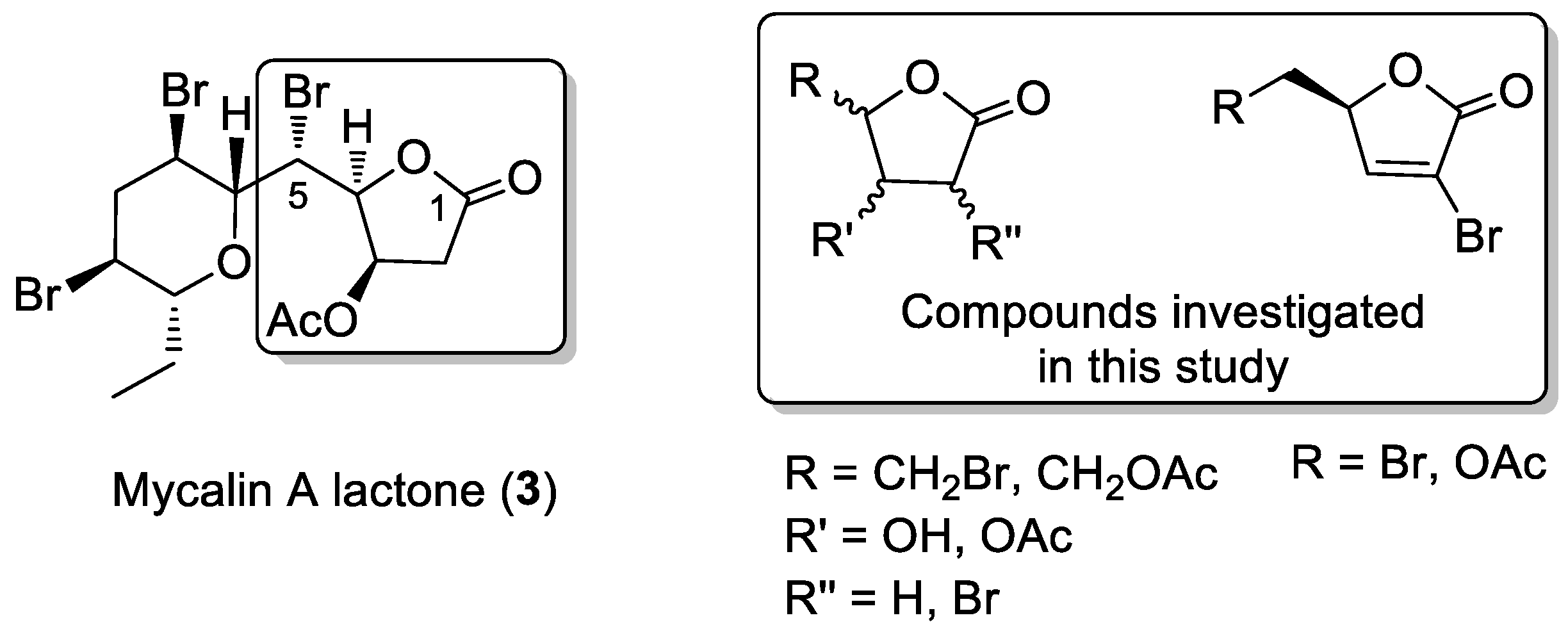
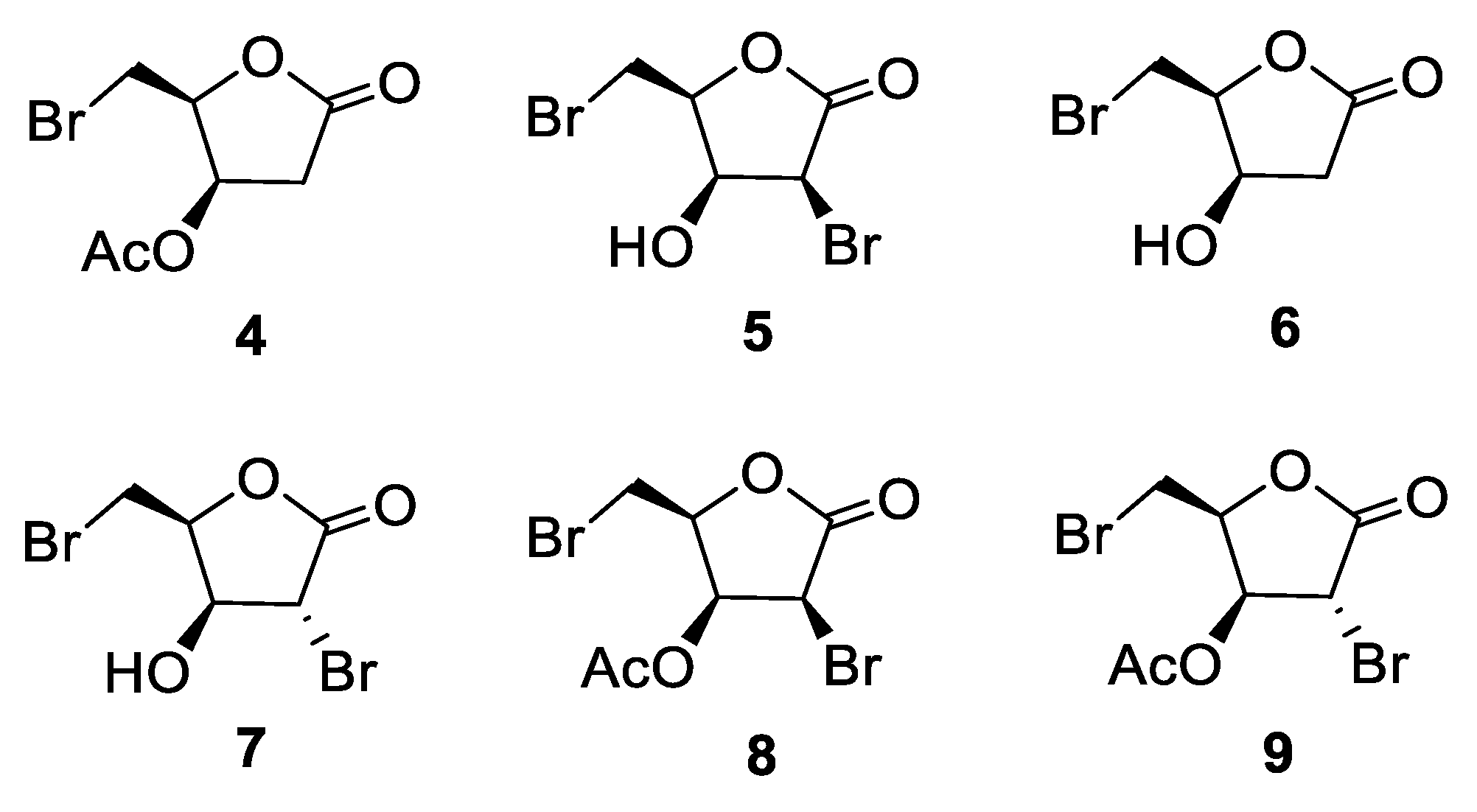
