chem. Soc. 2000, 65, 7792–7799. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Arenastatin A, a Potent Cytotoxic Depsipeptide from the Okinawan Marine Sponge Dysidea Arenaria. Tetrahedron Lett. 1994, 35, 7969–7972. [Google Scholar] [CrossRef]
- Koiso, Y.; Morita, K.; Kobayashi, M.; Wang, W.; Ohyabu, N.; Iwasaki, S. Effects of Arenastatin A and Its Synthetic Analogs on Microtubule Assembly. Chem. Biol. Interact. 1996, 102, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Purnima, K.V.; Subba Reddy, B.V.; Nagaiah, K.; Ghamdi, A.K. Total Synthesis of Cryptophycin-24 (Arenastatin A) via Prins Cyclization. Tetrahedron Lett. 2011, 52, 6709–6712. [Google Scholar] [CrossRef]
- Biard, J.-F.; Roussakis, C.; Kornprobst, J.-M.; Gouiffes-Barbin, D.; Verbist, J.-F. Bistramides A, B, C, D and K: A New Class of Bioactive Cyclic Polyethers from Lissoclinum Bistratum. J. Nat. Prod. 1994, 57, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Commandeur, M.; Commandeur, C.; Cossy, J. Synthesis of a Platform To Access Bistramides and Their Analogues. Org. Lett. 2011, 13, 6018–6021. [Google Scholar] [CrossRef] [PubMed]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin Is the Primary Cellular Receptor of Bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef]
- Statsuk, A.V.; Liu, D.; Kozmin, S.A. Synthesis of Bistramide A. J. Am. Chem. Soc. 2004, 126, 9546–9547. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Sawamura, M.; Hayashi, T. Asymmetric Aldol Reaction of an Isocyanoacetate with Aldehydes Bychiral Ferrocenylphosphine-Gold(I) Complexes: Design and Preparation of New Efficient Ferrocenylphosphine Ligands. Tetrahedron Lett. 1987, 28, 6215–6218. [Google Scholar] [CrossRef]
- Nolan, S.P. Catalytic Gold Rush. Nature 2007, 445, 496–497. [Google Scholar] [CrossRef] [PubMed]
- Collado, A.; Nelson, D.J.; Nolan, S.P. Optimizing Catalyst and Reaction Conditions in Gold(I) Catalysis–Ligand Development. Chem. Rev. 2021, 121, 8559–8612. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Bochmann, M. Recent Advances in Gold(III) Chemistry: Structure, Bonding, Reactivity, and Role in Homogeneous Catalysis. Chem. Rev. 2021, 121, 8364–8451. [Google Scholar] [CrossRef] [PubMed]
- Pflästerer, D.; Hashmi, A.S.K. Gold Catalysis in Total Synthesis—Recent Achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Dan, A.K.; Sahu, R.; Parida, S.; Das, D. Total Synthesis of Natural Products Using Gold Catalysis. Chem. Asian. J. 2022, 17, e202200896. [Google Scholar] [CrossRef] [PubMed]
- Higa, T.; Tanaka, J.; Kitamura, A.; Koyama, T.; Takahashi, M.; Uchida, T. Bioactive Compounds from Marine Sponges. Pure Appl. Chem. 1994, 66, 2227–2230. [Google Scholar] [CrossRef]
- Fernández, A.; Levine, Z.G.; Baumann, M.; Sulzer-Mossé, S.; Sparr, C.; Schläger, S.; Metzger, A.; Baxendale, I.R.; Ley, S.V.L. Setyternthesis of (–)-Hennoxazole A: Integrating Batch and Flow Chemistry Methods. Synlett 2013, 24, 514–518. [Google Scholar]
- Zampella, A.; D’Auria, M.V.; Minale, L.; Debitus, C.; Roussakis, C. Callipeltoside A: A Cytotoxic Aminodeoxy Sugar-Containing Macrolide of a New Type from the Marine Lithistida Sponge Callipelta sp. J. Am. Chem. Soc. 1996, 118, 11085–11088. [Google Scholar] [CrossRef]
- Zampella, A.; D’Auria, M.V.; Minale, L.; Debitus, C. Callipeltosides B and C, Two Novel Cytotoxic Glycoside Macrolides from a Marine Lithistida Sponge Callipelta sp. Tetrahedron 1997, 53, 3243–3248. [Google Scholar] [CrossRef]
- Frost, J.R.; Pearson, C.M.; Snaddon, T.N.; Booth, R.A.; Ley, S.V. Convergent Total Syntheses of Callipeltosides A, B, and C. Angew. Chem. 2012, 124, 9500–9505. [Google Scholar] [CrossRef]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a Potent Sponge-Derived Cytotoxin from Psammocinia Distantly Related to the Pederin Family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Xu, J.-P.; Chapuis, J.-C.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.A.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge Ircinia r Amosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Jiang, X.; De Brabander, J.K. Studies toward the Unique Pederin Family Member Psymberin: Full Structure Elucidation, Two Alternative Total Syntheses, and Analogs. J. Am. Chem. Soc. 2012, 134, 17083–17093. