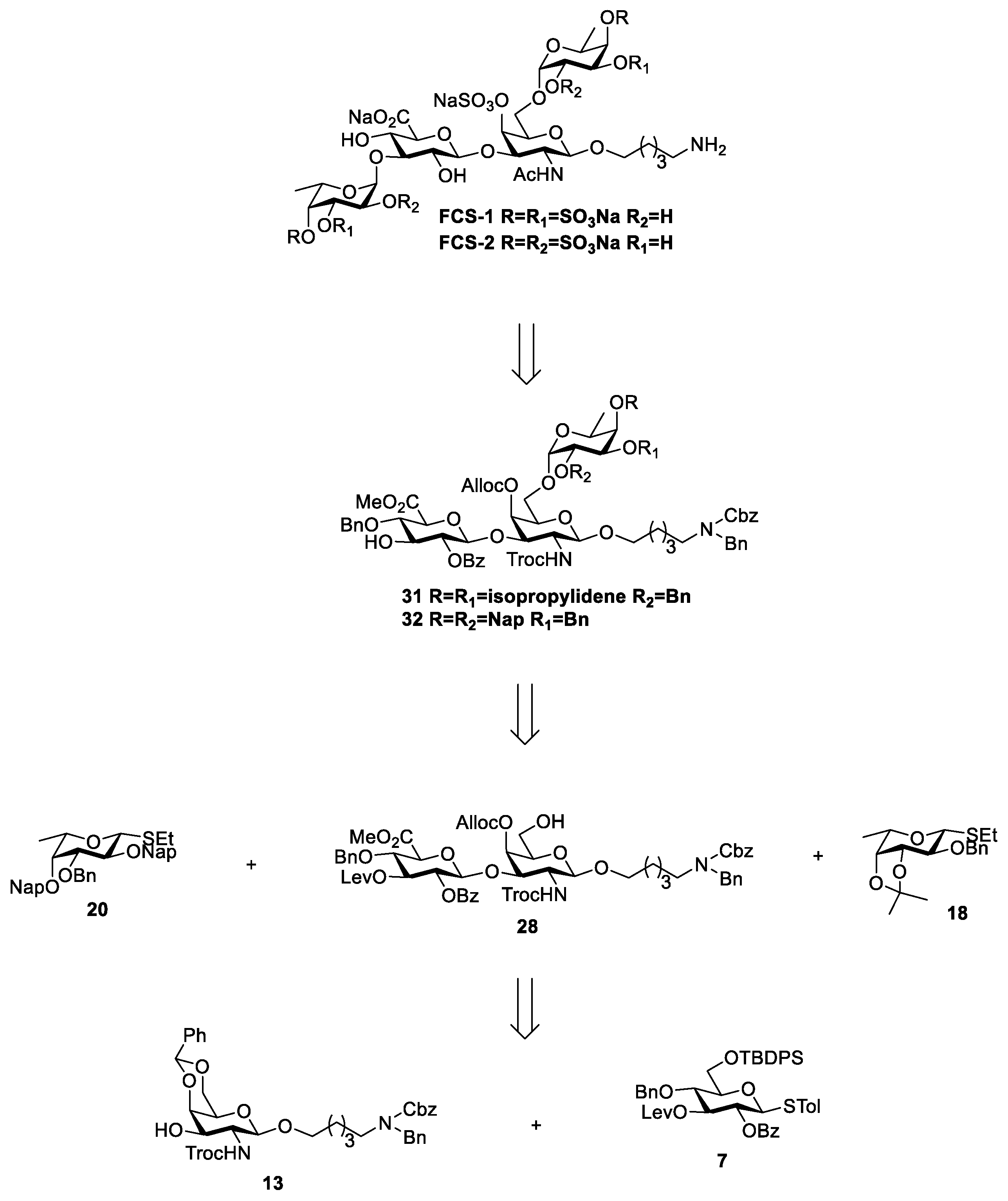
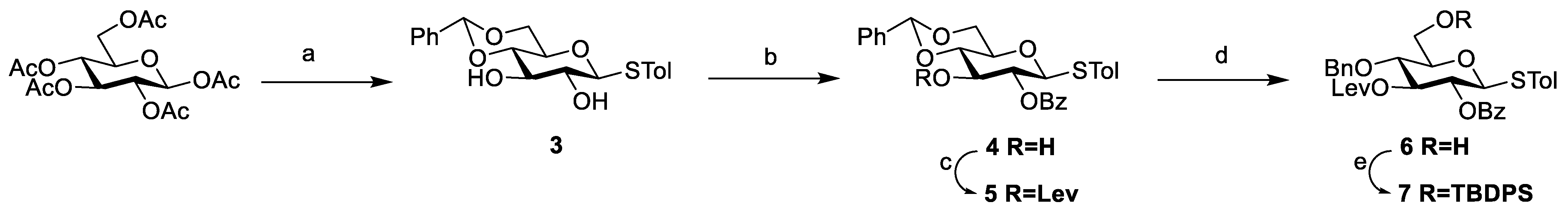
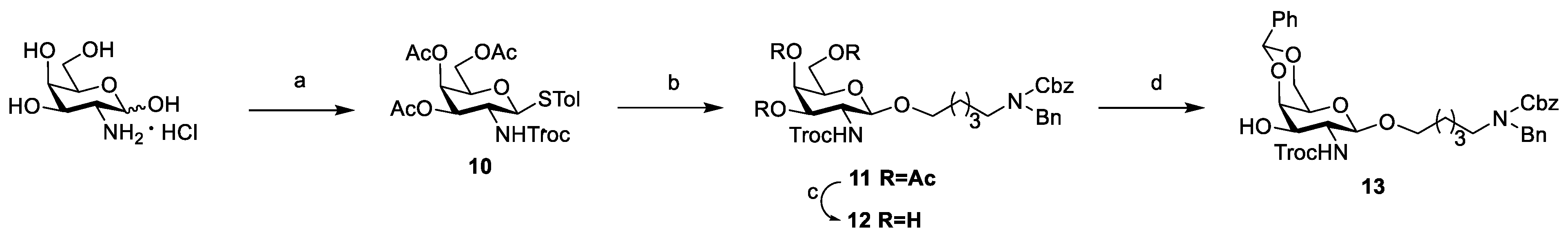
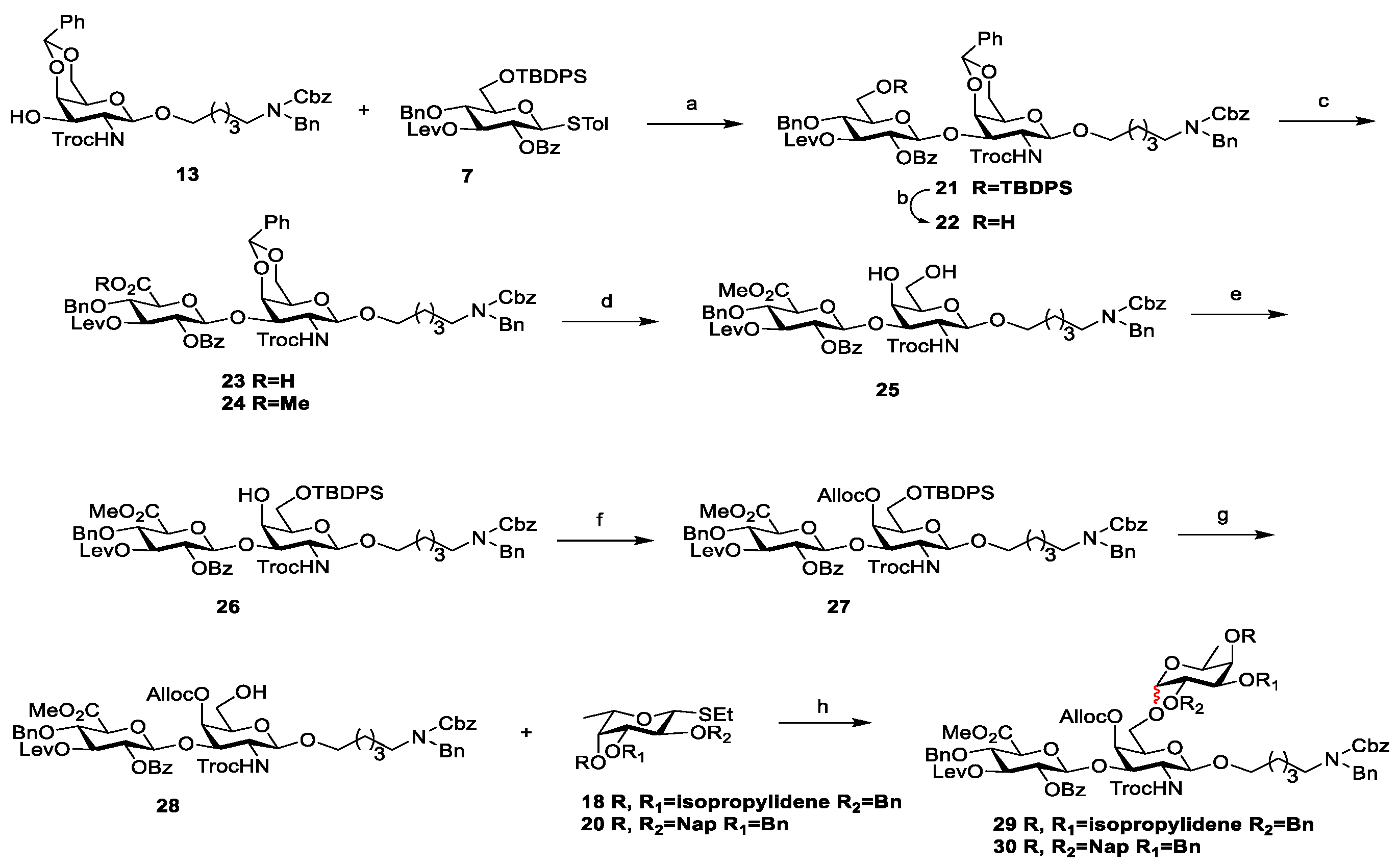
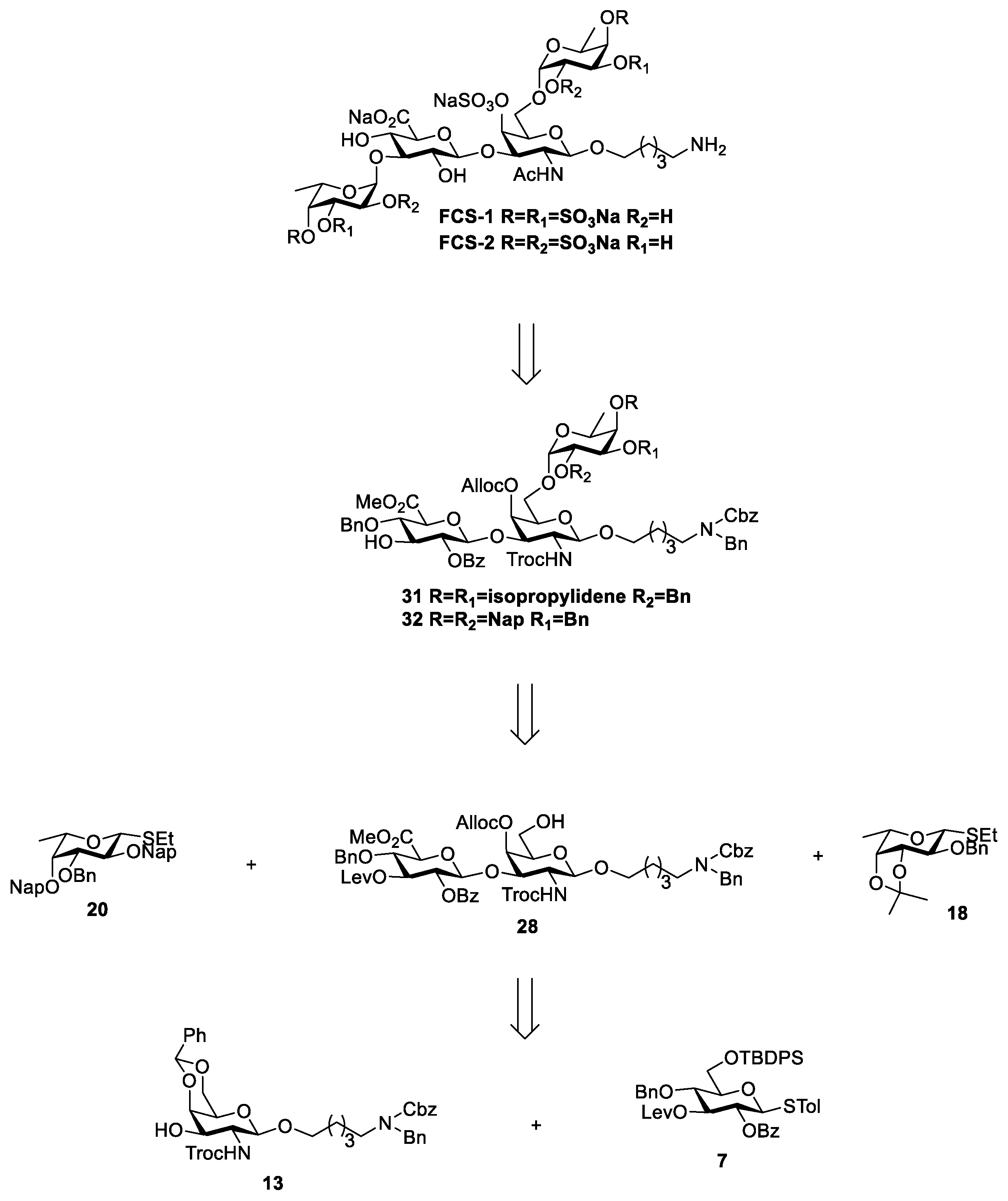
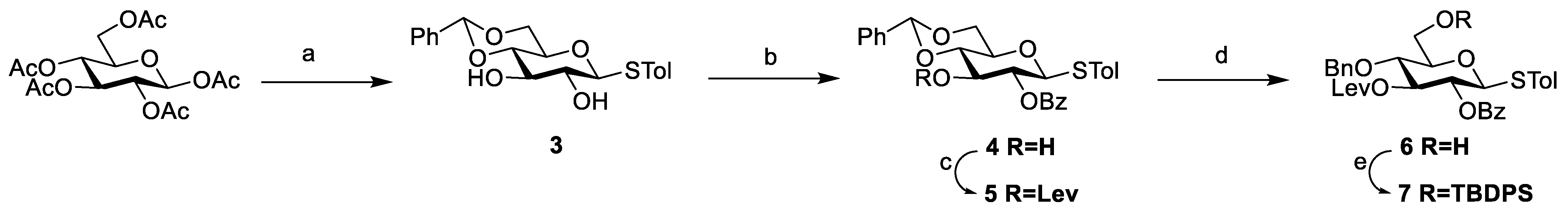
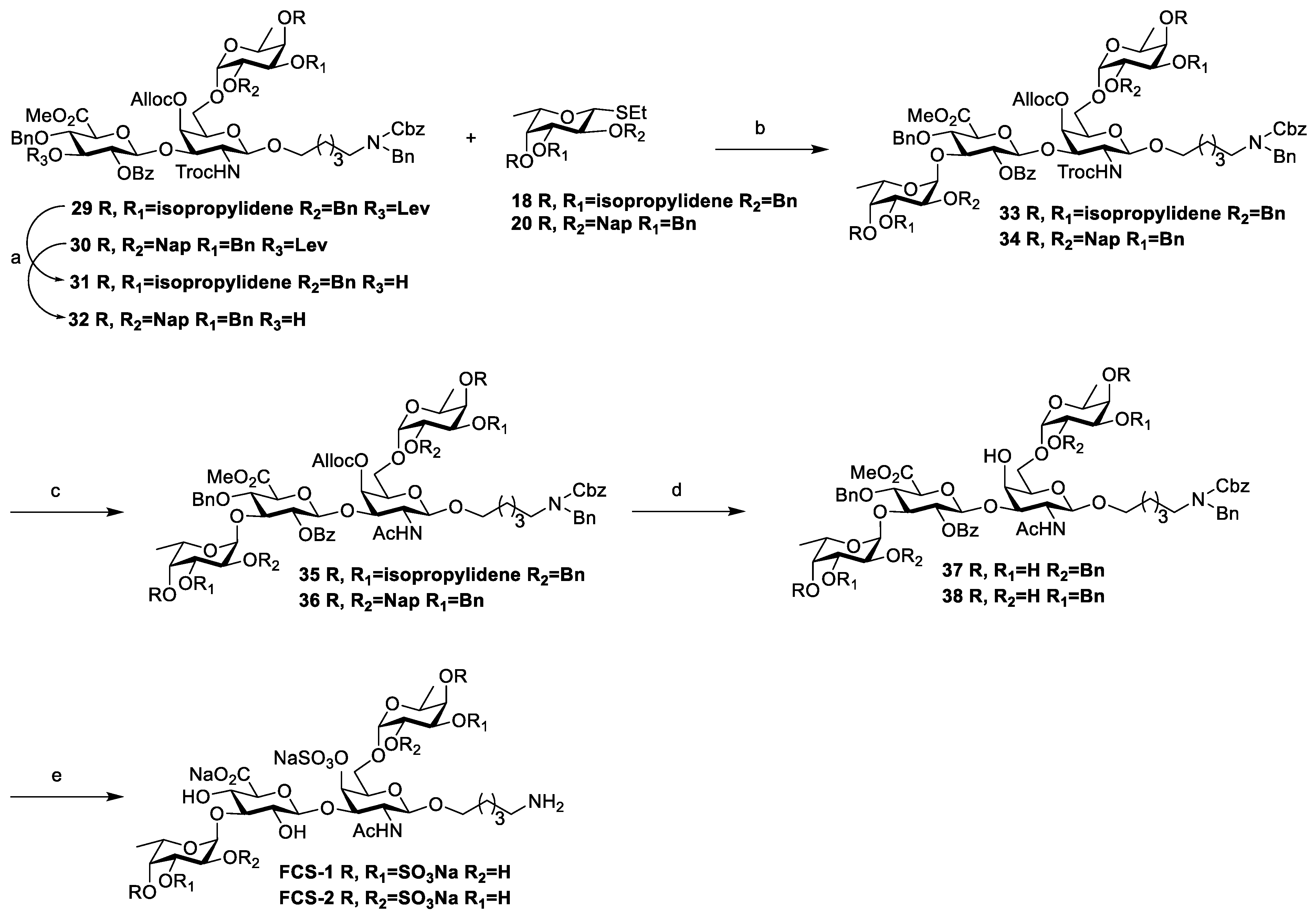
3.2. Chemical Synthesis
Compounds
7 [
22],
13 [
25], and
18 [
26,
27] were synthesized according to
Scheme 2,
Scheme 3, and
Scheme 4, respectively. Their NMR data were found to be consistent with the literature.
Compound
21: To a solution of glucose donor
7 [
22] (141 mg, 0.17 mmol) and acceptor
13 [
25] (100 mg, 0.13 mmol) in dry DCM (3 mL) dried 4 Å molecular sieves were added under a nitrogen atmosphere at room temperature. The mixture was stirred at room temperature for 1 h and then cooled to −25 °C. NIS (59 mg, 0.26 mmol) and TfOH (8.7 µL, 0.09 mmol) were added to the reaction solution and stirred for 20 min. The reaction was quenched with Et
3N and gradually warmed to room temperature. The mixture was filtered through celite and extracted with DCM. The organic phase was washed with saturated NaHCO
3 and brine, dried with anhydrous Na
2SO
4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 10:1,
v/
v) to afford white solid compound
21 (180 mg, 94%), R
f = 0.27 (PE/EtOAc = 2:1,
v/
v).
1H NMR (400 MHz, CDCl
3):
δ 8.02 (d,
J = 4.0 Hz, 2H, Ar-
H), 7.73–7.64 (m, 4H, Ar-
H), 7.55 (t,
J = 7.4 Hz, 1H, Ar-
H), 7.50–7.06 (m, 28H, Ar-
H), 5.42–5.33 (m, 2H, Ph-C
H,
H-3), 5.22 (dd,
J = 9.6, 7.9 Hz, 1H,
H-2), 5.19–5.08 (m, 2H, Cbz-
CH2), 4.87 (d,
J = 7.9 Hz, 1H,
Glu-H-1), 4.82 (d,
J = 7.1 Hz, 1H,
Gal-H-1′), 4.70 (d,
J = 12.3 Hz, 1H, Ph-C
H2), 4.61 (d,
J = 11.1 Hz, 1H, CCl
3CH2), 4.52–4.41 (m, 4H, N-Ph
CH2,
H-3′, CCl
3CH2), 4.36–4.27 (m, 2H,
H-4′, Ph-C
H2), 4.17 (d,
J = 11.9 Hz, 1H,
H-6a′), 4.02 (d,
J = 9.8 Hz, 1H,
H-6a), 3.87 (dd,
J = 11.2, 5.7 Hz, 1H,
H-6b), 3.80 (d,
J = 11.1 Hz, 2H,
H-6b′, OC
H2-a), 3.72 (t,
J = 9.4 Hz, 1H,
H-4), 3.64–3.56 (m, 1H,
H-5), 3.44–3.27 (m, 2H,
H-2′, OC
H2-b), 3.24 (s, 1H,
H-5′), 3.23–3.12 (m, 2H, NC
H2), 2.53–2.44 (m, 2H, Lev-C
H2), 2.44–2.32 (m, 2H, Lev-C
H2), 2.00 (s, 3H, Lev-C
H3), 1.55–1.37 (m, 4H, OCH
2C
H2, C
H2CH
2N), 1.29–1.15 (m, 2H, OCH
2CH
2C
H2), 1.10 (s, 9H, SiC(C
H3)
3).
13C NMR (100 MHz, CDCl
3):