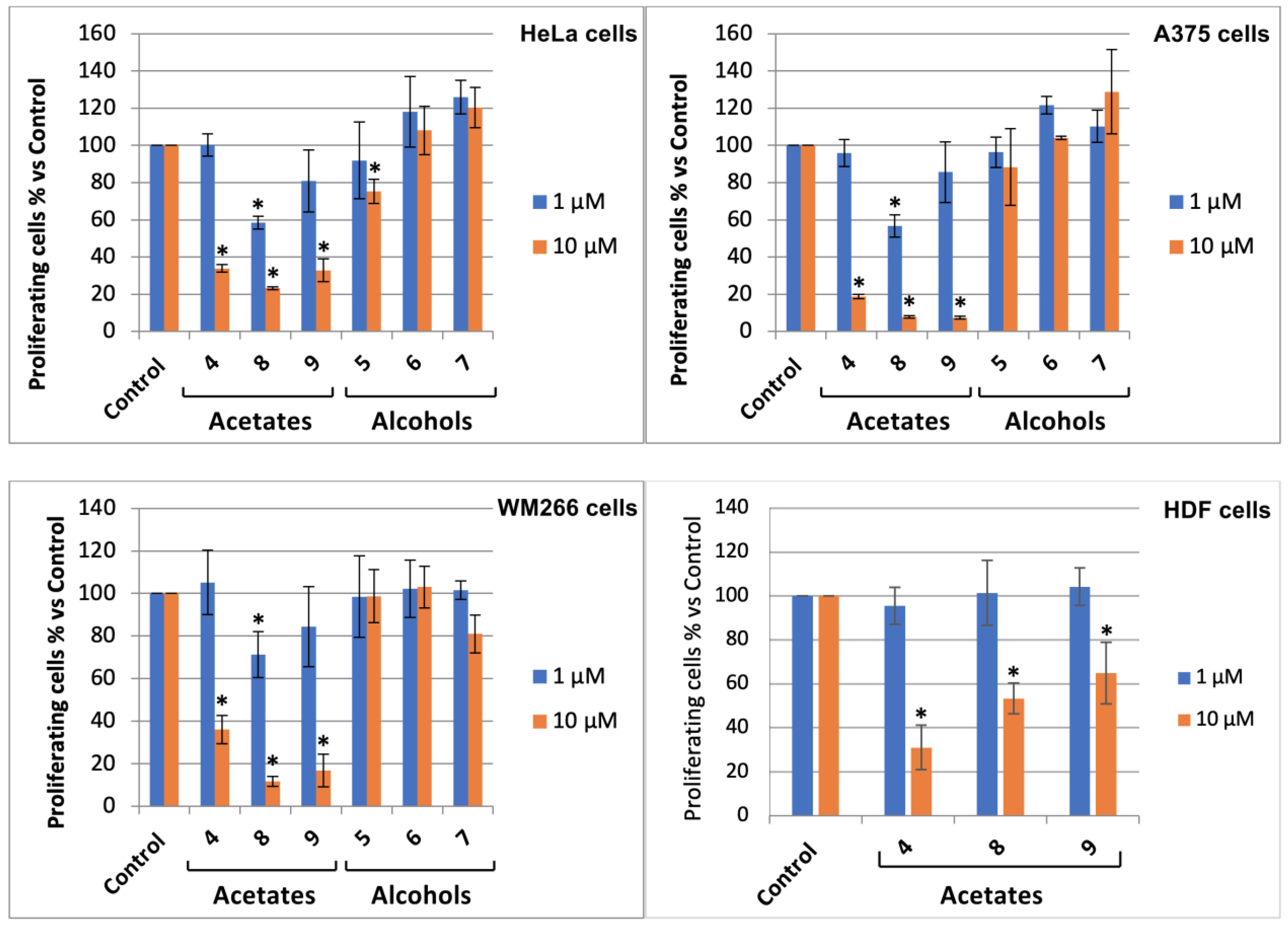
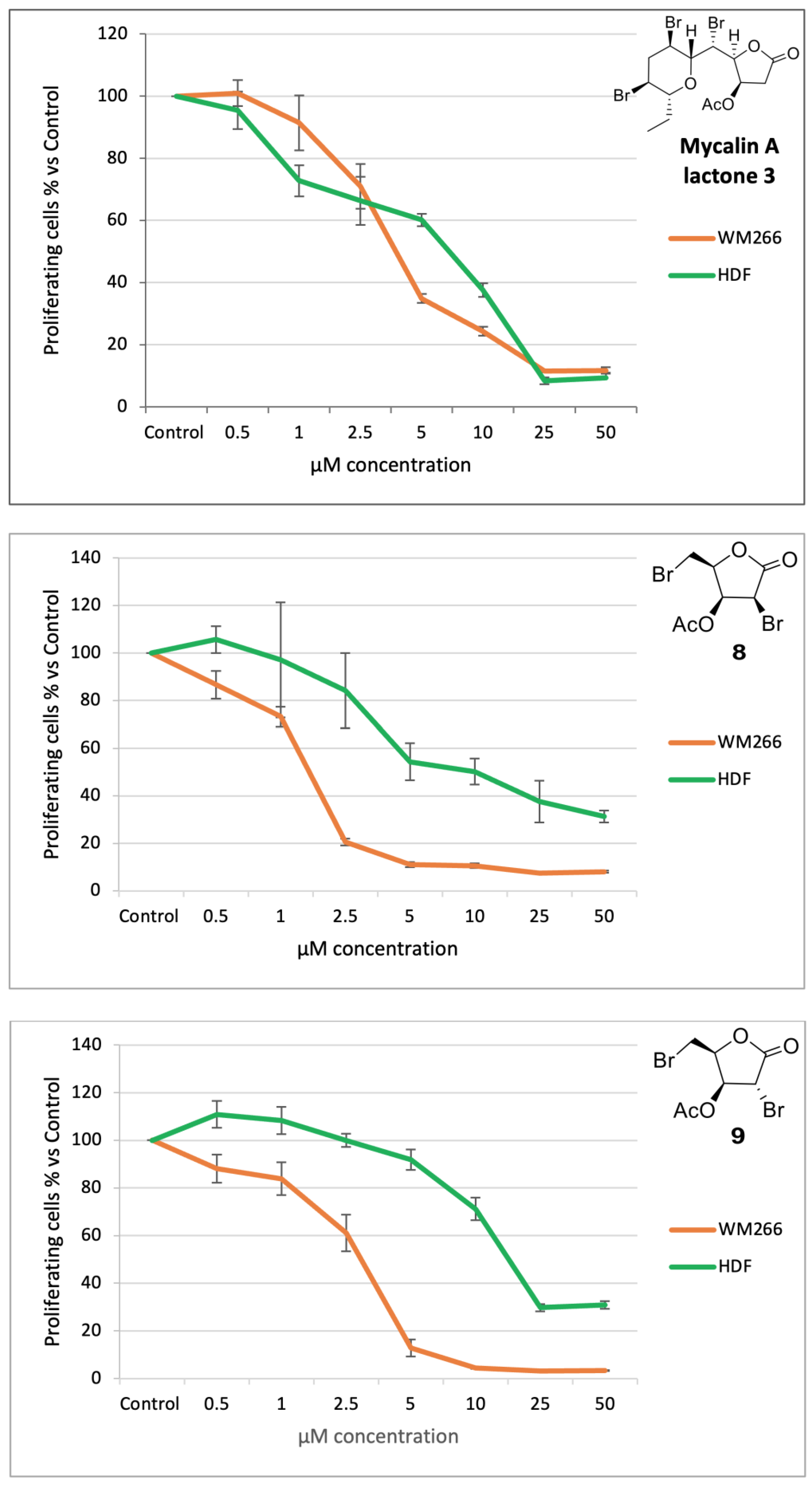