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, F.; Simbari, F.; Abad, J.L.; Casasampere, M.; Fabrias, G.; Futerman, A.H.; Casas, J. Jaspine B Induces Nonapoptotic Cell Death in Gastric Cancer Cells Independently of Its Inhibition of Ceramide Synthase. J. Lipid Res. 2017, 58, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, V.M.; Stefani, S.; Reissig, H.-U. Stereodivergent Synthesis of Jaspine B and Its Isomers Using a Carbohydrate-Derived Alkoxyallene as C3 -Building Block. Beilstein J. Org. Chem. 2013, 9, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Iwata, F.; Mukai, T.; Yamada, S.; Takeo, J.; Abe, A.; Kawahara, H. Indoxamycins A–F. Cytotoxic Tricycklic Polypropionates from a Marine-Derived Actinomycete. J. Org. Chem. 2009, 74, 5502–5509. [Google Scholar] [CrossRef]
- Jeker, O.F.; Carreira, E.M. Total Synthesis and Stereochemical Reassignment of (±)-Indoxamycin B. Angew. Chem. Int. Ed. 2012, 51, 3474–3477. [Google Scholar] [CrossRef]
- Lee, S.M.; Li, X.F.; Jiang, H.; Cheng, J.G.; Seong, S.; Choi, H.D.; Son, B.W. Terreusinone, a Novel UV-A Protecting Dipyrroloquinone from the Marine Algicolous Fungus Aspergillus Terreus. Tetrahedron Lett. 2003, 44, 7707–7710. [Google Scholar] [CrossRef]
- Wang, C.; Sperry, J. Total Synthesis of the Photoprotecting Dipyrrolobenzoquinone (+)-Terreusinone. Org. Lett. 2011, 13, 6444–6447. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, M.; Tong, C.; Yamada, K.; Chiba, T.; Hayashi, Y.; Uemura, D. Halichlorine, an Inhibitor of VCAM-1 Induction from the Marine Sponge Halichondria Okadai Kadata. Tetrahedron Lett. 1996, 37, 3867–3870. [Google Scholar] [CrossRef]
- Zhu, D.; Zhang, Z.; Mou, X.; Tu, Y.; Zhang, F.; Peng, J.; Wang, S.; Zhang, S. Gold(I)/Copper(II)-Cocatalyzed Tandem Cyclization/Semipinacol Reaction: Construction of 6- Aza/Oxa -Spiro [4.5]Decane Skeletons and Formal Synthesis of (±)-Halichlorine. Adv. Synth. Catal. 2015, 357, 747–752. [Google Scholar] [CrossRef]
- Marín-Luna, M.; Nieto Faza, O.; Silva López, C. Gold-Catalyzed Homogeneous (Cyclo)Isomerization Reactions. Front. Chem. 2019, 7, 296. [Google Scholar] [CrossRef] [PubMed]
- Amirzakariya, B.Z.; Shakeri, A. Bioactive Terpenoids Derived from Plant Endophytic Fungi: An Updated Review (2011–2020). Phytochemistry 2022, 197, 113130. [Google Scholar] [CrossRef] [PubMed]
- Hönig, M.; Carreira, E.M. Total Synthesis and Structural Revision of a Harziane Diterpenoid. Angew. Chem. Int. Ed. 2020, 59, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Wang, Z.; Zhang, Z.; Huang, J.; Yang, Z. Synthetic Strategy for Construction of Highly Congested Tetracyclic Core (6–5–7–4) of Harziane Diterpenoids. Org. Lett. 2021, 23, 4088–4093. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Tsuda, M. Amphidinolides, Bioactive Macrolides from Symbiotic Marine Dinoflagellates. Nat. Prod. Rep. 2004, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, I.; Lee, D. Asymmetric Total Synthesis of (−)-Amphidinolide V through Effective Combinations of Catalytic Transformations. J. Am. Chem. Soc. 2013, 135, 5324–5327. [Google Scholar] [CrossRef]
- Gribble, G. Biological Activity of Recently Discovered Halogenated Marine Natural Products. Mar. Drugs 2015, 13, 4044–4136. [Google Scholar] [CrossRef]
- Brandstätter, M.; Freis, M.; Huwyler, N.; Carreira, E.M. Total Synthesis of (−)-Merochlorin A. Angew. Chem. 2019, 131, 2512–2516. [Google Scholar] [CrossRef]
- Yu, B. Gold(I)-Catalyzed Glycosylation with Glycosyl o -Alkynylbenzoates as Donors. Acc. Chem. Res. 2018, 51, 507–516. [Google Scholar] [CrossRef]
- Xiao, G.; Yu, B. Total Synthesis of Starfish Saponin Goniopectenoside B. Chem. Eur. J. 2013, 19, 7708–7712. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.D.; Iorizzi, M.; Zollo, F.; Amsler, C.D.; Greer, S.P.; McClintock, J.B. Three New Asterosaponins from the Starfish Goniopecten demonstrans. Eur. J. Org. Chem. 2000, 2000, 4093–4098. [Google Scholar] [CrossRef]
- Kobayashi, S.; Hachiya, I.; Araki, M.; Ishitani, H. Scandium Trifluoromethanesulfonate (Sc(OTf)3). A Novel Reusable Catalyst in the Diels-Alder Reaction. Tetrahedron Lett. 1993, 34, 3755–3758. [Google Scholar] [CrossRef]