δ 206.0, 172.0, 165.3, 154.0, 138.0, 138.0, 137.6, 135.8, 135.6, 133.6, 133.4, 133.1, 130.1, 130.0, 129.7, 128.7, 128.5, 128.2, 128.1, 128.0, 127.3, 126.4, 126.4, 101.8(C-1), 100.7(Ph-CH), 99.4(C-1′), 95.8, 76.3(C-5), 76.2(C-4′), 76.0(C-4), 75.4(C-3), 75.3(C-3′), 74.7, 74.0, 72.6(C-2), 69.8(O-CH
2), 69.1(C-6′), 67.3(Cbz-CH
2), 66.6(C-5′), 63.4(C-6), 54.2(C-2′), 50.4(N-PhCH
2), 47.3(N-CH
2), 46.2(N-CH
2), 37.9(Lev-CH
2), 29.7(Lev-CH
3), 29.2, 28.1(Lev-CH
2), 27.5, 27.1(SiC(CH
3)
3), 23.3, 19.6. HRMS (ESI)
m/
z calcd for C
77H
86Cl
3N
2O
17Si [M+H]
+ 1443.4756, found 1443.4731.
Compound 22: To a solution of 21 (191 mg, 0.13 mmol) in THF/Py (3 mL/0.6 mL), HF·Py (400 μL) was added at 0 °C under a nitrogen atmosphere. After being warmed to room temperature, the mixture was stirred for 7 h. The resulting mixture was concentrated in vacuum and extracted with DCM. The organic phase was washed with 1 N HCl, saturated NaHCO3, and brine, dried with anhydrous Na2SO4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 1:1, v/v) to afford white solid compound 22 (135 mg, 85%), Rf = 0.13 (PE/EtOAc = 1:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 7.4 Hz, 2H, Ar-H), 7.55 (t, J = 7.3 Hz, 1H, Ar-H), 7.40 (t, J = 7.7 Hz, 2H, Ar-H), 7.37–7.19 (m, 19H, Ar-H), 7.15 (d, J = 6.1 Hz, 1H, Ar-H), 5.40 (s, 1H, Ph-CH), 5.36 (t, J = 9.4 Hz, 1H, H-3), 5.20 (t, J = 8.9 Hz, 1H, H-2), 5.17–5.12 (m, 2H, Cbz-CH2), 4.99 (d, J = 7.3 Hz, 1H, Glu-H-1), 4.82 (d, J = 7.8 Hz, 1H, Gal-H-1′), 4.73 (d, J = 12.3 Hz, 1H, CCl3CH2), 4.66 (q, J = 11.4 Hz, 2H, Ph-CH2), 4.56–4.41 (m, 3H, N-PhCH2, H-3′), 4.27 (d, J = 11.4 Hz, 2H, H-6a′, H-4′), 4.21–4.08 (m, 2H, CCl3CH2), 4.03 (d, J = 12.0 Hz, 1H, H-6b′), 3.92–3.71 (m, 4H, H-4, H-6a, H-6b, OCH2-a), 3.49 (d, J = 9.6 Hz, 2H, H-5, H-2′), 3.42 (s, 1H, H-5′), 3.40–3.28 (m, 1H, OCH2-b), 3.28–3.12 (m, 2H, NCH2), 2.55–2.45 (m, 2H, Lev-CH2), 2.39–2.28 (m, 2H, Lev-CH2), 2.00 (s, 3H, Lev-CH3), 1.57–1.39 (m, 4H, OCH2CH2, CH2CH2N), 1.29–1.23 (m, 2H, OCH2CH2CH2). 13C NMR (100 MHz, CDCl3): δ 205.9, 172.0, 165.4, 154.4, 138.0, 137.7, 133.6, 130.1, 129.4, 129.0, 128.7, 128.7, 128.7, 128.3, 128.2, 128.2, 128.1, 128.0, 127.4, 127.3, 126.4, 101.1, 100.3, 99.8, 95.71, 75.8, 75.7, 75.1, 75.0, 74.8, 74.1, 73.8, 72.2, 69.8, 69.3, 67.3, 66.5, 61.5, 54.1, 50.4, 50.4, 47.3, 46.1, 37.8, 29.7, 28.1, 23.4. HRMS (ESI) m/z calcd for C61H71Cl237ClN3O17 [M+NH4]+1224.3814, found: 1224.3849.