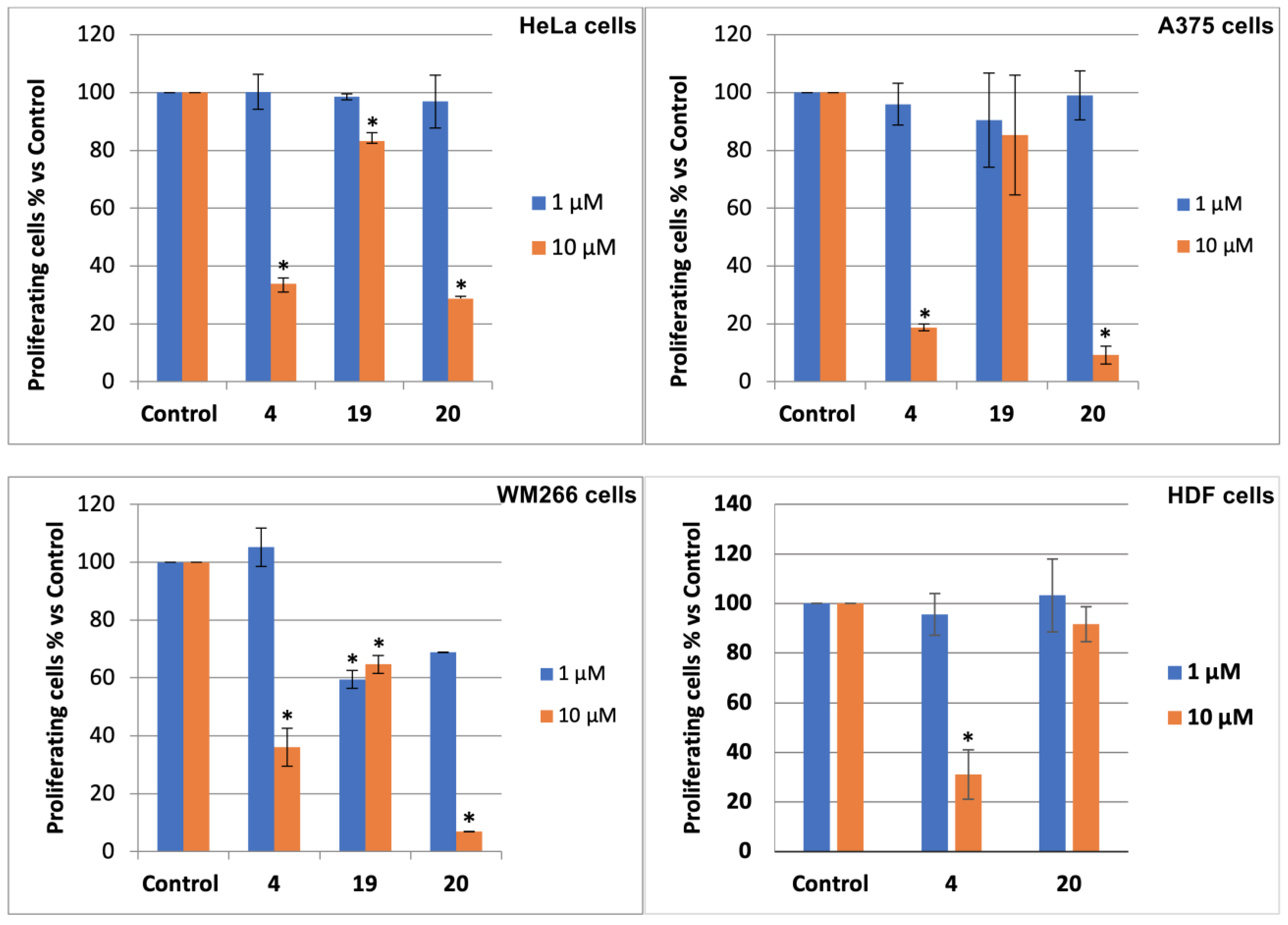
3.2. Bromolactones 4–9
3.2.1. Synthesis of Lactone 5
The following is a slightly modified procedure with respect to that reported in the literature [
10].