- Brennan, J.G.; Sella, A. Scandium, Yttrium and the Lanthanides. In Organometallic Chemistry; Fairlamb, I.J.S., Lynam, J.M., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2008; Volume 34, pp. 111–154. [Google Scholar]
- Pellissier, H. Recent Developments in Enantioselective Scandium–Catalyzed Transformations. Coord. Chem. Rev. 2016, 313, 1–37. [Google Scholar] [CrossRef]
- Custar, D.W.; Zabawa, T.P.; Hines, J.; Crews, C.M.; Scheidt, K.A. Total Synthesis and Structure−Activity Investigation of the Marine Natural Product Neopeltolide. J. Am. Chem. Soc. 2009, 131, 12406–12414. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a Macrolide from a Lithistid Sponge of the Family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.J.; Farmer, T.J.; Clark, J.H. Elemental Sustainability and the Importance of Scarce Element Recovery. In Element Recovery and Sustainability; Hunt, A., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2013; Volume 22, pp. 1–28. [Google Scholar]
- Egorova, K.S.; Ananikov, V.P. Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef]
- Collins, R.A.; Russell, A.F.; Mountford, P. Group 4 Metal Complexes for Homogeneous Olefin Polymerisation: A Short Tutorial Review. Appl. Petrochem. Res. 2015, 5, 153–171. [Google Scholar] [CrossRef]
- Sidambe, A. Biocompatibility of Advanced Manufactured Titanium Implants—A Review. Materials 2014, 7, 8168–8188. [Google Scholar] [CrossRef] [PubMed]
- Manßen, M.; Schafer, L.L. Titanium Catalysis for the Synthesis of Fine Chemicals—Development and Trends. Chem. Soc. Rev. 2020, 49, 6947–6994. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, T.; Narasaka, K.; Banno, K. New aldol type reaction. Chem. Lett. 1973, 2, 1011–1014. [Google Scholar] [CrossRef]
- Ramón, D.J.; Yus, M. In the Arena of Enantioselective Synthesis, Titanium Complexes Wear the Laurel Wreath. Chem. Rev. 2006, 106, 2126–2208. [Google Scholar] [CrossRef] [PubMed]
- Deems, R.A.; Lombardo, D.; Morgan, B.P.; Mihelich, E.D.; Dennis, E.A. The Inhibition of Phospholipase A2 by Manoalide and Manoalide Analogues. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1987, 917, 258–268. [Google Scholar] [CrossRef]
- de Silva, E.D.; Scheuer, P.J. Manoalide, an Antibiotic Sesterterpenoid from the Marine Sponge (Polejaeff). Tetrahedron Lett. 1980, 21, 1611–1614. [Google Scholar] [CrossRef]
- Grange, E.; Rabin, O.; Bell, J.; Chang, M.C.J. Manoalide, a Phospholipase A2 Inhibitor, Inhibits Arachidonate Incorporation and Turnover in Brain Phospholipids of the Awake Rat. Neurochem. Res. 1998, 23, 1251–1257. [Google Scholar] [CrossRef]
- Salam, K.A.; Furuta, A.; Noda, N.; Tsuneda, S.; Sekiguchi, Y.; Yamashita, A.; Moriishi, K.; Nakakoshi, M.; Tsubuki, M.; Tani, H.; et al. Inhibition of Hepatitis C Virus NS3 Helicase by Manoalide. J. Nat. Prod. 2012, 75, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Soriente, A.; De Rosa, M.; Apicella, A.; Scettri, A.; Sodano, G. First Enantioselective Synthesis of Manoalide: Application of Aldehyde–Dioxinone Enantioselective Condensation. Tetrahedron Asymmetry 1999, 10, 4481–4484. [Google Scholar] [CrossRef]
- Davis-Gilbert, Z.W.; Tonks, I.A. Titanium Redox Catalysis: Insights and Applications of an Earth-Abundant Base Metal. J. Chem. Soc. Dalton Trans. 2017, 46, 11522–11528. [Google Scholar] [CrossRef]
- Beaumier, E.P.; Pearce, A.J.; See, X.Y.; Tonks, I.A. Modern Applications of Low-Valent Early Transition Metals in Synthesis and Catalysis. Nat. Rev. Chem. 2018, 3, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Tonks, I.A. Ti-Catalyzed and -Mediated Oxidative Amination Reactions. Acc. Chem. Res. 2021, 54, 3476–3490. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Dunn, P.J.; Hayler, J.D.; Humphrey, G.R.; Leazer, J.L., Jr.; Linderman, R.J.; Lorenz, K.; Manley, J.; Pearlman, B.A.; Wells, A.; et al. Key Green Chemistry Research Areas—A Perspective from Pharmaceutical Manufacturers. Green Chem. 2007, 9, 411–420. [Google Scholar] [CrossRef]