Compound 24: To a solution of 22 (300 mg, 0.25 mmol) in DCM/H2O (1 mL/0.5 mL), TEMPO (19 mg, 0.12 mmol) and BAIB (200 mg, 0.62 mmol) were added under a nitrogen atmosphere. The reaction mixture was stirred for 7 h at room temperature. The reaction was quenched with saturated Na2S2O3, then extracted with DCM, and washed with brine. The organic phase was dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to give compound 23 as a yellow oil, which could be used in the next reaction without purification.
To a solution of 23 in dry DMF (2 mL), K2CO3 (44 mg, 0.32 mmol) and CH3I (69 μL, 1.11 mmol) were added at 50 °C under a nitrogen atmosphere. After being stirred for 5 h, the reaction was quenched using 1 N HCl, then extracted with EtOAc and washed with saturated Na2S2O3, water, and brine. The organic phase was dried with anhydrous Na2SO4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 1:1, v/v) to afford white solid compound 24 (227 mg, 74% for two steps), Rf = 0.84 (PE/EtOAc = 1:2, v/v). 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 7.5 Hz, 2H, Ar-H), 7.58–7.46 (m, 3H, Ar-H), 7.42 (t, J = 7.6 Hz, 2H, Ar-H), 7.39–7.18 (m, 19H, Ar-H), 7.15 (d, J = 4.7 Hz, 1H, Ar-H), 5.52 (s, 1H, Ph-CH), 5.33 (t, J = 7.8 Hz, 1H, H-3), 5.22 (t, J = 7.8 Hz, 1H, H-2), 5.19–5.09 (m, 2H, Cbz-CH2), 4.96 (d, J = 6.8 Hz, 1H, Glu-H-1), 4.83 (d, J = 8.3 Hz, 1H, Gal-H-1′), 4.65 (d, J = 11.8 Hz, 1H, CCl3CH2), 4.57 (q, J = 11.3 Hz, 2H, Ph-CH2), 4.52–4.41 (m, 3H, N-PhCH2, H-3′), 4.34 (d, J = 3.1 Hz, 1H, H-4′), 4.28 (d, J = 11.7 Hz, 1H, H-6a′), 4.22–4.13 (m, 1H, CCl3CH2), 4.10 (d, J = 7.0 Hz, 2H, H-4, H-5), 4.05 (d, J = 12.2 Hz, 1H, H-6b′), 3.89–3.76 (m, 1H, OCH2-a), 3.72 (s, 3H, COOCH3), 3.43 (s, 1H, H-5′), 3.43–3.27 (m, 2H, H-2′, OCH2-b), 3.26–3.11 (m, 2H, NCH2), 2.55–2.47 (m, 2H, Lev-CH2), 2.43–2.33 (m, 2H, Lev-CH2), 2.01 (s, 3H, Lev-CH3), 1.53–1.40 (m, 4H, OCH2CH2, CH2CH2N), 1.29–1.23 (m, 2H, OCH2CH2CH2). 13C NMR (100 MHz, CDCl3): δ 206.0, 171.8, 168.7, 165.0, 138.0, 137.6, 133.5, 130.1, 129.4, 128.9, 128.7, 128.7, 128.6, 128.6, 128.2, 128.1, 128.0, 127.4, 127.4, 126.4, 101.2, 100.8, 100.3, 99.5, 95.8, 77.4, 75.7, 74.9, 74.7, 74.2, 74.0, 74.0, 72.1, 69.8, 69.7, 69.3, 67.3, 66.6, 54.1, 52.8, 50.6, 37.9, 29.8, 29.7, 29.2, 29.1, 28.1, 23.3. HRMS (ESI) m/z calcd for C62H71Cl2N3O1837Cl [M+NH4]+1252.3763, found 1252.3799.
Compound 25: Compound 24 (58 mg, 0.05 mmol) was dissolved in AcOH/H2O (2 ml/0.5 ml) and stirred for 6 h at room temperature. The reaction was quenched with saturated NaHCO3, then extracted with DCM and washed with brine. The organic phase was dried with anhydrous Na2SO4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (CH2Cl2/CH3OH = 40:1, v/v) to afford white solid compound 25 (47 mg, 89%), Rf = 0.40 (CH2Cl2/CH3OH = 20:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 7.6 Hz, 2H, Ar-H), 7.57 (t, J = 7.3 Hz, 1H, Ar-H), 7.45 (t, J = 7.6 Hz, 2H, Ar-H), 7.39–7.12 (m, 18H, Ar-H), 5.38 (t, J = 8.9 Hz, 1H, H-3), 5.22 (d, J = 8.3 Hz, 1H, H-2), 5.19–5.10 (m, 2H, Cbz-CH2), 4.84 (d, J = 7.1 Hz, 1H, Glu-H-1), 4.72 (d, J = 8.5 Hz, 1H, Gal-H-1′), 4.67–4.58 (m, 2H, Ph-CH2), 4.56–4.42 (m, 3H, CCl3CH2, N-PhCH2), 4.36 (d, J = 9.5 Hz, 1H, H-3′), 4.09 (d, J = 9.1 Hz, 2H, H-4′, H-5), 4.04 (t, J = 8.9 Hz, 1H, H-4), 4.00–3.87 (m, 2H, H-6a′, CCl3CH2), 3.86–3.79 (m, 2H, OCH2-a, H-6b′), 3.76 (s, 3H, COOCH3), 3.54 (t, J = 4.9 Hz, 1H, H-5′), 3.42–3.24 (m, 2H, H-2′, OCH2-b), 3.24–3.11 (m, 2H, NCH2), 2.54–2.46 (m, 2H, Lev-CH2), 2.42–2.34 (m, 2H, Lev-CH2), 2.00 (s, 3H, Lev-CH3), 1.55–1.40 (m, 4H, OCH2CH2, CH2CH2N), 1.29–1.22 (m, 2H, OCH2CH2CH2). 13C NMR (100 MHz, CDCl3): δ 205.8, 171.8, 168.5, 165.1, 138.0, 137.4, 133.7, 130.1, 128.8, 128.7, 128.6, 128.2, 128.1, 128.0, 127.4, 127.3, 101.8, 100.9, 99.4, 95.7, 95.3, 78.6, 77.4, 74.9, 74.5, 73.9, 73.8, 71.9, 69.8, 69.0, 68.9, 68.9, 67.3, 62.8, 54.3, 53.0, 50.6, 47.3, 37.8, 29.7, 28.0, 23.2. HRMS (ESI) m/z calcd for C55H67Cl237ClN3O18 [M+NH4]+1164.3450, found 1164.3483.
Compound 26: To a solution of 25 (47 mg, 0.04 mmol) in dry pyridine (2 mL), DMAP (3 mg, 0.02 mmol) and TBDPSCl (32 μL, 0.12 mmol) were added under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 5 h, concentrated in vacuo, and extracted with DCM. The organic phase was washed with 1 N HCl, saturated NaHCO3, and brine, dried with anhydrous Na2SO4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 4:1, v/v) to afford white solid compound 26 (50 mg, 88%), Rf = 0.56 (PE/EtOAc = 1:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 7.5 Hz, 2H, Ar-H), 7.66 (t, J = 6.9 Hz, 4H, Ar-H), 7.57 (t, J = 7.4 Hz, 1H, Ar-H), 7.50–7.11 (m, 26H, Ar-H), 5.39 (t, J = 8.8 Hz, 1H, H-3), 5.24 (dd, J = 9.1, 7.7 Hz, 1H, H-2), 5.13 (d, J = 10.0 Hz, 2H, Cbz-CH2), 4.87 (d, J = 7.6 Hz, 1H, Glu-H-1), 4.69 (d, J = 8.1 Hz, 1H, Gal-H-1′), 4.67–4.57 (m, 2H, Ph-CH2), 4.53 (d, J = 11.5 Hz, 1H, CCl3CH2), 4.43 (s, 2H, N-PhCH2), 4.41–4.33 (m, 1H, H-3′), 4.19 (s, 1H, H-4′), 4.12–4.00 (m, 2H, H-4, H-5), 3.95 (dd, J = 9.9, 7.2 Hz, 1H, H-6a′), 3.90–3.80 (m, 1H, CCl3CH2), 3.83–3.77 (m, 1H, H-6b′), 3.76–3.68 (m, 1H, OCH2-a), 3.65 (s, 3H, COOCH3), 3.57 (t, J = 6.4 Hz, 1H, H-5′), 3.33–3.07 (m, 4H, H-2′, OCH2-b, NCH2), 2.53–2.45 (m, 2H, Lev-CH2), 2.42–2.33 (m, 2H, Lev-CH2), 2.00 (s, 3H, Lev-CH3), 1.50–1.35 (m, 4H, OCH2CH2, CH2CH2N), 1.22–1.10 (m, 2H, OCH2CH2CH2), 1.03 (s, 9H, SiC(CH3)3). 13C NMR (100 MHz, CDCl3): δ 206.1, 205.8, 171.8, 168.4, 165.1, 138.0, 137.4, 135.7, 135.7, 133.6, 133.5, 133.4, 133.4, 130.1, 129.9, 129.3, 128.8, 128.7, 128.6, 128.2, 128.2, 128.0, 128.0, 127.9, 127.4, 127.3, 101.7, 99.3, 95.8, 78.9, 77.4, 74.9, 74.4, 74.2, 74.0, 73.7, 72.4, 71.9, 69.5, 67.4, 67.3, 62.4, 54.5, 52.9, 50.6, 37.8, 35.6, 29.7, 28.0, 26.9, 19.3. HRMS (ESI) m/z calcd for C71H85Cl237ClN3O18Si [M+NH4]+1402.4628, found 1402.4628.
Compound 27: To a solution of 26 (66 mg, 0.05 mmol) in dry DCM (2 mL), DMAP (12 mg, 0.10 mmol) and AllocCl (20 μL, 0.19 mmol) were added under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 4 h, then concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 1.5:1, v/v) to afford white solid compound 27 (60 mg, 86%), Rf = 0.51 (PE/EtOAc = 1:1, v/v). 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 7.5 Hz, 2H, Ar-H), 7.64 (t, J = 5.8 Hz, 4H, Ar-H), 7.55 (t, J = 7.4 Hz, 1H, Ar-H), 7.48–7.17 (m, 22H, Ar-H), 7.17–7.11 (m, 1H, Ar-H), 5.91 (ddt, J = 16.3, 11.0, 5.7 Hz, 1H, CH=CH2), 5.41–5.29 (m, 3H, CH=CH2trans, H-3, H-4′), 5.23 (d, J = 10.4 Hz, 1H, CH=CH2cis), 5.20–5.09 (m, 3H, Cbz-CH2, H-2), 4.85 (d, J = 7.4 Hz, 1H, Glu-H-1), 4.74 (d, J = 6.4 Hz, 1H, Gal-H-1′), 4.66–4.53 (m, 6H, Ph-CH2, CCl3CH2, H-3′, CH2-CH=CH2), 4.44 (s, 2H, N-PhCH2), 4.19 (d, J = 15.5 Hz, 1H, CCl3CH2), 4.12 (t, J = 9.3 Hz, 1H, H-4), 4.03 (d, J = 9.8 Hz, 1H, H-5), 3.80–3.61 (m, 7H, COOCH3, H-6′, OCH2-a, H-5′), 3.36–3.06 (m, 4H, H-2′, OCH2-b, NCH2), 2.58–2.45 (m, 2H, Lev-CH2), 2.41–2.29 (m, 2H, Lev-CH2), 2.01 (s, 3H, Lev-CH3), 1.53–1.35 (m, 4H, OCH2CH2, CH2CH2N), 1.25–1.12 (m, 2H, OCH2CH2CH2), 1.03 (s, 9H, SiC(CH3)3). 13C NMR (100 MHz, CDCl3): δ 205.9, 171.8, 168.3, 165.0, 154.2, 138.0, 137.8, 137.0, 135.7, 135.7,133.5, 133.3, 132.0, 130.1, 129.8, 129.5, 128.7, 128.6, 128.5, 128.1, 128.1, 128.0, 128.0, 127.8, 127.4, 127.3, 118.7, 101.2, 99.3, 95.7, 77.4, 74.9, 74.7, 74.6, 74.4, 74.4, 74.0, 73.9, 73.0, 72.3, 69.8, 69.7, 68.7, 67.3, 62.1, 55.2, 52.8, 50.6, 50.4, 47.3, 46.2, 37.9, 29.8, 29.7, 28.0, 26.9, 23.3, 22.8, 19.3. HRMS (ESI) m/z calcd for C75H89Cl3N3O20Si [M+NH4]+ 1484.4869, found 1484.4868.