D-xylose (1.0 g, 6.66 mmol) was dissolved in water (2.7 mL) and the solution was cooled to 5 °C (ice-bath). Then, K
2CO
3 (1.13 g, 8.18 mmol) was added in portions under stirring, maintaining the temperature below 20 °C. To the clear solution, bromine was dropwise added (1.23 g, 400 μL); during the addition, the temperature was maintained in the range 5–10 °C. The cloudy, yellow-orange mixture was stirred for 30 min at this temperature and then overnight at room temperature. The process was quenched by addition of formic acid (65 μL) and the mixture was stirred for 20 min. Acetic acid (20 mL) was added and the aqueous suspension was taken to dryness under an air stream at 45–50 °C, furnishing a syrupy product.
1H-NMR analysis confirmed the formation of the D-xylono-1,4-lactone [
10].
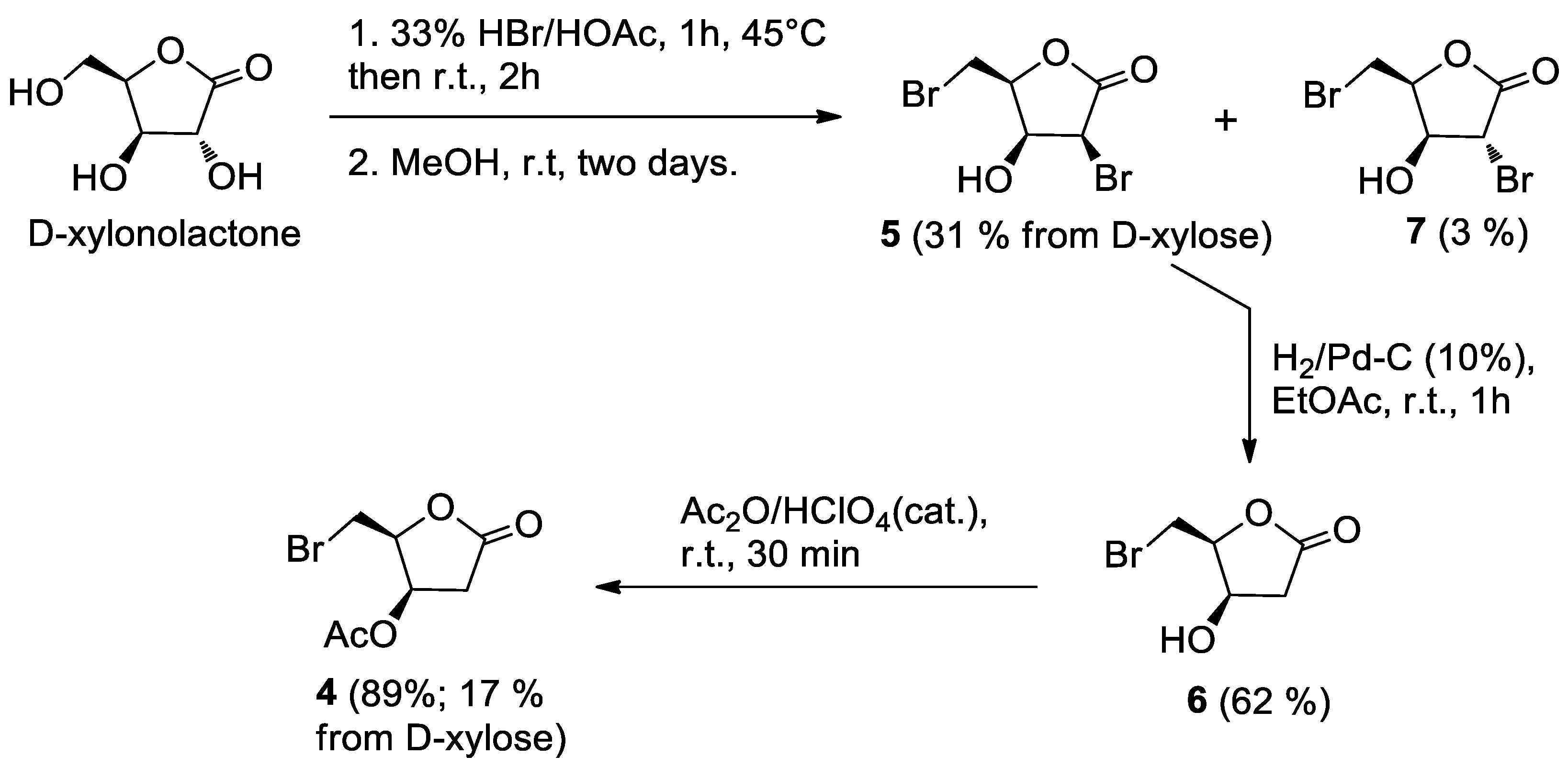
Acetic acid (3 mL) was added to the crude D-xylono-1,4-lactone obtained as above, followed by 33% HBr/AcOH (5.2 mL), and the mixture was stirred at 45 °C for 1 h and then at room temperature for 2 h. The TLC analysis (hexane-EtOAc, 1:1) showed the formation of mainly two products at RF = 0.5 (major product) and RF = 0.6 (minor product) attributable to the alcohol 5 and the corresponding acetate 8, respectively.
MeOH (8.5 mL) was added in portions to the above mixture at 0 °C over a period of 30 min. After stirring for two days at room temperature, TLC analysis showed the complete conversion of the acetate 8 into the corresponding alcohol 5. The mixture was filtered through a Büchner funnel to remove KBr, giving a clear solution. The MeOH was evaporated under reduced pressure and the mixture was co-evaporated twice with water (2 × 3 mL). The resulting solid was partitioned between water and EtOAc and