- Chiu, H.C.; Tonks, I.A. Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem. Int. Ed. 2018, 57, 6090–6094. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Hobbs, L.; Hooper, J.; Capon, R. Lamellarins Q and R: New Aromatic Metabolites From an Australian Marine Sponge, Dendrilla Cactos. Aust. J. Chem. 1995, 48, 1491. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer Properties of Lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, J.; Fan, A.; Cui, Y.; Jia, Y. Total Synthesis of Lamellarins D, H, and R and Ningalin B. Org. Lett. 2011, 13, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Knölker, H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [PubMed]
- Czaplik, W.M.; Mayer, M.; Cvengroš, J.; von Wangelin, A.J. Coming of Age: Sustainable Iron-Catalyzed Cross-Coupling Reactions. ChemSusChem 2009, 2, 396–417. [Google Scholar] [CrossRef]
- Tamura, M.; Kochi, J.K. Vinylation of Grignard Reagents. Catalysis by Iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. [Google Scholar] [CrossRef]
- Corriu, R.J.P.; Masse, J.P. Activation of Grignard Reagents by Transition-Metal Complexes. A New and Simple Synthesis of Trans-Stilbenes and Polyphenyls. J. Chem. Soc. Chem. Commun. 1972, 3, 144. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective Carbon-Carbon Bond Formation by Cross-Coupling of Grignard Reagents with Organic Halides. Catalysis by Nickel-Phosphine Complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Alam, N.; Hong, J.; Lee, C.O.; Im, K.S.; Son, B.W.; Choi, J.S.; Choi, W.C.; Jung, J.H. Montipyridine, a New Pyridinium Alkaloid from the Stony Coral Montipora Species. J. Nat. Prod. 2001, 64, 956–957. [Google Scholar] [CrossRef]
- Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. Iron-Catalyzed Cross-Coupling Reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Fields, E.K. Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides. J. Am. Chem. Soc. 1941, 63, 2316–2320. [Google Scholar] [CrossRef]
- Borthakur, I.; Sau, A.; Kundu, S. Cobalt-Catalyzed Dehydrogenative Functionalization of Alcohols: Progress and Future Prospect. Coord. Chem. Rev. 2022, 451, 214257. [Google Scholar] [CrossRef]
- Lukasevics, L.; Cizikovs, A.; Grigorjeva, L. C–H Bond Functionalization by High-Valent Cobalt Catalysis: Current Progress, Challenges and Future Perspectives. Chem. Commun. 2021, 57, 10827–10841. [Google Scholar] [CrossRef]
- Kojima, M.; Matsunaga, S. The Merger of Photoredox and Cobalt Catalysis. Trends Chem. 2020, 2, 410–426. [Google Scholar] [CrossRef]
- Gao, K.; Yoshikai, N. Low-Valent Cobalt Catalysis: New Opportunities for C–H Functionalization. Acc. Chem. Res. 2014, 47, 1208–1219. [Google Scholar] [CrossRef]
- Kyne, S.H.; Lefèvre, G.; Ollivier, C.; Petit, M.; Ramis Cladera, V.-A.; Fensterbank, L. Iron and Cobalt Catalysis: New Perspectives in Synthetic Radical Chemistry. Chem. Soc. Rev. 2020, 49, 8501–8542. [Google Scholar] [CrossRef]
- Yang, Z. Navigating the Pauson–Khand Reaction in Total Syntheses of Complex Natural Products. Acc. Chem. Res. 2021, 54, 556–568. [Google Scholar] [CrossRef]
- Momeni, T.; Zadsirjan, V.; Hadi Meshkatalsadat, M.; Pourmohammadi-Mahunaki, M. Applications of Cobalt-Catalyzed Reactions in the Total Synthesis of Natural Products. Chem. Sel. 2022, 7, e20220281. [Google Scholar] [CrossRef]
- Nazari, M.; Serrill, J.D.; Sikorska, J.; Ye, T.; Ishmael, J.E.; McPhail, K.L. Discovery of Mandelalide E and Determinants of Cytotoxicity for the Mandelalide Series. Org. Lett. 2016, 18, 1374–1377. [Google Scholar] [CrossRef]
- Mattos, D.; Wan, X.; Serrill, J.; Nguyen, M.; Humphreys, I.; Viollet, B.; Smith, A.; McPhail, K.; Ishmael, J. The Marine-Derived Macrolactone Mandelalide A is an indirect activator of AMPK. Mar. Drugs 2022, 20, 418. [Google Scholar] [CrossRef]
- Willwacher, J.; Fürstner, A. Catalysis-Based Total Synthesis of Putative Mandelalide A. Angew. Chem. 2014, 126, 4301–4305. [Google Scholar] [CrossRef]
- Kubota, T.; Tsuda, M.; Kobayashi, J. Absolute Stereochemistry of Amphidinolide E. J. Org. Chem. 2002, 67, 1651–1656. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kubota, T. Bioactive Macrolides and Polyketides from Marine Dinoflagellates of the Genus Amphidinium. J. Nat. Prod. 2007, 70, 451–460. [Google Scholar] [CrossRef]