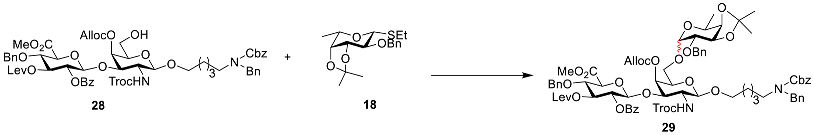
Compound 29: To a solution of 27 (277 mg, 0.19 mmol) in THF/Py (1 mL/0.2 mL), HF·Py (200 μL) was added at 0 °C under a nitrogen atmosphere. After being warmed to room temperature, the mixture was stirred for 2 h. The resulting mixture was concentrated in vacuum and extracted with DCM. The organic phase was washed with 1 N HCl, saturated NaHCO3, and brine, then dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to afford crude product 28, which could be used in the next reaction without purification.
To a solution of disaccharide acceptor
28 (232 mg, 0.19 mmol) and fucose donor
18 [
26,
27] (96 mg, 0.28 mmol) in dry DCM/Et
2O (1 mL/1 mL), dried 4 Å molecular sieves were added under a nitrogen atmosphere at room temperature. The mixture was stirred at room temperature for 1 h and then cooled to −15 °C. NIS (64 mg, 0.28 mmol) and TfOH (5.7 µL, 0.06 mmol) were added to the reaction solution and stirred for 30 min. The reaction was quenched with Et
3N and gradually warmed to room temperature. The mixture was filtered through celite and extracted with DCM. The organic phase was washed with saturated NaHCO
3 and brine, dried with anhydrous Na
2SO
4, and filtered and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 1.5:1,
v/
v) to afford white solid compounds
29alpha (164 mg, 58% for two steps) and
29beta (91 mg, 32% for two steps), R
f = 0.33 (PE/EtOAc = 1.5:1,
v/
v). Data for alpha anomer:
1H NMR (400 MHz, CDCl
3):
δ 7.98 (d,
J = 7.4 Hz, 2H, Ar-
H), 7.55 (t,
J = 7.3 Hz, 1H, Ar-
H), 7.43 (t,
J = 7.5 Hz, 2H, Ar-
H), 7.38–7.18 (m, 22H, Ar-
H), 7.14 (d,
J = 6.5 Hz, 1H, Ar-
H), 5.95 (ddt,
J = 16.1, 11.1, 5.7 Hz, 1H, C
H=CH
2), 5.41–5.36 (m, 1H, CH=C
H2trans), 5.34 (d,
J = 10.4 Hz, 1H,
H-3), 5.28 (d,
J = 10.9 Hz, 2H,
H-4′, CH=C
H2cis), 5.18–5.09 (m, 3H,
H-2, Cbz-
CH2), 4.83–4.66 (m, 5H,
Fuc-H-1″,
Gal-H-1′,
Glu-H-1, Ph-C
H2), 4.64 (d,
J = 5.5 Hz, 2H, C
H2-CH=CH
2), 4.64–4.57 (m, 2H, Ph-C
H2), 4.57 (d,
J = 7.0 Hz, 1H, CCl
3CH2), 4.50 (d,
J = 9.0 Hz, 1H,
H-3′), 4.45 (s, 2H, N-Ph
CH2), 4.36–4.28 (m, 1H,
H-3″), 4.17–3.98 (m, 5H,
H-4″,
H-5″,
H-4,
H-5, CCl
3CH2), 3.81 (t,
J = 6.1 Hz, 1H,
H-5′), 3.77 (s, 3H, COO
Me), 3.77–3.64 (m, 2H,
H-6a′, OC
H2-a), 3.49 (dd,
J = 7.9, 3.3 Hz, 1H,
H-2″), 3.45 (d,
J = 6.7 Hz, 1H,
H-6b′), 3.34–3.08 (m, 4H,
H-2′, OC
H2-b, NC
H2), 2.51–2.44 (m, 2H, Lev-C
H2), 2.39–2.31 (m, 2H, Lev-C
H2), 2.00 (s, 3H, Lev-C
H3), 1.52–1.41 (m, 4H, OCH
2C
H2, C
H2CH
2N), 1.40 (s, 3H, C
H3), 1.34 (s, 3H, C
H3), 1.34–1.29 (m, 2H, OCH
2CH
2C
H2), 1.26 (d,
J = 6.5 Hz, 3H,
H-6″).
13C NMR (100 MHz, CDCl
3):
δ 205.9, 171.8, 168.3, 165.0, 154.4, 153.9, 138.5, 138.0, 137.7, 133.5, 131.9(CH=CH
2), 130.1, 129.5, 128.7, 128.6, 128.5, 128.1, 128.0, 128.0, 127.9, 127.9, 127.4, 127.3, 118.9(CH=CH
2), 108.9, 101.1(C-1), 99.6(C-1′), 98.0(C-1″), 95.8, 77.4(C-4), 76.5(C-2″), 76.3(C-4″), 75.8(C-3″), 74.9, 74.6(C-3′), 74.5(C-5), 74.3(C-3), 74.2, 73.9, 73.3(C-4′), 72.4, 72.1(C-5′, C-2), 69.8(O-CH
2), 68.9(CH
2-CH=CH
2), 67.3(Cbz-CH
2), 66.3(C-6′), 63.5(C-5″), 55.0(C-2′), 52.8(COOMe), 50.4(N-PhCH
2), 47.4(N-CH
2), 46.2(N-CH
2), 37.8(Lev-CH
2), 32.1, 31.6, 29.8, 29.7(Lev-CH
3), 29.5, 28.3(CH
3), 28.0(Lev-CH
2), 26.5(CH
3), 23.3, 22.8, 16.4(C-6″), 14.3. HRMS (ESI)
m/
z calcd for C
75H
91Cl
237ClN
3O
24 [M+NH
4]
+1524.5023, found: 1524.5039; Data for beta anomer:
1H NMR (400 MHz, CDCl
3):
δ 7.99 (d,
J = 7.0 Hz, 2H, Ar-
H), 7.56 (t,
J = 7.0 Hz, 1H, Ar-
H), 7.49–7.17 (m, 22H, Ar-
H), 7.14 (d,
J = 5.7 Hz, 1H, Ar-
H), 5.93 (ddt,
J = 16.2, 10.8, 5.6 Hz, 1H, C
H=CH
2), 5.33 (dd,
J = 17.7, 7.0 Hz, 3H,
H-3,
H-4′, CH=C
H2trans), 5.25 (d,
J = 10.5 Hz, 1H, CH=C
H2cis), 5.20–5.09 (m, 3H,
H-2, Cbz-
CH2), 4.78 (q,
J = 11.9 Hz, 4H,
Gal-H-1′,
Glu-H-1, Ph-C
H2), 4.67–4.48 (m, 6H, C
H2-CH=CH
2, Ph-C
H2, CCl
3CH2,
H-3′), 4.44 (s, 2H, N-Ph
CH2), 4.29 (d,
J = 8.0 Hz, 1H,
Fuc-H-1″), 4.11 (dt,
J = 12.0, 6.6 Hz, 2H,
H-3″,
H-4), 4.07–3.95 (m, 3H,
H-4″,
H-5, CCl
3CH2), 3.86 (dd,
J = 9.7, 4.9 Hz, 1H,
H-6a′), 3.83–3.70 (m, 4H,
H-5″,
H-5′,
H-6b′, OC
H2-a), 3.70 (s, 3H, COO
Me), 3.33 (t,
J = 7.5 Hz, 1H,
H-2″), 3.30–3.07 (m, 4H,
H-2′, OC
H2-b, NC
H2), 2.556–2.41 (m, 2H, Lev-C
H2), 2.41–2.25 (m, 2H, Lev-C
H2), 2.00 (s, 3H, Lev-C
H3), 1.36 (d,
J = 6.4 Hz, 3H,
H-6″), 1.31 (s, 3H, C
H3), 1.30 (s, 3H, C
H3), 1.28–1.25 (m, 6H, OCH
2C
H2, C
H2CH
2N, OCH
2CH
2C
H2).
13C NMR (151 MHz, CDCl
3):
δ 205.9, 177.5, 171.8, 165.0, 154.3, 138.6, 138.0, 137.6, 133.5, 131.9(CH=CH
2), 130.1, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 128.0, 127.6, 127.4, 127.4, 127.3, 118.7(CH=CH
2), 109.7, 109.7, 103.2(C-1″), 101.2(C-1), 99.2(C-1′), 79.0(C-3″, C-2″), 76.4(C-4″), 74.8, 74.4(C-5), 74.2(C-3), 73.7, 73.5, 73.3(C-4′), 72.3(C-5′), 72.0(C-2), 69.8(O-CH
2), 68.9(CH
2-CH=CH
2), 68.8(C-5″), 67.7(C-6′), 67.3(Cbz-CH
2), 60.6, 54.9(C-2′), 52.8(COOMe), 50.6(N-PhCH
2), 50.4, 47.3(N-CH
2), 46.2(N-CH
2), 37.8(Lev-CH
2), 32.0, 31.6, 29.7(Lev-CH
3), 28.0(Lev-CH
2), 27.9(CH
3), 26.5(CH
3), 23.2, 22.8, 21.2, 16.7(C-6″), 14.3, 14.3. TOF-HRMS (ESI)
m/
z: calcd for C
75H
91Cl
237ClN
3O
24 [M+NH
4]
+1524.5023, found: 1524.5039.