the aqueous phase was extracted twice with EtOAc. The combined organic phases were dried, filtered, and taken to dryness. Residual acetic acid was azeotropically removed with n-heptane (2 × 5 mL) furnishing a brown oil (1.16 g). Column chromatography on silica gel, eluting with n-hexane-EtOAc (2:8), gave alcohol 5 (564 mg) contaminated by a small amount (by 1H-NMR) of 7 (5: 31%; 7: 3%). Pure compounds 5 and 7, suitable for biological assays, were obtained by HPLC (analytical silica column, mobile phase: n-hexane-EtOAc, 65:35).
5 (3S,4S,5S)-3-bromo-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]
D20 = –19.7 (c = 1.59, CHCl
3). IR (neat) ν
max 3420 (br), 1770 cm
−1.
1H-NMR [
10,
24] (500 MHz, CDCl
3): δ 4.80 (1H, d,
J = 4.4, H-2), 4.69 (1H, m, H-4), 4.63 (1H, bdd,
J = 3.5, 3.5, H-3), 3.73 (1H, A part of an AB system further coupled,
J = 10.1, 8.7, Ha-5), 3.67 (1H, B part of an AB system further coupled,
J = 10.1, 5.8, Hb-5), 2.60 (1H, br, OH).
13C-NMR: (125 MHz, CDCl
3): [
25] δ 169.4, 80.3, 68.8, 48.2, 25.9. HRESIMS (High resolution electrospray ionization mass spectrometry)
m/
z: calcd for C
5H
679Br
2NaO
3 294.8581 [M + Na]
+, found: 294.8566 [M + Na]
+, 296.8547 [M + Na + 2]
+, 298.8523 [M + Na + 4]
+.
7 (3R,4S,5S)-3-bromo-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = −7.80 (c = 1.73, CHCl3). IR (neat) νmax 3420 (br), 1770 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ: 4.99 (1H, dt, J = 7.5, 7.5, 3.4, H-4), 4.66 (1H, d, J = 3.4, H-3), 4.24 (1H, s, H-2), 3.67 (2H, d, J = 3.4, H2-5), 3.4–2.7 (1H, br, OH). 13C-NMR: (125 MHz, CDCl3): δ 171.2, 80.6, 73.9, 41.1, 25.5. HRESIMS m/z: calcd for C5H679Br2NaO3 294.8581 [M + Na]+, found: 294.8570 [M + Na]+, 296.8548 [M + Na + 2]+, 298.8530 [M + Na + 4]+.