- Morra, N.A.; Pagenkopf, B.L. Gram Scale Synthesis of the C(1)–C(9) Fragment of Amphidinolide C. Tetrahedron 2013, 69, 8632–8644. [Google Scholar] [CrossRef]
- Williams, D.R.; Meyer, K.G. Total Synthesis of (+)-Amphidinolide K. J. Am. Chem. Soc. 2001, 123, 765–766. [Google Scholar] [CrossRef]
- Ananikov, V.P. Nickel: The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. [Google Scholar] [CrossRef]
- Gao, R.; Sun, W.-H.; Redshaw, C. Nickel Complex Pre-Catalysts in Ethylene Polymerization: New Approaches to Elastomeric Materials. Catal. Sci. Technol. 2013, 3, 1172. [Google Scholar] [CrossRef]
- Lavoie, C.M.; MacQueen, P.M.; Rotta-Loria, N.L.; Sawatzky, R.S.; Borzenko, A.; Chisholm, A.J.; Hargreaves, B.K.V.; McDonald, R.; Ferguson, M.J.; Stradiotto, M. Challenging Nickel-Catalysed Amine Arylations Enabled by Tailored Ancillary Ligand Design. Nat. Commun. 2016, 7, 11073. [Google Scholar] [CrossRef]
- Pan, Q.; Ping, Y.; Kong, W. Nickel-Catalyzed Ligand-Controlled Selective Reductive Cyclization/Cross-Couplings. Acc. Chem. Res. 2023, 56, 515–535. [Google Scholar] [CrossRef]
- Tamao, K.; Kodama, S.; Nakajima, I.; Kumada, M.; Minato, A.; Suzuki, K. Nickel-Phosphine Complex-Catalyzed Grignard Coupling—II. Tetrahedron 1982, 38, 3347–3354. [Google Scholar] [CrossRef]
- Netherton, M.R.; Fu, G.C. Nickel-Catalyzed Cross-Couplings of Unactivated Alkyl Halides and Pseudohalides with Organometallic Compounds. Adv. Synth. Catal. 2004, 346, 1525–1532. [Google Scholar] [CrossRef]
- Frisch, A.C.; Beller, M. Catalysts for Cross-Coupling Reactions with Non-activated Alkyl Halides. Angew. Chem. Int. Ed. 2005, 44, 674–688. [Google Scholar] [CrossRef]
- Glorius, F. Asymmetric Cross-Coupling of Non-Activated Secondary Alkyl Halides. Angew. Chem. Inter. Ed. 2008, 47, 8347–8349. [Google Scholar] [CrossRef]
- Hu, X. Nickel-Catalyzed Cross Coupling of Non-Activated Alkyl Halides: A Mechanistic Perspective. Chem. Sci. 2011, 2, 1867. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C−S, C−Se, and C−Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef]
- Park, N.H.; Teverovskiy, G.; Buchwald, S.L. Development of an Air-Stable Nickel Precatalyst for the Amination of Aryl Chlorides, Sulfamates, Mesylates, and Triflates. Org. Lett. 2014, 16, 220–223. [Google Scholar] [CrossRef]
- Tasker, S.Z.; Gutierrez, A.C.; Jamison, T.F. Nickel-Catalyzed Mizoroki–Heck Reaction of Aryl Sulfonates and Chlorides with Electronically Unbiased Terminal Olefins: High Selectivity for Branched Products. Angew. Chem. Inter. Ed. 2014, 53, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Ghosh, T. Emerging Nickel Catalysis in Heck Reactions: Recent Developments. Adv. Synth. Catal. 2020, 362, 5257–5274. [Google Scholar] [CrossRef]
- Huang, D.; Newhouse, T.R. Dehydrogenative Pd and Ni Catalysis for Total Synthesis. Acc. Chem. Res. 2021, 54, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, J. Nickel-Catalyzed Reductive Cyclizations and Couplings. Angew. Chem. Int. Ed. 2004, 43, 3890–3908. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-K.; Cha, B.-Y.; Fujiwara, T.; Kanamoto, A.; Woo, J.-T.; Ojika, M.; Imokawa, G. Arenarol Isolated from a Marine Sponge Abrogates Endothelin-1-Stimulated Melanogenesis by Interrupting MEK Phosphorylation in Normal Human Melanocytes. Cytotechnology 2013, 65, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.T.; Park, K.; Wiemer, D.F.; Scott, W.J. Application of the Nickel-Mediated Neopentyl Coupling in the Total Synthesis of the Marine Natural Product Arenarol. J. Org. Chem. 1995, 60, 5102–5106. [Google Scholar] [CrossRef]
- Corey, E.J.; Semmelhack, M.F. Organonickel Compounds as Reagents for Selective Carbon-Carbon Bond Formation between Unlike Groups. J. Am. Chem. Soc. 1967, 89, 2755–2757. [Google Scholar] [CrossRef]
- Johnson, C.R.; Dutra, G.A. Reactions of Lithium Diorganocuprates(I) with Tosylates. I. Synthetic Aspects. J. Am. Chem. Soc. 1973, 95, 7777–7782. [Google Scholar] [CrossRef]
- Capon, R.J.; Macleod, J.K.; Scammells, P.J. The Trikentrins: Novel Indoles from the Sponge. Tetrahedron 1986, 42, 6545–6550. [Google Scholar] [CrossRef]
- Macleod, J.; Monahan, L. The Total Synthesis of (±)-Cis-Trans-Trikentrin A. Aust. J. Chem. 1990, 43, 329. [Google Scholar] [CrossRef]