Compound 30: Following the synthesis of compound 29, compounds 28 (23 mg, 0.02 mmol) and 20 (16 mg, 0.03 mmol) were reacted under the catalytic reaction of NIS (6 mg, 0.03 mmol) and TfOH (0.6 μL, 0.01 mmol) to obtain the corresponding product, which was purified using flash chromatography (PE/EtOAc = 15:1, v/v) to afford white solid compounds 30alpha (16 mg, 67% for two steps) and 30beta (6 mg, 25% for two steps), Rf = 0.51 (PE/EtOAc = 1:1, v/v). Data for alpha anomer: 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 7.5 Hz, 2H, Ar-H), 7.83–7.67 (m, 8H, Ar-H), 7.54 (t, J = 7.5 Hz, 1H, Ar-H), 7.50–7.17 (m, 29H, Ar-H), 7.13 (d, J = 6.9 Hz, 1H, Ar-H), 5.89 (ddt, J = 16.2, 11.3, 5.7 Hz, 1H, CH=CH2), 5.36–5.27 (m, 3H, CH=CH2trans, H-3, H-4′), 5.20 (d, J = 10.3 Hz, 1H, CH=CH2cis), 5.18–5.07 (m, 4H, Ph-CH2, H-2, Cbz-CH2), 4.94 (dd, J = 11.8, 1.9 Hz, 2H, Ph-CH2), 4.88 (d, J = 3.4 Hz, 1H, Fuc-H-1″), 4.86–4.78 (m, 4H, Glu-H-1, Ph-CH2), 4.65 (d, J = 7.9 Hz, 1H, Gal-H-1′), 4.58 (dd, J = 10.2, 5.8 Hz, 5H, CH2-CH=CH2, Ph-CH2, CCl3CH2), 4.49 (d, J = 12.7 Hz, 1H, H-3′), 4.43 (s, 2H, N-PhCH2), 4.24–4.16 (m, 1H, CCl3CH2), 4.13 (dd, J = 10.2, 3.5 Hz, 1H, H-2″), 4.08–4.01 (m, 2H, H-3″, H-4), 3.99 (d, J = 9.6 Hz, 1H, H-5), 3.87–3.80 (m, 2H, H-5″, H-5′), 3.73 (dd, J = 9.8, 4.5 Hz, 2H, H-6a′, H-4″), 3.66 (s, 4H, OCH2-a, COOMe), 3.48 (dd, J = 9.8, 6.0 Hz, 1H, H-6b′), 3.33–3.03 (m, 4H, H-2′, OCH2-b, NCH2), 2.57–2.41 (m, 2H, Lev-CH2), 2.41–2.27 (m, 2H, Lev-CH2), 2.00 (s, 3H, Lev-CH3), 1.44–1.29 (m, 4H, OCH2CH2, CH2CH2N), 1.20–1.09 (m, 2H, OCH2CH2CH2), 1.06 (d, J = 6.5 Hz, 3H, H-6″). 13C NMR (100 MHz, CDCl3): δ 205.8, 171.8, 168.2, 165.0, 154.5, 139.2, 138.0, 137.7, 136.3, 136.2, 133.5, 133.4, 133.3, 133.1, 131.8(CH=CH2), 130.1, 129.5, 128.7, 128.6, 128.6, 128.5, 128.3, 128.1, 128.1, 128.1, 128.0, 127.8, 127.7, 127.4, 127.3, 127.2, 126.8, 126.7, 126.2, 126.1, 126.0, 125.9, 118.8(CH=CH2), 101.1(C-1), 99.3(C-1′), 98.4(C-1″), 95.8, 83.3, 79.6(C-4), 77.7(C-4″), 77.4(C-3″), 76.6(C-2″), 75.0, 74.9, 74.5(C-5), 74.4(C-3′), 74.3(C-3), 74.0, 73.6, 73.4, 73.4(C-4′), 72.4(C-5′), 72.2(C-2), 69.6(O-CH2), 68.8(CH2-CH=CH2), 67.3(Cbz-CH2), 66.6(C-5″), 66.2(C-6′), 65.7, 55.0(C-2′), 52.7(COOMe), 50.6(N-PhCH2), 50.4, 47.3(N-CH2), 46.3(N-CH2), 37.8(Lev-CH2), 29.7(Lev-CH3), 28.0(Lev-CH2), 23.3, 16.9(C-6″). HRMS (ESI) m/z calcd for C94H103Cl3N3O24 [M+NH4]+ 1762.5992, found 1762.6027; Data for beta anomer: 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 7.7 Hz, 2H, Ar-H), 7.86–7.67 (m, 8H, Ar-H), 7.58–7.39 (m, 9H, Ar-H), 7.37–7.18 (m, 22H, Ar-H), 7.13 (d, J = 6.4 Hz, 1H, Ar-H), 5.87 (ddt, J = 16.4, 10.9, 5.7 Hz, 1H, CH=CH2), 5.42–5.27 (m, 3H, CH=CH2trans, H-3, H-4′), 5.23 (d, J = 10.3 Hz, 1H, CH=CH2cis), 5.19–5.07 (m, 5H, Ph-CH2, H-2, Cbz-CH2), 4.91 (d, J = 12.7 Hz, 2H, Ph-CH2), 4.88–4.79 (m, 1H, Ph-CH2), 4.74 (d, J = 11.8 Hz, 2H, Ph-CH2, Glu-H-1), 4.66 (d, J = 8.1 Hz, 1H, Gal-H-1′), 4.62–4.50 (m, 5H, CH2-CH=CH2, Ph-CH2, CCl3CH2), 4.43 (d, J = 7.2 Hz, 3H, H-3′, N-PhCH2), 4.37 (d, J = 7.6 Hz, 1H, Fuc-H-1″), 4.02 (d, J = 9.0 Hz, 2H, CCl3CH2, H-4), 3.97 (d, J = 9.7 Hz, 1H, H-5), 3.89 (t, J = 8.7 Hz, 2H, H-6a′, H-2″), 3.83–3.63 (m, 3H, OCH2-a, H-6b′, H-5′), 3.60 (s, 4H, H-4″, COOMe), 3.57–3.49 (m, 1H, H-3″), 3.45 (q, J = 6.4 Hz, 1H, H-5″), 3.32–3.02 (m, 4H, H-2′, OCH2-b, NCH2), 2.56–2.41 (m, 2H, Lev-CH2), 2.41–2.26 (m, 2H, Lev-CH2), 2.00 (s, 3H, Lev-CH3), 1.42–1.30 (m, 6H, OCH2CH2, CH2CH2N, OCH2CH2CH2), 1.18 (d, J = 6.3 Hz, 3H, H-6″). 13C NMR (151 MHz, CDCl3): δ 205.9, 171.8, 164.9, 138.7, 138.0, 137.7, 136.7, 136.0, 133.5, 133.2, 133.1, 133.0, 131.8(CH=CH2), 130.1, 128.6, 128.5, 128.5, 128.2, 128.1, 128.0, 128.0, 127.8, 127.7, 127.7, 127.4, 126.9, 126.8, 126.6, 126.1, 126.0, 125.9, 125.8, 118.7(CH=CH2), 104.1(C-1″), 101.1(C-1), 99.5(C-1′), 82.6(C-3″), 79.3(C-2″), 76.1(C-4″), 75.1, 74.8, 74.7(C-3′), 74.4(C-5), 74.3(C-3), 73.8, 73.5, 73.3(C-4′), 72.3(C-5′), 72.1(C-2), 70.6(C-5″), 69.7(O-CH2), 68.8(CH2-CH=CH2), 67.7(C-6′), 67.3(Cbz-CH2), 55.0(C-2′), 52.7(COOMe), 50.6(N-PhCH2), 50.4, 47.3(N-CH2), 46.2(N-CH2), 37.8(Lev-CH2), 30.3, 29.8, 29.7(Lev-CH3), 29.5, 29.2, 28.0(Lev-CH2), 23.2, 22.8, 17.1(C-6″), 14.3. TOF-HRMS (ESI) m/z: calcd for C94H103Cl3N3O24 [M+NH4]+ 1762.5992, found 1762.6027.
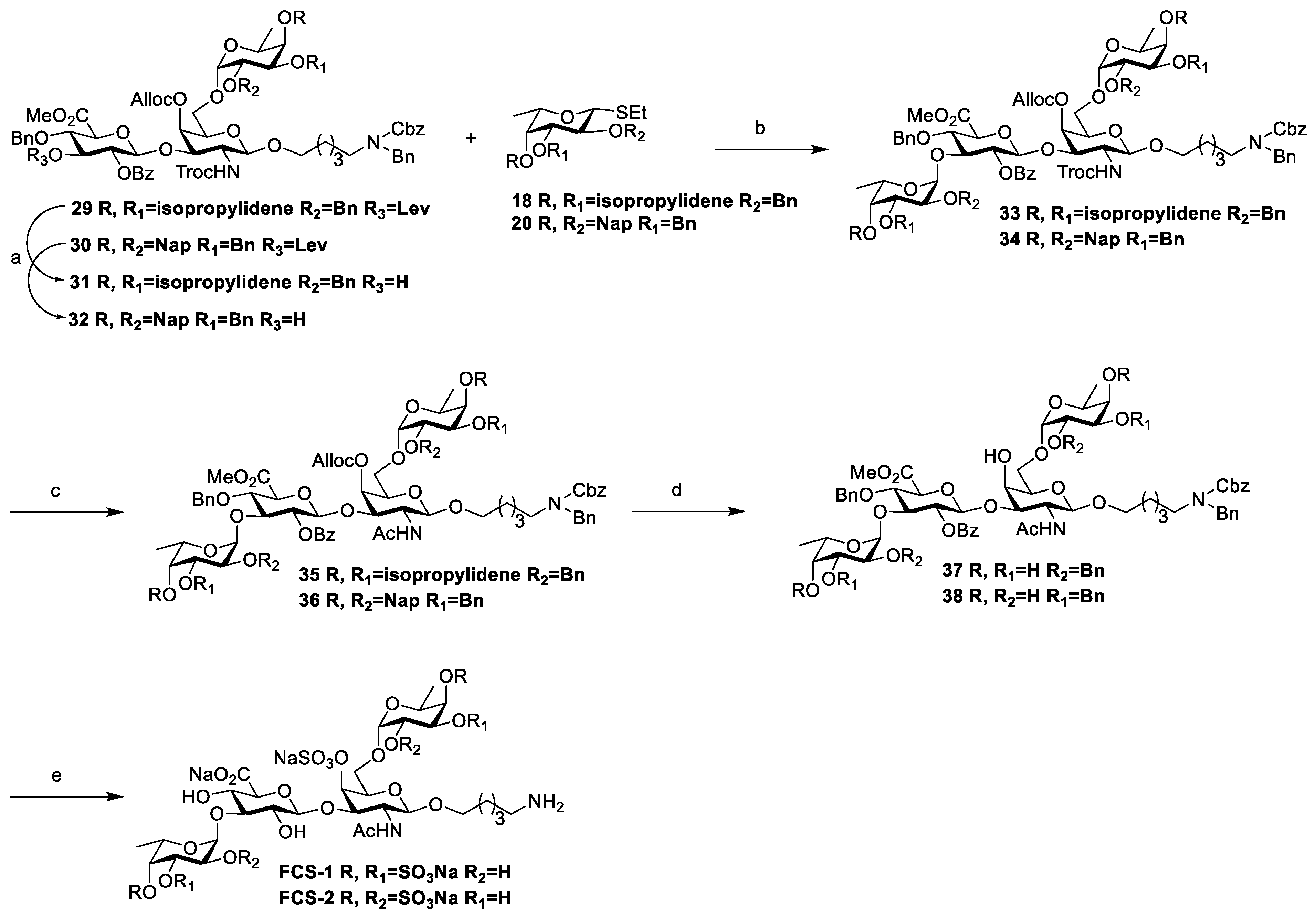
Compound 33: To a solution of 29 (33 mg, 0.02 mmol) in DCM/CH3OH (1 mL/1 mL), Hydrazine acetate (92 mg, 0.22 mmol) was added at room temperature under a nitrogen atmosphere. The reaction was quenched with acetone after 2 h and then extracted with DCM. The organic phase was washed with saturated NaHCO3 and brine, dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to afford crude product 31. This compound was suitable for the next step without purification.
Following the synthesis of compound 29, compounds 31 (27 mg, 0.02 mmol) and 18 (10 mg, 0.03 mmol) were reacted under the catalytic reaction of NIS (6 mg, 0.03 mmol) and TfOH (1.2 μL, 0.01 mmol) to obtain the corresponding product after 15 min, which was purified using flash chromatography (PE/EtOAc = 2:1, v/v) to afford white solid compound 33 (17 mg, 72% for two steps, only α), Rf = 0.29 (PE/EtOAc = 2:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.05 (d, J = 7.3 Hz, 3H, Ar-H), 7.57 (t, J = 7.4 Hz, 1H, Ar-H), 7.43 (dd, J = 17.3, 9.5 Hz, 3H, Ar-H), 7.38–7.20 (m, 34H, Ar-H), 7.16–7.10 (m, 5H, Ar-H), 7.09–7.01 (m, 2H, Ar-H), 5.98 (ddt, J = 16.6, 11.3, 5.9 Hz, 1H, CH=CH2), 5.43–5.33 (m, 2H, CH=CH2trans, H-2), 5.33–5.25 (m, 2H, CH=CH2cis, H-4′), 5.19–5.09 (m, 3H, Cbz-CH2, Fuc-H-1‴), 4.83–4.71 (m, 5H, Gal-H-1′, Fuc-H-1″, Ph-CH2, Glu-H-1), 4.69 (d, J = 10.9 Hz, 4H, CH2-CH=CH2, Ph-CH2), 4.56 (dd, J = 12.4, 6.2 Hz, 2H, Troc-CH2, Ph-CH2), 4.50 (d, J = 7.4 Hz, 1H, H-3′), 4.46 (s, 2H, N-PhCH2), 4.35–4.28 (m, 3H, Troc-CH2, Ph-CH2, H-3″), 4.26 (dd, J = 6.4, 1.7 Hz, 1H, H-5‴), 4.17–4.13 (m, 2H, H-3‴), 4.08–4.01 (m, 5H, H-4″, H-5″, H-4, H-5, H-3), 3.82 (t, J = 5.5 Hz, 1H, H-5′), 3.76 (s, 5H, COOMe, H-6a′, OCH2-a), 3.49 (dd, J = 7.8, 3.4 Hz, 3H, H-6b′, H-2″, H-4‴), 3.27 (d, J = 3.7 Hz, 2H, H-2‴, OCH2-b), 3.23–3.09 (m, 3H, H-2′, NCH2), 1.51–1.41 (m, 4H, OCH2CH2, CH2CH2N), 1.39 (s, 3H, CH3), 1.34 (s, 3H, CH3), 1.26 (d, J = 5.4 Hz, 5H, OCH2CH2CH2, H-6″), 1.20 (s, 3H, CH3), 1.15 (s, 3H, CH3), 1.04 (d, J = 6.5 Hz, 3H, H-6‴). 13C NMR (125 MHz, CDCl3): δ 138.5, 138.0, 137.9, 133.5, 131.9(CH=CH2), 130.0, 128.9, 128.7, 128.6, 128.6, 128.5, 128.4, 128.3, 128.0, 127.9, 127.8, 127.5, 127.4, 127.3, 118.9(CH=CH2), 108.9, 108.7, 101.2(C-1), 99.5(C-1′), 97.9(C-1″), 96.2(C-1‴), 78.1(C-4), 76.4(C-2″), 76.3(C-4″), 75.9(C-3″), 75.8(C-4‴), 75.7(C-3), 75.3(C-3‴), 75.1(C-3′), 75.0, 74.8(C-2‴), 74.7(C-5), 74.4(C-2), 73.9, 73.9, 73.2(C-4′), 72.4, 72.0(C-5′), 72.0, 71.7, 70.1, 69.8(C-6′), 68.9(CH2-CH=CH2), 67.3(Cbz-CH2), 66.2(C-6′), 63.6(C-5‴), 63.4(C-5″), 55.1(C-2′), 52.8(COOMe), 50.4(N-PhCH2), 47.3(N-CH2), 46.2(N-CH2), 32.1, 31.6, 28.3(CH3), 27.8(CH3), 26.5(CH3), 26.2(CH3), 22.8, 22.1, 16.4(C-6″), 16.1(C-6‴), 14.3. HRMS (ESI) m/z: calcd for C86H101N2O26Cl3Na [M+Na]+1705.5600, found: 1705.5641.