3.2.2. Alcohol 6
Pd/C (10% w/w, 8.0 mg) and Et3N (30 μL) were added to compound 5 (53.0 mg, 0.193 mmol) in EtOAc (1.5 mL), and the mixture was stirred under a hydrogen atmosphere. A vacuum-fill technique was employed using a hydrogen balloon and a three-way vacuum adapter. After 1 h, the reaction mixture was filtered over celite, and the filtrate was taken to dryness under reduced pressure to give a mixture of the starting product 5 and the corresponding C-2 debrominated lactone 6 (39.4 mg). Column chromatography on silica gel (eluent EtOAc) gave unreacted 5 (16.2 mg) and the desired compound 6 (23.4 mg, 62%), as a collarless oil. Further HPLC purification on an analytical silica column (eluent: n-hexane-EtOAc, 3:7) afforded a sample of 6 suitable for biological assays.
6 (4R,5S)-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = −23.9 (c = 0.27, CHCl3). IR (neat) νmax 3430 (br), 1770 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 4.73 (1H, bdd, J = 5.6, 3.8, H-3), 4.61 (1H, m, H-4), 3.69–3.62 (2H, m, H2-5), 2.84 (1H, dd, J = 17.9, 5.6, Ha-2) 2.65 (1H, d, J = 17.9, Hb-2), 2.4–2.0 (1H, broad, OH). 13C-NMR: (125 MHz, CDCl3): δ 174.7, 82.1, 67.7, 38.6, 26.4. HRESIMS m/z: calcd for C5H779BrNaO3 216.9476 [M + Na]+, found: 216.9461 [M + Na]+, 218.9449 [M + Na + 2]+.
3.2.3. Acetate 4
A 65% HClO4 solution (two drops) was added to alcohol 6 (11.8 mg, 0.0605 mmol) in acetic anhydride (1 mL). After 30 min, TLC analysis (n-hexane-EtOAc, 2:8) showed the complete conversion of 6 (RF = 0.5) into a less polar product (RF = 0.7). Ice was added and the mixture was extracted with CH2Cl2 (3 × 3 mL). The organic phase was washed with water, dried and taken to dryness. Filtration through a short pad of silica gel gave crude 4 (14.5 mg) as an oil. Further purification by HPLC on an analytical silica column (n-hexane-EtOAc, 65:35) gave pure acetate 4 (12.7 mg, 89%) as a white solid which crystallized on standing.
4 (2S,3R)-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = −47.8 (c = 0.21, CHCl3). IR (neat) νmax 1796 (s), 1736 (s), 1237 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 5.58 (1H, ddd, J = 5.8, 4.4, 1.1, H-3), 4.76 (1H, ddd, J = 8.3, 5.9, 4.3, H-4), 3.59 (2H, AB system further coupled, A part: J = 10.3, 5.9; B part: J = 10.3, 8.3), 2.93 (1H, dd, J = 18.3, 6.0, Ha-2), 2.67 (1H, dd, J = 18.3, 1.1, Hb-2), 2.13 (3H, s, acetate). 13C-NMR: (175 MHz, CDCl3): δ 173.0, 169.6, 80.3, 69.3, 36.8, 25.7, 20.7. HRESIMS m/z: calcd for C7H979BrNaO4 258.9582 [M + Na]+, found: 258.9560 [M + Na]+, 260.9539 [M + Na + 2]+.