- MacLeod, J.K.; Ward, A.; Willis, A.C. Total Synthesis of (±)-Iso-Trans-Trikentrin B. Aust. J. Chem. 1998, 51, 177. [Google Scholar] [CrossRef]
- Arp, F.O.; Fu, G.C. Catalytic Enantioselective Negishi Reactions of Racemic Secondary Benzylic Halides. J. Am. Chem. Soc. 2005, 127, 10482–10483. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, F.; Bielecki, J. Ueber Synthesen in Der Biphenylreihe. Ber. Dtsch. Chem. Ges. 1901, 34, 2174–2185. [Google Scholar] [CrossRef]
- Kochi, J.K.; Tamura, M. Alkylcopper(I) in the Coupling of Grignard Reagents with Alkyl Halides. J. Am. Chem. Soc. 1971, 93, 1485–1487. [Google Scholar] [CrossRef]
- Evano, G.; Theunissen, C.; Pradal, A. Impact of Copper-Catalyzed Cross-Coupling Reactions in Natural Product Synthesis: The Emergence of New Retrosynthetic Paradigms. Nat. Prod. Rep. 2013, 30, 1467. [Google Scholar] [CrossRef] [PubMed]
- Tlili, A.; Taillefer, M. Ullmann Condensation Today: Arylation of Alcohols and Thiols with Aryl Halides. In Copper-Mediated Cross-Coupling Reactions; Evano, G., Blanchard, N., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 41–91. [Google Scholar]
- Monnier, F.; Taillefer, M. Catalytic C-C, C-N, and C-O Ullmann-Type Coupling Reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef]
- Cheng, L.-J.; Mankad, N.P. C–C and C–X Coupling Reactions of Unactivated Alkyl Electrophiles Using Copper Catalysis. Chem. Soc. Rev. 2020, 49, 8036–8064. [Google Scholar] [CrossRef]
- Kobayashi, J.; Ishibashi, M.; Nakamura, H.; Ohizumi, Y.; Yamasu, T.; Sasaki, T.; Hirata, Y. Amphidinolide-A, a Novel Antineoplastic Macrolide from the Marine Dinoflagellate Amphidinium sp. Tetrahedron Lett. 1986, 27, 5755–5758. [Google Scholar] [CrossRef]
- Maleczka, R.E.; Robert, E.; Terrell, L.R.; Geng, F.; Ward, J.S. Total Synthesis of Proposed Amphidinolide A via a Highly Selective Ring-Closing Metathesis. Org. Lett. 2002, 4, 2841–2844. [Google Scholar] [CrossRef]
- Wu, X.; Neumann, H. Zinc-Catalyzed Organic Synthesis: C-C, C-N, C-O Bond Formation Reactions. Adv. Synth. Catal. 2012, 354, 3141–3160. [Google Scholar] [CrossRef]
- González, M.J.; López, L.A.; Vicente, R. Zinc Reagents as Non-Noble Catalysts for Alkyne Activation. Tetrahedron Lett. 2015, 56, 1600–1608. [Google Scholar] [CrossRef]
- Reber, S.; Knöpfel, T.F.; Carreira, E.M. Enantioselective Total Synthesis of (R)-Strongylodiols A and B. Tetrahedron 2003, 59, 6813–6817. [Google Scholar] [CrossRef]
- Chinkov, N.; Warm, A.; Carreira, E.M. Asymmetric Autocatalysis Enables an Improved Synthesis of Efavirenz. Angew. Chem. Int. Ed. 2011, 50, 2957–2961. [Google Scholar] [CrossRef]
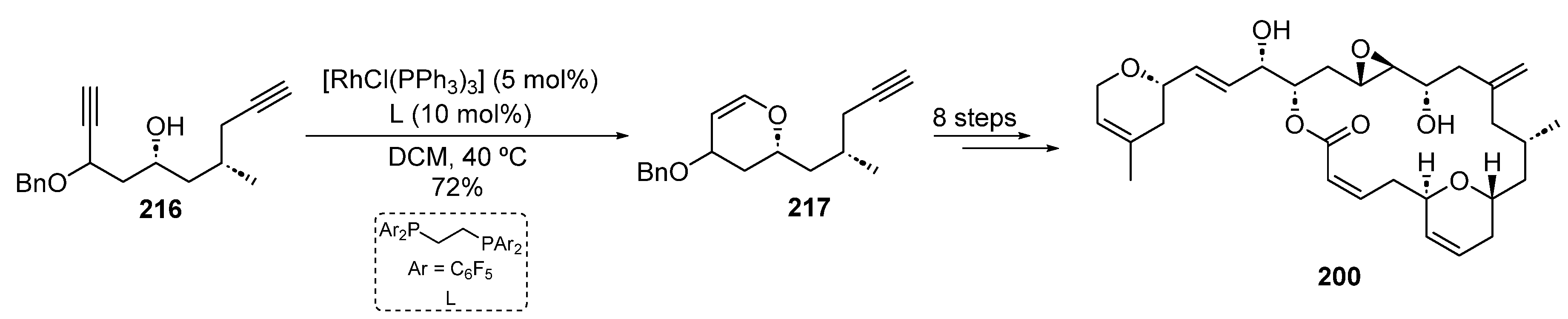
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and Isolaulimalide, New Paclitaxel-Like Microtubule-Stabilizing Agents. Cancer Res. 1999, 59, 653–660. [Google Scholar]
- Trost, B.M.; Amans, D.; Seganish, W.M.; Chung, C.K. Evaluating Transition-Metal-Catalyzed Transformations for the Synthesis of Laulimalide. J. Am. Chem. Soc. 2009, 131, 17087–17089. [Google Scholar] [CrossRef]
- Khurana, J.M.; Chauhan, S.; Agrawal, A. Molybdenum in organic synthesis. A review. Org. Prep. Proced. Int. 2004, 36, 201–276. [Google Scholar] [CrossRef]
- Khusnutdinov, R.I.; Oshnyakova, T.M.; Dzhemilev, U.M. Molybdenum Compounds in Organic Synthesis. Russ. Chem. Rev. 2017, 86, 128–163. [Google Scholar] [CrossRef]
- Zhang, W.; Moore, J.S. Alkyne Metathesis: Catalysts and Synthetic Applications. Adv. Synth. Catal. 2007, 349, 93–120. [Google Scholar] [CrossRef]
- Wölfl, B.; Mata, G.; Fürstner, A. Total Synthesis of Callyspongiolide, Part 2: The Ynoate Metathesis/Cis -Reduction Strategy. Chem. Eur. J. 2019, 25, 255–259. [Google Scholar] [CrossRef]
- Ha, J.; Park, S.B. Callyspongiolide Kills Cells by Inducing Mitochondrial Dysfunction via Cellular Iron Depletion. Commun. Biol. 2021, 4, 1123. [Google Scholar] [CrossRef]
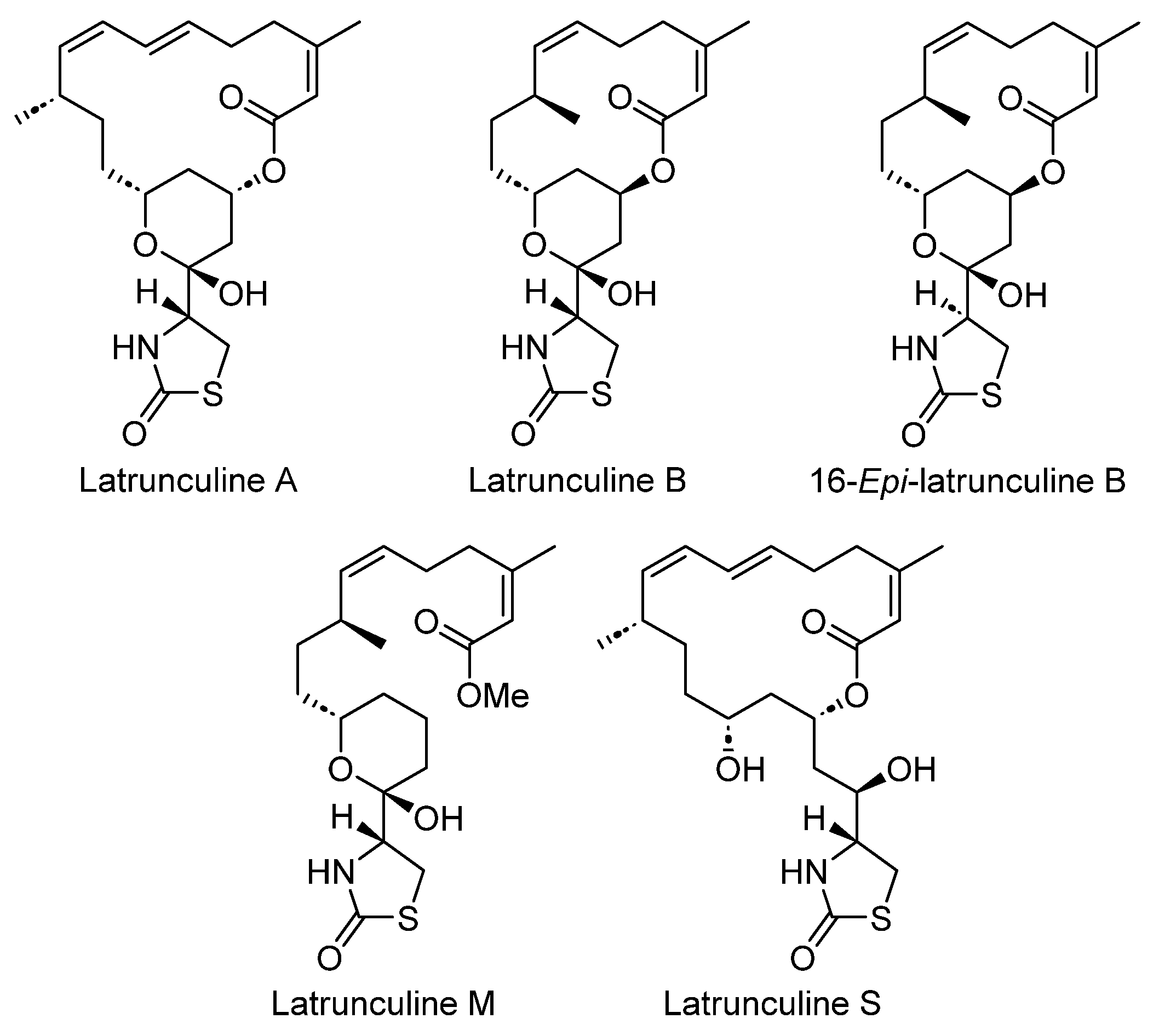
- Khalifa, S.; Ahmed, S.; Mesbah, M.; Youssef, D.; Hamann, M. Quantitative Determination of Latrunculins A and B in the Red Sea Sponge Negombata Magnifica by High Performance Liquid Chromatography. J. Chromatogr. B 2006, 832, 47–51. [Google Scholar] [CrossRef]