Compound 34: To a solution of 30 (93 mg, 0.05 mmol) in DCM/CH3OH (1 mL/1 mL), Hydrazine acetate (50 mg, 0.54 mmol) was added at room temperature under a nitrogen atmosphere. The reaction was quenched with acetone after 2 h and then extracted with DCM. The organic phase was washed with saturated NaHCO3 and brine, dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to afford crude product 32. This compound was suitable for the next step without purification.
Following the synthesis of compound 29, compounds 32 (87 mg, 0.05 mmol) and 20 (50 mg, 0.08 mmol) were reacted under the catalytic reaction of NIS (18 mg, 0.08 mmol) and TfOH (3.2 μL, 0.03 mmol) to obtain the corresponding product after 15 min, which was purified using flash chromatography (PE/EtOAc = 2:1, v/v) to afford white solid compound 34 (95 mg, 83% for two steps, only α), Rf = 0.24 (PE/EtOAc = 2:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 7.4 Hz, 2H, Ar-H), 7.82–7.64 (m, 11H, Ar-H), 7.64–7.56 (m, 4H, Ar-H), 7.53 (d, J = 7.4 Hz, 2H, Ar-H), 7.52–7.25 (m, 29H, Ar-H), 7.12 (t, J = 7.3 Hz, 3H, Ar-H), 7.04 (dd, J = 13.6, 7.1 Hz, 3H, Ar-H), 5.92 (ddt, J = 16.3, 11.0, 5.8 Hz, 1H, CH=CH2), 5.43 (t, J = 7.6 Hz, 1H, H-2), 5.31 (d, J = 9.2 Hz, 1H, CH=CH2trans), 5.28 (dd, J = 7.7, 4.4 Hz, 2H, Fuc-H-1‴, H-4′), 5.19 (d, J = 10.4 Hz, 2H, CH=CH2cis), 5.14 (d, J = 8.3 Hz, 2H, Cbz-CH2), 5.09 (d, J = 11.8 Hz, 1H, Ph-CH2), 4.94 (dd, J = 11.9, 3.2 Hz, 3H, Ph-CH2), 4.87 (d, J = 3.5 Hz, 1H, Fuc-H-1″), 4.81 (q, J = 12.3 Hz, 4H, Ph-CH2), 4.76–4.69 (m, 2H, Gal-H-1′, Glu-H-1), 4.68 (d, J = 11.0 Hz, 3H, Ph-CH2, CH2-CH=CH2), 4.65–4.57 (m, 4H, Ph-CH2, Troc-CH2, CH2-CH=CH2), 4.57–4.50 (m, 3H, Ph-CH2, Troc-CH2, H-3′), 4.46–4.35 (m, 3H, N-PhCH2, Ph-CH2), 4.15–4.10 (m, 1H, H-2″), 4.09–4.01 (m, 3H, H-4, H-3″, H-3), 3.97 (d, J = 9.0 Hz, 1H, H-5), 3.91 (dq, J = 10.5, 5.0, 3.5 Hz, 2H, H-5‴, H-2‴), 3.87–3.76 (m, 3H, H-3‴, H-5″, H-5′), 3.73 (dd, J = 8.9, 4.7 Hz, 2H, H-6a′, H-4″), 3.66 (s, 4H, COOMe, OCH2-a), 3.47 (dd, J = 9.7, 5.2 Hz, 1H, H-6b′), 3.22 (s, 1H, H-4‴), 3.21–3.10 (m, 3H, NCH2-a, OCH2-b, H-2′), 3.10–3.03 (m, 1H, NCH2-b), 1.25–1.19 (m, 4H, OCH2CH2, CH2CH2N), 1.18–1.11 (m, 2H, OCH2CH2CH2), 1.06 (d, J = 6.4 Hz, 3H, H-6″), 0.71 (d, J = 6.4 Hz, 3H, H-6‴). 13C NMR (150 MHz, CDCl3): δ 168.9, 164.7, 154.5, 139.2, 139.1, 138.0, 137.7, 136.3, 136.2, 135.9, 135.7, 133.5, 133.4, 133.3, 133.3, 133.2, 133.1, 133.1, 132.0(CH=CH2), 130.0, 128.9, 128.7, 128.6, 128.4, 128.3, 128.2, 128.1, 128.0, 127.8, 127.8, 127.7, 127.7, 127.6, 127.4, 127.3, 127.2, 126.9, 126.9, 126.8, 126.2, 126.1, 126.0, 126.0, 125.9, 125.8, 118.8(CH=CH2), 101.3(C-1), 99.4(C-1′), 98.3(C-1″), 96.8(C-1‴), 79.6(C-3″), 78.5(C-4), 78.3(C-3‴), 77.7(C-4″), 77.5(C-4‴), 76.5(C-2″), 75.7(C-2‴), 75.3(C-3), 75.2(Troc-CH2), 75.0, 75.0, 74.8(C-3), 74.6(C-2), 74.0, 73.8, 73.6(C-3′), 73.5, 73.3, 73.2(C-4′), 72.4, 72.2(C-5′), 69.7, 69.7(O-CH2), 69.6, 68.9(CH2-CH=CH2), 67.3(Cbz-CH2), 66.7(C-5‴), 66.6(C-5″), 66.0(C-6′), 66.0, 60.5, 55.2(C-2′), 52.7(COOMe), 50.6(N-PhCH2), 50.4, 47.3(N-CH2), 46.2(N-CH2), 44.4, 32.1, 31.6, 31.6, 30.3, 29.8, 29.8, 29.5, 29.2, 29.1, 23.2, 22.8, 21.2, 16.8(C-6″), 16.3(C-6‴), 14.3, 14.3. HRMS (ESI) m/z calcd for C124H125Cl3N2O26NH4 [M+ NH4]+ 2180.7930, found 2180.7883.
Compound 35: To a solution of 33 (23 mg, 0.01 mmol) in THF/Ac2O/AcOH (1.8 mL/0.3 mL/0.3 mL), zinc dust (72 mg, 1.09 mmol) was added at room temperature under a nitrogen atmosphere. After being stirred for 10 h, the mixture was filtered through celite and concentrated in vacuo. The residue was purified using flash chromatography (PE/EtOAc = 1:1, v/v) to afford white solid compound 35 (17 mg, 80%), Rf = 0.23 (PE/EtOAc = 1:1, v/v). 1H NMR (400 MHz, CDCl3): δ 8.03 (d, J = 7.3 Hz, 2H, Ar-H), 7.58 (t, J = 7.3 Hz, 1H, Ar-H), 7.48–7.02 (m, 37H, Ar-H), 5.97 (ddt, J = 16.2, 11.2, 5.7 Hz, 1H, CH=CH2), 5.43–5.33 (m, 2H, CH=CH2trans, H-2), 5.31–5.27 (m, 2H, CH=CH2cis, H-4′), 5.18–5.10 (m, 3H, Cbz-CH2, Fuc-H-1‴), 4.97 (d, J = 8.3 Hz, 1H, Gal-H-1′), 4.82–4.73 (m, 4H, Fuc-H-1″, Ph-CH2, H-3′), 4.68 (t, J = 6.4 Hz, 6H, CH2-CH=CH2, Ph-CH2, Glu-H-1), 4.56 (d, J = 12.6 Hz, 1H, Ph-CH2), 4.45 (s, 2H, N-PhCH2), 4.32 (dd, J = 7.7, 5.6 Hz, 2H, Ph-CH2, H-3″), 4.28 (dd, J = 6.6, 2.2 Hz, 1H, H-5‴), 4.15 (ddd, J = 8.9, 6.5, 2.3 Hz, 2H, H-3‴, H-3), 4.12–4.02 (m, 4H, H-4″, H-5″, H-4, H-5), 3.84 (t, J = 6.0 Hz, 1H, H-5′), 3.76 (s, 3H, COOMe), 3.76–3.70 (m, 2H, H-6a′, OCH2-a), 3.52 (dd, J = 5.2, 1.8 Hz, 1H, H-4‴), 3.49 (dd, J = 7.8, 3.4 Hz, 2H, H-6b′, H-2″), 3.34–3.25 (m, 2H, H-2‴, OCH2-b), 3.17–3.04 (m, 3H, H-2′, NCH2), 1.39 (s, 3H, CH3), 1.38–1.34 (m, 4H, OCH2CH2, CH2CH2N), 1.33 (s, 3H, CH3), 1.26 (d, J = 5.1 Hz, 5H, OCH2CH2CH2, H-6″), 1.20 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.03 (d, J = 6.6 Hz, 3H, H-6‴). 13C NMR (100 MHz, CDCl3): δ 168.8, 164.7, 154.4, 138.6, 138.1, 138.0, 133.6, 132.0, 130.0, 129.0, 128.7, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.9, 127.8, 127.5, 127.5, 127.4, 118.8, 108.8, 108.7, 101.4, 98.8, 98.0, 96.4, 78.2, 77.4, 76.5, 76.3, 76.2, 75.8, 75.7, 75.3, 75.0, 74.9, 74.5, 73.5, 72.4, 72.3, 72.2, 71.8, 69.9, 68.8, 67.3, 63.7, 63.4, 55.6, 52.8, 50.4, 47.1, 46.1, 32.1, 31.6, 30.3, 29.8, 29.5, 28.3, 27.8, 26.5, 26.2, 23.1, 22.8, 16.4, 16.1, 14.3, 10.4. HRMS (ESI) m/z calcd for C85H106N3O25 [M+NH4]+1568.7110, found: 1568.7158.