- Jin, Z. Muscarine, Imidazole, Oxazole and Thiazole Alkaloids. Nat. Prod. Rep. 2009, 26, 382. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Sun, Y.; Wang, W.; Song, X.; Zhang, D. Chemical Diversity and Biological Activities of Marine-Derived Sulphur Containing Alkaloids: A Comprehensive Update. Arab. J. Chem. 2023, 16, 105011. [Google Scholar] [CrossRef]
- Varlet, T.; Portmann, S.; Fürstner, A. Total Synthesis of Njaoamine C by Concurrent Macrocycle Formation. J. Am. Chem. Soc. 2023, 145, 21197–21202. [Google Scholar] [CrossRef]
- Reyes, F.; Fernández, R.; Urda, C.; Francesch, A.; Bueno, S.; de Eguilior, C.; Cuevas, C. Njaoamines A–F, New Cytotoxic Polycyclic Alkaloids from the Haplosclerid Sponge Reniera sp. Tetrahedron 2007, 63, 2432–2438. [Google Scholar] [CrossRef]
- Osborn, J.A.; Wilkinson, G.; Mrowca, J.J. Tris(Triphenylphosphine)Halorhodium(I). In Inorganic Syntheses; Muetterties, E.L., Ed.; Mc-Graw Hill: New York, NY, USA, 1967; Volume 10, pp. 67–71. [Google Scholar]
- Blieck, R.; Taillefer, M.; Monnier, F. Metal-Catalyzed Intermolecular Hydrofunctionalization of Allenes: Easy Access to Allylic Structures via the Selective Formation of C–N, C–C, and C–O Bonds. Chem. Rev. 2020, 120, 13545–13598. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Nájera, C.; Yus, M. Catalysis and Regioselectivity in Hydrofunctionalization Reactions of Unsaturated Carbon Bonds. Part I. Russ. Chem. Rev. 2020, 89, 250–274. [Google Scholar] [CrossRef]
- Chen, Z.; Dong, V.M. Rhodium(I)-Catalyzed Hydroformylation and Hydroamination. In Rhodium Catalysis in Organic Synthesis; Tanaka, K., Ed.; Wiley: Weinheim, Germany, 2019; pp. 49–62. [Google Scholar]
- Stoffels, M.A.; Klauck, F.J.R.; Hamadi, T.; Glorius, F.; Leker, J. Technology Trends of Catalysts in Hydrogenation Reactions: A Patent Landscape Analysis. Adv. Synth. Catal. 2020, 362, 1258–1274. [Google Scholar] [CrossRef]
- Dickson, R.S. Oxidation Reactions. In Homogeneous Catalysis with Compounds of Rhodium and Iridium; Dickson, R.S., Ed.; Springer: Dordrecht, The Netherlands, 1985; Volume 8, pp. 159–167. [Google Scholar]
- Fujiwara, M.; Ojima, I. Rhodium(I)-Catalyzed Cycloisomerization and Cyclotrimerization Reactions. In Modern Rhodium-Catalyzed Organic Reactions; Evans, P.A., Ed.; Wiley-VCH: Weinheim, Germany, 2005; pp. 129–149. [Google Scholar]
- Burnie, A.J.; Evans, P.A. Recent Developments in Rhodium-Catalyzed Cyclocarbonylation Reactions. In Rhodium Catalysis; Claver, C., Ed.; Springer International Publishing: Cham, Switherland, 2018; Volume 61, pp. 167–230. [Google Scholar]
- Hayashi, T.; Yamasaki, K. Rhodium-Catalyzed Asymmetric 1,4-Addition and Its Related Asymmetric Reactions. Chem. Rev. 2003, 103, 2829–2844. [Google Scholar] [CrossRef]
- Jean, M.; Casanova, B.; Gnoatto, S.; van de Weghe, P. Strategy of Total Synthesis Based on the Use of Rh-Catalyzed Stereoselective 1,4-Addition. Org. Biomol. Chem. 2015, 13, 9168–9175. [Google Scholar] [CrossRef]
- Koschker, P.; Breit, B. Branching Out: Rhodium-Catalyzed Allylation with Alkynes and Allenes. Acc. Chem. Res. 2016, 49, 1524–1536. [Google Scholar] [CrossRef]
- Turnbull, B.W.H.; Evans, P.A. Asymmetric Rhodium-Catalyzed Allylic Substitution Reactions: Discovery, Development and Applications to Target-Directed Synthesis. J. Org. Chem. 2018, 83, 11463–11479. [Google Scholar] [CrossRef]
- Panahi, F.; Bauer, F.; Breit, B. Rhodium-Catalyzed Allylic Addition as an Atom-Efficient Approach in Total Synthesis. Acc. Chem. Res. 2023, 56, 3676–3693. [Google Scholar] [CrossRef]
- Colby, D.A.; Bergman, R.G.; Ellman, J.A. Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation. Chem. Rev. 2010, 110, 624–655. [Google Scholar] [CrossRef]
- Brady, P.B.; Bhat, V. Recent Applications of Rh- and Pd-Catalyzed C(sp3)–H Functionalization in Natural Product Total Synthesis. Eur. J. Org. Chem. 2017, 2017, 5179–5190. [Google Scholar] [CrossRef]
- Padwa, A. Domino Reactions of Rhodium(II) Carbenoids for Alkaloid Synthesis. Chem. Soc. Rev. 2009, 38, 3072. [Google Scholar] [CrossRef]
- Li, Y.; Yang, H.; Zhai, H. The Expanding Utility of Rhodium-Iminocarbenes: Recent Advances in the Synthesis of Natural Products and Related Scaffolds. Chem. Eur. J. 2018, 24, 12757–12766. [Google Scholar] [CrossRef]
- Zhu, W.; Gunnoe, T.B. Advances in Rhodium-Catalyzed Oxidative Arene Alkenylation. Acc. Chem. Res. 2020, 53, 920–936. [Google Scholar] [CrossRef]
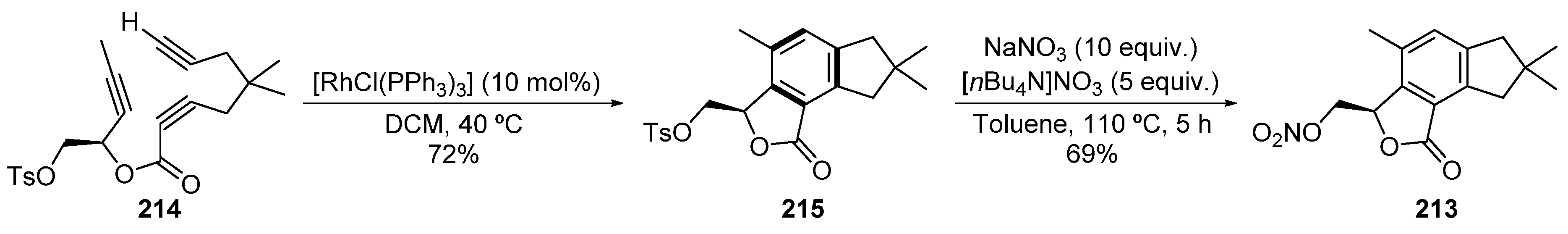
- Witulski, B.; Zimmermann, A.; Gowans, N.D. First total synthesis of the marine illudalane sesquiterpenoid Alcyopterosin E. Chem. Commun. 2002, 24, 2984–2985. [Google Scholar] [CrossRef]
- Palermo, J.A.; Rodríguez Brasco, M.F.; Spagnuolo, C.; Seldes, A.M. Illudalane Sesquiterpenoids from the Soft Coral Alcyonium Paessleri: The First Natural Nitrate Esters. J. Org. Chem. 2000, 65, 4482–4486. [Google Scholar] [CrossRef]
- Finkielsztein, L.M.; Bruno, A.M.; Renou, S.G.; Iglesias, G.Y.M. Design, Synthesis, and Biological Evaluation of Alcyopterosin A and Illudalane Derivatives as Anticancer Agents. Bioorg. Med. Chem. 2006, 14, 1863–1870. [Google Scholar] [CrossRef]
- Limon, A.-C.D.; Patabendige, H.M.L.W.; Azhari, A.; Sun, X.; Kyle, D.E.; Wilson, N.G.; Baker, B.J. Chemistry and Bioactivity of the Deep-Water Antarctic Octocoral Alcyonium sp. Mar. Drugs 2022, 20, 576. [Google Scholar] [CrossRef]
- Trost, B.M.; Amans, D.; Seganish, W.M.; Chung, C.K. Total Synthesis of Laulimalide: Assembly of the Fragments and Completion of the Synthesis of the Natural Product and a Potent Analogue. Chem. Eur. J. 2012, 18, 2961–2971. [Google Scholar] [CrossRef]
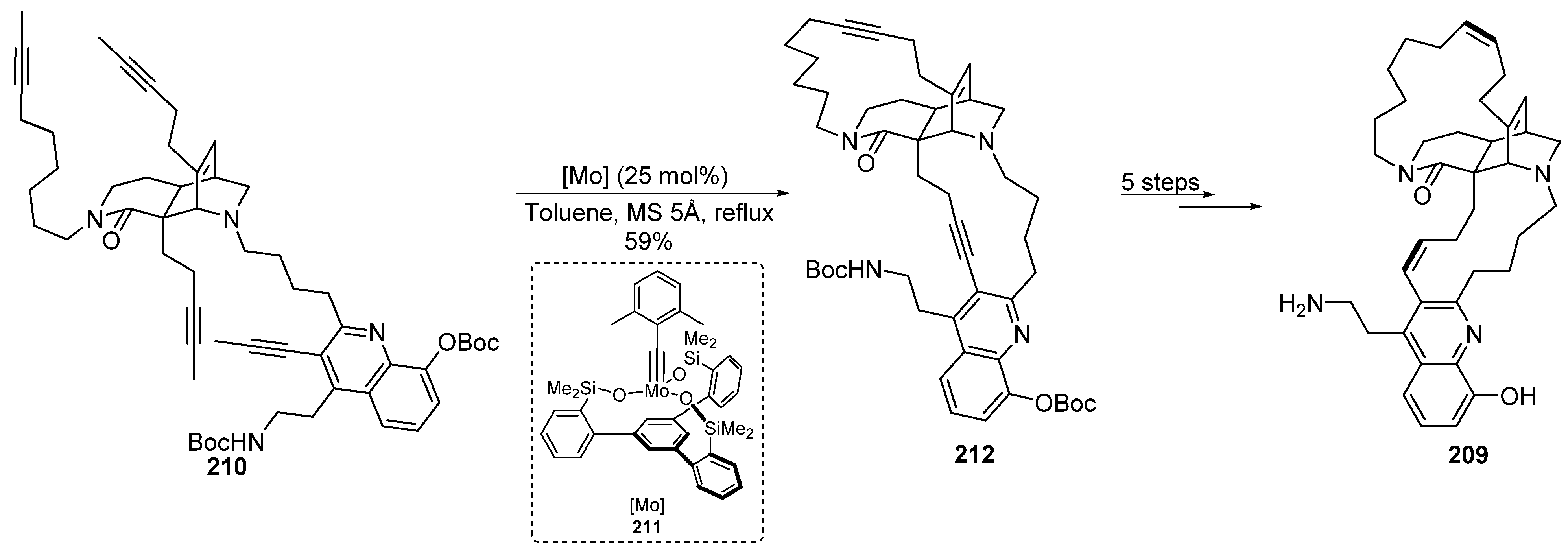
- Crabtree, R.H. Introduction and History. In Iridium Catalysis. Topics in Organometallic Chemistry; Andersson, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 34, pp. 1–10. [Google Scholar]
- Yuan, C.; Liu, B. Total Synthesis of Natural Products via Iridium Catalysis. Org. Chem. Front. 2018, 5, 106–131. [Google Scholar] [CrossRef]
- Hayashi, H. Bioactive Alkaloids of Fungal Origin. In Studies of Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Karachi, Pakistan, 2005; Volume 32, Part L; pp. 549–609. [Google Scholar]
- Liang, X.; Zhang, T.-Y.; Zeng, X.-Y.; Zheng, Y.; Wei, K.; Yang, Y.-R. Ir-Catalyzed Asymmetric Total Synthesis of (−)-Communesin F. J. Am. Chem. Soc. 2017, 139, 3364–3367. [Google Scholar] [CrossRef]
- Cipres, A.; O’Malley, D.P.; Li, K.; Finlay, D.; Baran, P.S.; Vuori, K. Sceptrin, a Marine Natural Compound, Inhibits Cell Motility in a Variety of Cancer Cell Lines. ACS Chem. Biol. 2010, 5, 195–202. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Jamison, T.F. Total Synthesis of (±)-Sceptrin. Org. Lett. 2020, 22, 6698–6702. [Google Scholar] [CrossRef]
- Chen, G.; Wang, H.F.; Pei, Y.H. Secondary Metabolites from Marine-Derived Microorganisms. J. Asian Nat. Prod. Res. 2014, 16, 105–122. [Google Scholar] [CrossRef]
- Zhang, P.P.; Yan, Z.M.; Li, Y.H.; Gong, J.X.; Yang, Z. Enantioselective Total Synthesis of (−)-Pavidolide B. J. Am. Chem. Soc. 2017, 139, 13989–13992. [Google Scholar] [CrossRef]
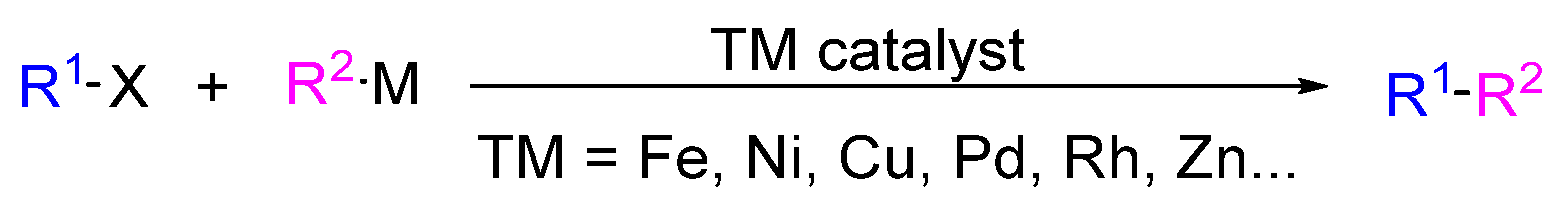













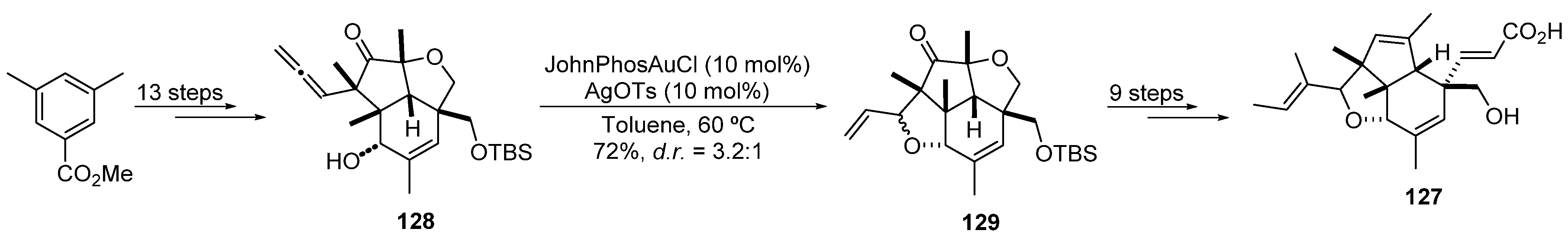











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Type | Scheme | |
|---|---|---|
| 1 | CM 1 |  |
| 2 | RCM 2 |  |
| 3 | ROCM 3 |  |
| 4 | ADMEP 4 |  |
| 5 | ROMP 5 |  |
| 6 | RCAM 6 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parte, L.G.; Fernández, S.; Sandonís, E.; Guerra, J.; López, E. Transition-Metal-Catalyzed Transformations for the Synthesis of Marine Drugs. Mar. Drugs 2024, 22, 253. https://doi.org/10.3390/md22060253
Parte LG, Fernández S, Sandonís E, Guerra J, López E. Transition-Metal-Catalyzed Transformations for the Synthesis of Marine Drugs. Marine Drugs. 2024; 22(6):253. https://doi.org/10.3390/md22060253
Chicago/Turabian StyleParte, Lucía G., Sergio Fernández, Eva Sandonís, Javier Guerra, and Enol López. 2024. "Transition-Metal-Catalyzed Transformations for the Synthesis of Marine Drugs" Marine Drugs 22, no. 6: 253. https://doi.org/10.3390/md22060253
APA StyleParte, L. G., Fernández, S., Sandonís, E., Guerra, J., & López, E. (2024). Transition-Metal-Catalyzed Transformations for the Synthesis of Marine Drugs. Marine Drugs, 22(6), 253. https://doi.org/10.3390/md22060253