Compound 36: Following the synthesis of compound 35, compound 34 (86 mg, 0.04 mmol) was reacted under zinc dust (104 mg, 1.59 mmol) to give the corresponding product, which was purified using flash chromatography (PE/EtOAc = 1:1, v/v) to afford white solid compound 36 (74 mg, 91%), Rf = 0.26 (PE/EtOAc = 1.5:1, v/v). 1H NMR (500 MHz, CDCl3): δ 7.88 (d, J = 7.3 Hz, 2H, Ar-H), 7.74–7.56 (m, 11H, Ar-H), 7.55–7.46 (m, 4H, Ar-H), 7.46–7.40 (m, 2H, Ar-H), 7.40–7.29 (m, 11H, Ar-H), 7.29–7.15 (m, 18H, Ar-H), 7.15–7.06 (m, 3H, Ar-H), 7.03 (t, J = 7.5 Hz, 3H, Ar-H), 6.95 (dd, J = 14.7, 7.2 Hz, 3H, Ar-H), 5.83 (ddt, J = 17.3, 10.4, 5.7 Hz, 1H, CH=CH2), 5.35 (t, J = 8.1 Hz, 1H, H-2), 5.23 (dq, J = 17.3, 1.6 Hz, 1H, CH=CH2trans), 5.20–5.17 (m, 2H, Fuc-H-1‴, H-4′), 5.12 (dq, J = 10.4, 1.3 Hz, 1H, CH=CH2cis), 5.06–4.97 (m, 3H, Cbz-CH2, Ph-CH2), 4.84 (td, J = 10.0, 8.8, 4.0 Hz, 4H, Gal-H-1′, Ph-CH2), 4.80 (d, J = 3.6 Hz, 1H, Fuc-H-1″), 4.78–4.70 (m, 3H, Ph-CH2), 4.65 (d, J = 11.9 Hz, 2H, Ph-CH2, H-3′), 4.58 (d, J = 11.6 Hz, 2H, Ph-CH2, Glu-H-1), 4.56–4.47 (m, 5H, CH2-CH=CH2, Ph-CH2), 4.43 (d, J = 10.7 Hz, 1H, Ph-CH2), 4.36–4.29 (m, 3H, N-PhCH2, Ph-CH2), 4.07–4.01 (m, 2H, H-3, H-2″), 3.97–3.92 (m, 2H, H-4, H-3″), 3.90 (d, J = 4.3 Hz, 1H, H-5), 3.86–3.75 (m, 4H, H-2‴, H-5″, H-5′, H-5‴), 3.70 (dd, J = 10.2, 2.7 Hz, 1H, H-3‴), 3.67–3.63 (m, 1H, H-6a′), 3.62 (d, J = 2.7 Hz, 1H, H-4″), 3.57 (s, 3H, COOMe), 3.54–3.45 (m, 1H, OCH2-a), 3.44–3.37 (m, 1H, H-6b′), 3.17–3.12 (m, 1H, H-4‴), 3.12–3.03 (m, 2H, NCH2-a, OCH2-b), 3.02–2.94 (m, 2H, NCH2-b, H-2′), 1.20–1.13 (m, 4H, OCH2CH2, CH2CH2N), 1.06–0.99 (m, 1H, OCH2CH2CH2), 0.98 (d, J = 6.4 Hz, 3H, H-6″), 0.97–0.90 (m, 1H, OCH2CH2CH2), 0.60 (d, J = 6.4 Hz, 3H, H-6‴). 13C NMR (150 MHz, CDCl3): δ 168.8, 164.7, 154.5, 139.2, 139.1, 138.0, 137.7, 136.3, 136.2, 135.9, 135.7, 133.6, 133.4, 133.3, 133.3, 133.2, 133.1, 133.0, 131.9, 130.0, 129.8, 128.9, 128.7, 128.5, 128.4, 128.2, 128.1, 128.1, 128.0, 128.0, 128.0, 127.8, 127.8, 127.7, 127.7, 127.6, 127.6, 127.4, 127.3, 127.2, 126.9, 126.8, 126.7, 126.1, 126.1, 126.0, 126.0, 125.9, 125.9, 125.8, 118.8, 101.5, 98.8, 98.4, 97.0, 79.5, 78.5, 78.5, 77.8, 77.5, 76.5, 75.7, 75.4, 75.1, 75.0, 75.0, 74.8, 74.7, 74.5, 73.6, 73.5, 73.4, 73.2, 72.6, 72.4, 69.7, 69.4, 68.8, 67.3, 66.7, 66.5, 55.6, 52.6, 50.5, 50.3, 47.2, 46.1, 42.1, 32.1, 31.6, 30.3, 29.5, 23.1, 22.8, 16.9, 16.3, 14.3. HRMS (ESI) m/z calcd for C123H127N2O25 [M+H]+2031.8722, found: 2031.8665.
Compound 37: To a solution of 35 (36 mg, 0.02 mmol) in AcOH/H2O (2 mL/0.5 mL), and stirred for 10 h at 40 °C. The reaction was quenched with saturated NaHCO3, then extracted with DCM and washed with brine. The organic phase was dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to give crude product as a yellow oil, which could be used in the next reaction without purification.
To a solution of crude product in dry THF (2 mL), PPh3 (2 mg, 0.007 mmol), Pd(PPh3)4 (3 mg, 0.002 mmol), and Ammonium formate (3 mg, 0.05 mmol) were added at room temperature under a nitrogen atmosphere. After being stirred for 3 h, the mixture was filtered through celite and concentrated in vacuo. The residue was purified using flash chromatography (DCM/MeOH = 40:1, v/v) to afford white solid compound 37 (25 mg, 78% for two steps), Rf = 0.32 (DCM/MeOH = 15:1, v/v). 1H NMR (400 MHz, Methanol-d4): δ 8.06 (d, J = 7.1 Hz, 2H, Ar-H), 7.64 (t, J = 7.4 Hz, 1H, Ar-H), 7.50 (t, J = 7.8 Hz, 2H, Ar-H), 7.41 (d, J = 7.3 Hz, 2H, Ar-H), 7.38–7.10 (m, 26H, Ar-H), 5.34 (t, J = 7.9 Hz, 1H, H-2), 5.23 (d, J = 3.8 Hz, 1H, Fuc-H-1‴), 5.13 (d, J = 7.8 Hz, 2H, Cbz-CH2), 4.88 (d, J = 7.9 Hz, 1H, Glu-H-1), 4.81 (d, J = 3.6 Hz, 1H, Fuc-H-1″), 4.74 (d, J = 11.9 Hz, 1H, Ph-CH2), 4.68–4.59 (m, 3H, Ph-CH2), 4.46 (s, 2H, N-PhCH2), 4.41 (d, J = 11.6 Hz, 1H, Ph-CH2), 4.38–4.32 (m, 1H, Gal-H-1′), 4.26 (dd, J = 18.1, 10.4 Hz, 3H, Ph-CH2, H-3′, H-3), 4.13 (s, 1H, H-4′), 4.01 (p, J = 6.9 Hz, 2H, H-5‴, H-5″), 3.92 (t, J = 8.9 Hz, 1H, H-4), 3.90–3.74 (m, 8H, H-3‴, H-3″, H-5, H-6a′, H-2′, COOMe), 3.75–3.66 (m, 2H, H-4″, H-2″), 3.65 (dd, J = 10.9, 4.9 Hz, 2H, H-5′, OCH2-a), 3.47 (dd, J = 8.6, 4.6 Hz, 1H, H-6b′), 3.43 (d, J = 3.8 Hz, 1H, H-2‴), 3.27 (d, J = 2.9 Hz, 2H, H-4‴, OCH2-b), 3.22–3.11 (m, 2H, NCH2), 1.34–1.25 (m, 4H, OCH2CH2, CH2CH2N), 1.21 (d, J = 6.5 Hz, 3H, H-6″), 1.19–1.16 (m, 2H, OCH2CH2CH2), 0.85 (d, J = 6.5 Hz, 3H, H-6‴). 13C NMR (150 MHz, Methanol-d4): δ 171.2, 166.2, 140.2, 139.3, 139.1, 134.9, 131.3, 131.0, 130.0, 129.6, 129.4, 129.2, 129.2, 129.2, 128.9, 128.9, 128.8, 128.7, 128.6, 128.3, 103.3, 102.6, 98.7, 97.3, 82.1, 79.7, 78.0, 76.6, 75.9, 75.3, 74.0, 73.8, 73.7, 73.5, 73.3, 70.9, 70.6, 70.0, 68.9, 68.4, 67.5, 67.4, 67.0, 54.8, 53.5, 52.8, 51.4, 47.5, 33.1, 30.7, 30.5, 30.2, 28.8, 24.1, 23.7, 22.6, 16.7, 16.2, 14.4. HRMS (ESI) m/z calcd for C75H94N3O23 [M+NH4]+1404.6273, found: 1404.6294.
Compound 38: To a solution of 36 (31 mg, 0.01 mmol) in DCM/Phosphate Buffered Saline (PBS, pH 7.4) (2 mL/0.11 mL), DDQ (21 mg, 0.09 mmol) was added at room temperature. The reaction mixture was stirred for 1 h and then extracted with DCM. The organic phase was washed with saturated NaHCO3 and brine, dried with anhydrous Na2SO4, and filtered and concentrated in vacuo to give crude product as a yellow oil, which could be used in the next reaction without purification.
To a solution of this crude product in dry THF (2 mL), PPh3 (1.2 mg, 0.005 mmol), Pd(PPh3)4 (1.8 mg, 0.002 mmol), and Ammonium formate (2 mg, 0.03 mmol) were added at room temperature under a nitrogen atmosphere. After stirring for 7 h, the mixture was filtered through celite and concentrated in vacuo. The residue was purified using flash chromatography (DCM/MeOH = 30:1, v/v) to afford white solid compound 38 (12.7 mg, 60% for two steps), Rf = 0.23 (DCM/MeOH = 20:1, v/v). 1H NMR (500 MHz, Methanol-d4): δ 8.07–8.02 (m, 1H, Ar-H), 7.61 (t, J = 7.4 Hz, 1H, Ar-H), 7.51–7.42 (m, 4H, Ar-H), 7.40 (d, J = 7.5 Hz, 2H, Ar-H), 7.38–7.10 (m, 17H, Ar-H), 5.31 (t, J = 7.6 Hz, 1H, H-2), 5.17–5.09 (m, 2H, Cbz-CH2), 5.10 (d, J = 3.9 Hz, 1H, Fuc-H-1‴), 4.95 (d, J = 7.3 Hz, 1H, Glu-H-1), 4.83 (d, J = 3.8 Hz, 1H, Fuc-H-1″), 4.76 (d, J = 11.9 Hz, 1H, Ph-CH2), 4.70 (d, J = 11.8 Hz, 1H, Ph-CH2), 4.65 (d, J = 12.1 Hz, 1H, Ph-CH2), 4.59 (dd, J = 11.4, 6.1 Hz, 2H, Ph-CH2), 4.52 (d, J = 11.0 Hz, 1H, Ph-CH2), 4.47 (s, 2H, N-PhCH2), 4.38 (d, J = 5.9 Hz, 1H, Gal-H-1′), 4.27 (d, J = 8.7 Hz, 1H, H-3′), 4.19 (t, J = 8.2 Hz, 1H, H-3), 4.11 (s, 1H, H-4′), 4.00 (q, J = 6.7, 6.1 Hz, 1H, H-5″), 3.96–3.90 (m, 2H, H-2″, H-4), 3.90–3.78 (m, 5H, H-5, H-4″, H-2′, H-5‴, H-6a′), 3.76–3.70 (m, 2H, H-5′, OCH2-a), 3.70–3.61 (m, 6H, COOMe, H-2‴, H-3″, H-6b′), 3.49 (dd, J = 10.0, 3.1 Hz, 1H, H-3‴), 3.46–3.42 (m, 1H, H-4‴), 3.39–3.33 (m, 1H, OCH2-b), 3.24–3.14 (m, 2H, NCH2), 1.31–1.29 (m, 4H, OCH2CH2, CH2CH2N), 1.28–1.25 (m, 2H, OCH2CH2CH2), 1.23 (d, J = 6.6 Hz, 3H, H-6″), 0.82 (d, J = 6.5 Hz, 3H, H-6‴). 13C NMR (125 MHz, Methanol-d4): δ 171.1, 166.3, 140.2, 140.1, 139.0, 134.6, 131.2, 131.0, 129.7, 129.6, 129.4, 129.4, 129.3, 129.1, 129.0, 128.9, 128.9, 128.7, 128.6, 128.5, 128.4, 103.0, 102.6, 100.9, 99.8, 81.9, 79.8, 79.4, 78.6, 76.7, 75.7, 75.5, 74.1, 72.6, 72.5, 70.6, 70.6, 70.1, 69.2, 68.9, 68.8, 68.4, 67.8, 67.6, 53.4, 52.8, 51.3, 47.5, 33.1, 31.8, 30.8, 30.8, 30.7, 30.6, 30.5, 30.3, 30.2, 24.2, 23.7, 22.7, 16.8, 16.3, 14.4. HRMS (ESI) m/z calcd for C75H94N3O23 [M+NH4]+ 1404.6273, found 1404.6259.
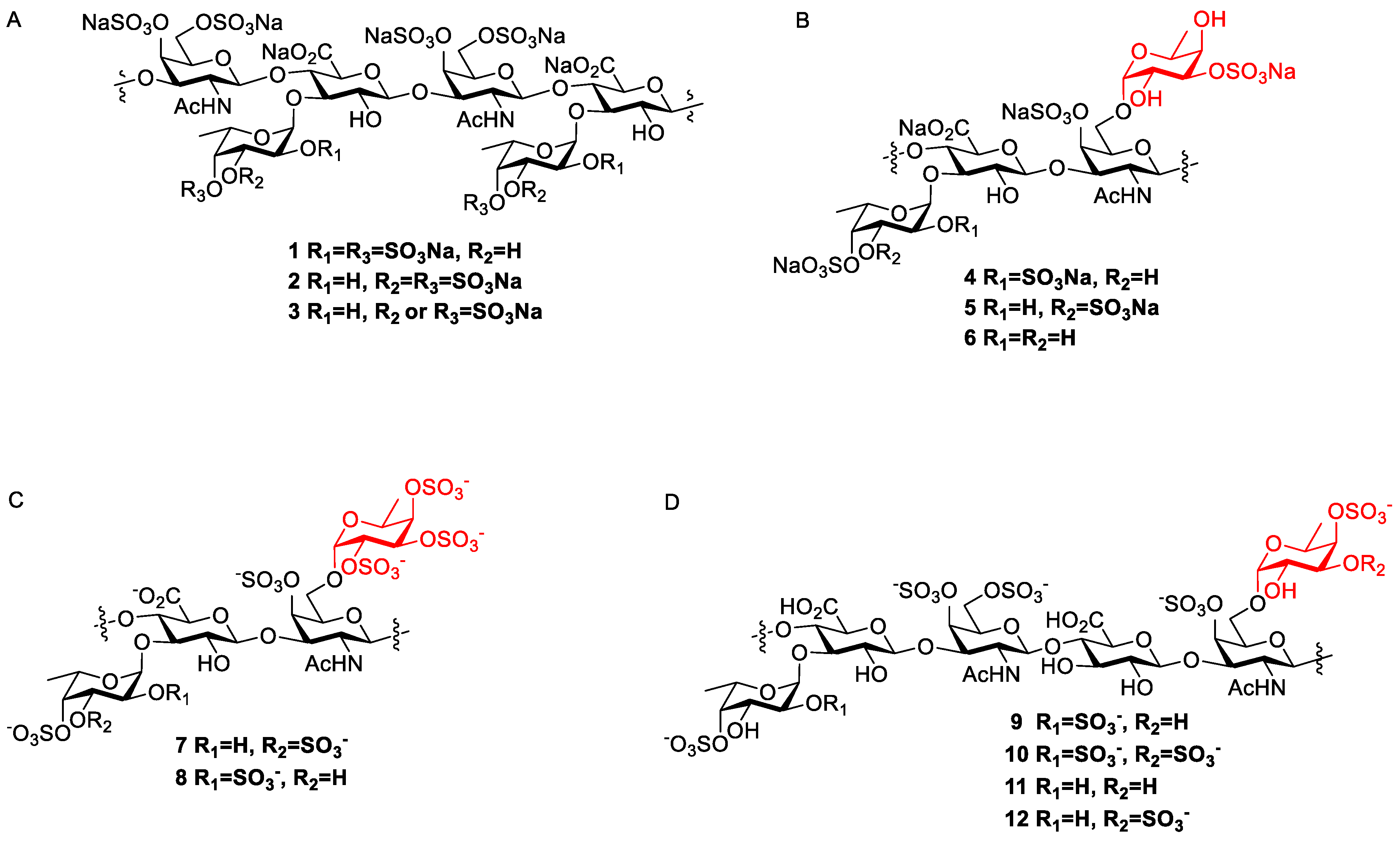
5-amino-pentanyl(6-O-(3,4-di-O-sulfo-α-L-fucopyranoside)-4-O-sulfo-3-O-(3-O-(3,4-di-O-sulfo-α-L-fucopyranoside)-β-D-glucopyranosyluronate)-2-acetamino-2-deoxy-β-D-galactopyranoside) (FCS-1).
To a solution of 37 (22 mg, 0.02 mmol) in dry DMF (1.5 mL), SO3·Me3N (221 mg, 1.59 mmol) was added at room temperature. The reaction mixture was heated to 70 °C in a microwave synthesizer and stirred for 2 h. Et3N and MeOH quenched the reaction, and it was concentrated in vacuo to give crude product as yellow oil. This product could be used in the next step without purification.
This product was dissolved in THF/H2O (1.6 mL/0.2 mL), and LiOH aqueous solution (1 M, 1 mL) was added. The reaction mixture was stirred overnight. After being concentrated in vacuo, it was dissolved in MeOH/DCM (1.1 mL/0.2 mL), and a NaOH aqueous solution (0.5 M, 2 mL) was added. The reaction was stirred for 8 h and the pH was adjusted to neutral by the addition of IR-120 H+ cation exchange resin. It was concentrated in vacuo to give crude product as yellow oil. This product could be used in the next step without purification.
To a solution of this crude product in H2O/MeOH (1 mL/1.5 mL), Pd/C (10%, 190 mg) and Pd(OH)2/C (10%, 190 mg) were added at 30 °C under a hydrogen atmosphere. After being stirred for 12 h, the mixture was filtered through celite and concentrated in vacuo. The residue was exchanged with Amberlite IR-120 (Na+) ion-exchange resin and purified with Sephadex LH-20 (CH3OH) to give the white solid compound FCS-1 (13 mg, 63% for three steps), Rf = 0.16 (EtOAc/EtOH/H2O = 2:1:1, v/v). 1H NMR (400 MHz, D2O): δ 5.33 (d, J = 4.0 Hz, 1H, Fuc-H-1‴), 5.04 (d, J = 3.9 Hz, 1H, Fuc-H-1″), 4.85 (t, J = 3.4 Hz, 2H, H-4‴, H-4″), 4.76 (s, 1H, H-4′), 4.57 (ddd, J = 16.3, 10.5, 2.9 Hz, 2H, H-3‴, H-3″), 4.48 (dt, J = 11.5, 6.0 Hz, 3H, Gal-H-1′, Glu-H-1, H-5‴), 4.21 (q, J = 6.6 Hz, 1H, H-5″), 4.08–3.98 (m, 3H, H-3′, H-2′, H-5′), 3.92 (dt, J = 9.4, 5.2 Hz, 4H, H-2‴, H-2″, H-6′), 3.86 (dd, J = 11.3, 5.0 Hz, 1H, OCH2-a), 3.68–3.59 (m, 3H, H-5, H-4, OCH2-b), 3.59–3.55 (m, 2H, H-2, H-3), 2.00 (s, 3H, Ac-CH3), 1.62 (dp, J = 20.9, 7.1, 6.6 Hz, 4H, OCH2CH2, CH2CH2N), 1.39 (p, J = 7.5, 6.9 Hz, 2H, OCH2CH2CH2), 1.27 (d, J = 6.6 Hz, 3H, H-6″), 1.22 (d, J = 6.5 Hz, 3H, H-6‴). 13C NMR (100 MHz, D2O): δ 103.2(C-1), 101.2(C-1′), 99.8(C-1″), 98.8(C-1‴), 81.3(C-3), 79.1(C-4″), 78.8(C-4‴), 77.0(C-5), 76.5(C-4′), 75.3(C-3″, C-3‴), 74.9(C-3′), 74.3, 73.6(C-5′), 73.0(C-2), 70.2(O-CH2), 70.1(C-4), 69.1(C-6′), 66.5(C-2″), 66.4(C-5‴), 66.3(C-5″), 66.1(C-5‴), 51.7(C-2′), 39.3(N-CH2), 28.0, 26.2, 22.2(Ac-CH3), 22.0, 16.0(C-6″), 15.8(C-6‴). HRMS (ESI) m/z calcd for C31H50N2O35S5 [M-6Na+2H]4− 292.5205, found 292.5213.
5-amino-pentanyl(6-O-(2,4-di-O-sulfo-α-L-fucopyranoside)-4-O-sulfo-3-O-(3-O-(2,4-di-O-sulfo-α-L-fucopyranoside)-β-D-glucopyranosyluronate)-2-acetamino-2-deoxy-β-D-galactopyranoside) (FCS-2).
Following the synthesis of compound FCS-1, compound 38 (23 mg, 0.02 mmol) was converted to the corresponding product, which was purified with Sephadex LH-20 (CH3OH) to give the white solid compound FCS-2 (14 mg, 65% for three steps), Rf = 0.16 (EtOAc/EtOH/H2O = 2:1:1, v/v). 1H NMR (500 MHz, D2O): δ 5.55 (d, J = 3.6 Hz, 1H, Fuc-H-1‴), 5.34 (d, J = 3.4 Hz, 1H, Fuc-H-1″), 4.88 (s, 1H, H-4′), 4.67 (dd, J = 5.4, 2.1 Hz, 2H, H-4″, H-4‴), 4.54 (dq, J = 13.5, 6.8, 5.6 Hz, 3H, Glu-H-1, Gal-H-1′, H-5‴), 4.42 (ddd, J = 17.7, 10.4, 3.4 Hz, 2H, H-2″, H-2‴), 4.25 (q, J = 6.3 Hz, 1H, H-5″), 4.12 (ddd, J = 13.8, 10.5, 2.7 Hz, 2H, H-3‴, H-3″), 4.04–3.92 (m, 4H, H-2′, H-3′, H-5′, H-3), 3.95–3.90 (m, 1H, H-6a′), 3.91–3.86 (m, 1H, OCH2-a), 3.82 (dd, J = 12.1, 8.1 Hz, 1H, H-6b′), 3.78–3.71 (m, 2H, H-4, H-5), 3.62 (ddt, J = 14.5, 8.9, 3.6 Hz, 4H, H-2, H-3, OCH2-b), 2.99 (t, J = 7.4 Hz, 2H, NCH2), 2.01 (s, 3H, Ac-CH3), 1.67 (dt, J = 15.3, 7.8 Hz, 2H, OCH2CH2), 1.61 (dt, J = 14.1, 6.3 Hz, 2H, CH2CH2N), 1.41 (dt, J = 15.4, 7.3 Hz, 2H, OCH2CH2CH2), 1.28 (d, J = 6.5 Hz, 3H, H-6″), 1.24 (d, J = 6.5 Hz, 3H, H-6‴). 13C NMR (150 MHz, D2O): δ 103.9(C-1), 101.0(C-1′), 97.1(C-1‴), 96.9(C-1″), 81.4(C-3), 80.8(C-4″), 80.6(C-4‴), 77.3(C-3′), 77.2(C-5), 76.8(C-4), 75.2(C-1″), 75.1(C-1‴), 74.7(C-3), 73.7(C-5′), 72.8(C-2), 70.2(O-CH2), 69.3(C-4), 67.3(C-6′), 66.5(C-3″), 66.6(C-3‴), 66.2(C-5‴), 66.1(C-5″), 51.5(C-2′), 39.4(N-CH2), 28.0, 26.3, 22.3(Ac-CH3), 22.1, 15.9(C-6″), 15.6(C-6‴). HRMS (ESI) m/z calcd for C31H51N2O35S5 [M-6Na+3H]3− 390.3620, found 390.3611.



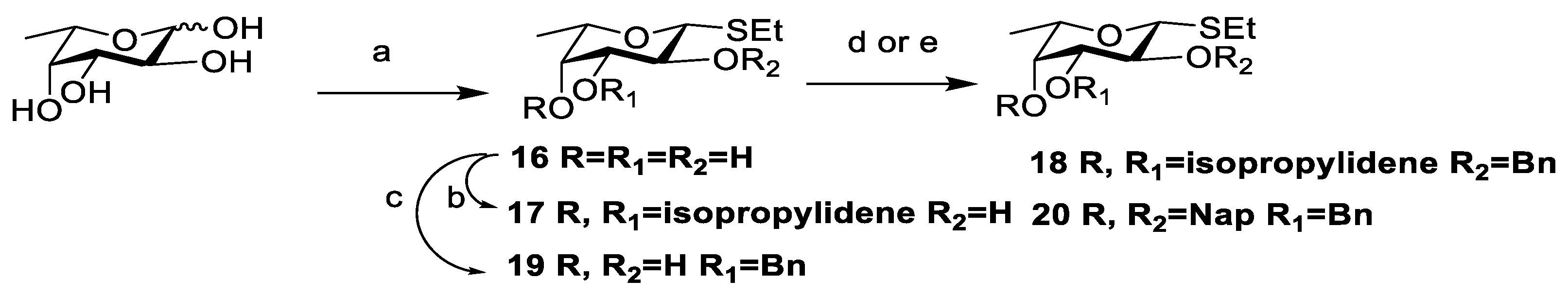

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}