3.2.4. Lactone 7
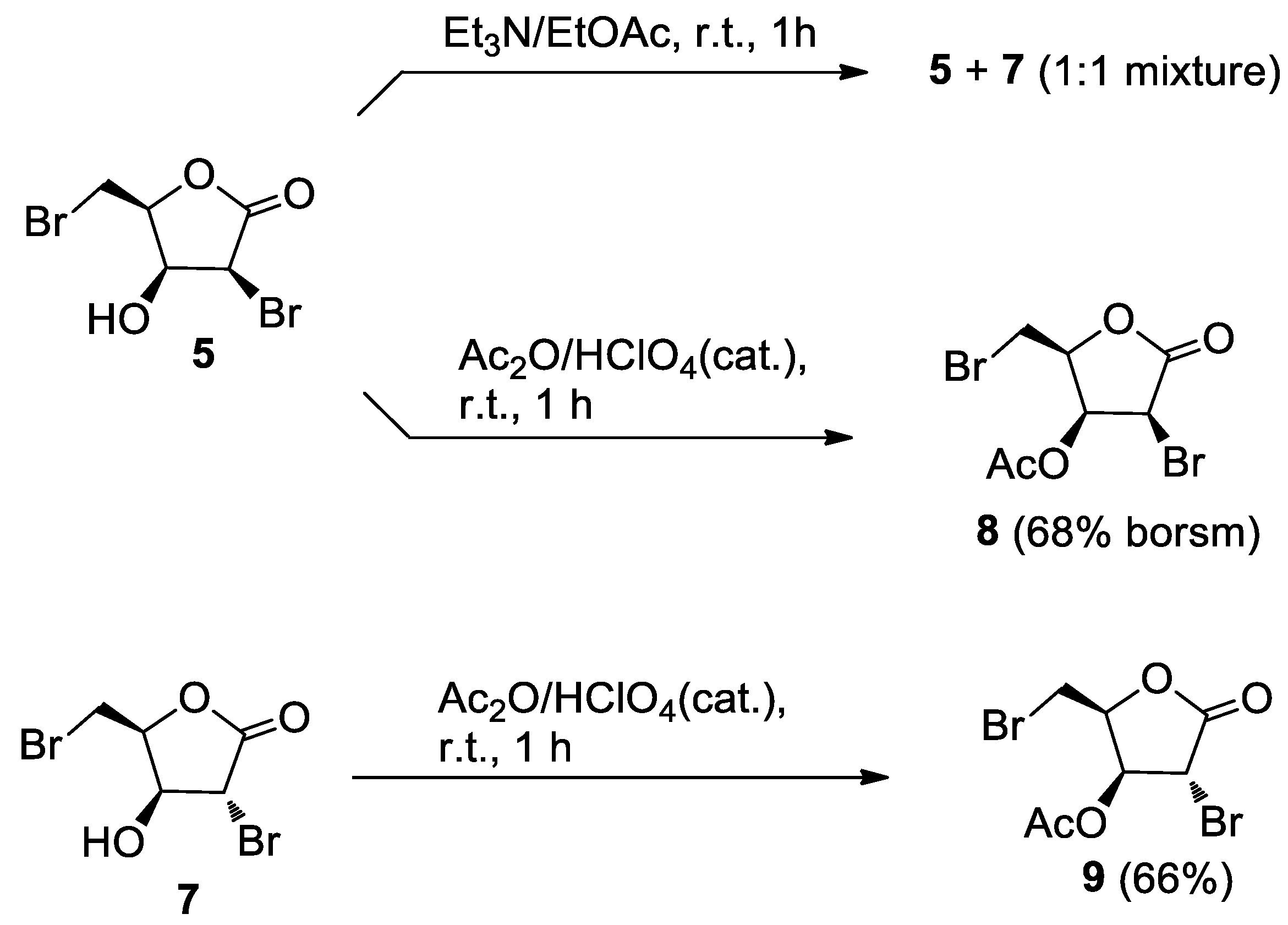
Et3N (four drops) was added to a stirred solution of 5 (5.5 mg, 0.0201 mmol) in EtOAc (1 mL). After 1h, the reaction mixture was taken to dryness and the residue was filtered on a short pad of silica gel eluting with EtOAc to give 5.3 mg of an oil. TLC analysis showed that it was essentially composed by two products at RF = 0.6 and RF = 0.65. HPLC separation on an analytical silica column (n-hexane/EtOAc, 8:2) furnished the starting products 5 (2.5 mg) and its C-2 epimeric alcohol 7 (2.5 mg).
3.2.5. Acetate 8
A 65% HClO4 solution (two drops) was added to alcohol 5 (22.8 mg, 0.0832 mmol) in acetic anhydride (0.5 mL). After 1 h, TLC analysis (n-hexane-EtOAc, 1:1) showed the almost complete conversion of 5 (RF = 0.4) into a less polar product (RF = 0.6). Usual work-up gave a white solid (22.3 mg). Further separation by analytical HPLC (n-hexane-EtOAc, 8:2) gave the starting product 5 (3.0 mg) and acetate 8 (15.1 mg, 68% borsm), as a crystalline solid.
8 (2S,3S,4S)-4-bromo-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = −61.9 (c = 0.78, CHCl3). IR (neat) νmax 1786 (s), 1758 (s), 1197 cm−1. 1H-NMR (500 MHz, CDCl3): δ 5.90 (1H, dd, J = 5.4, 4.4, H-3), 4.83 (1H, m, H-4), 4.79 (1H, d, J = 5.5, H-2), 3.63 (1H, A part of an AB system further coupled, J = 10.2, 6.2, Ha-5), 3.67 (1H, B part of an AB system further coupled, J = 10.2, 8.8, Hb-5), 2.20 (3H, s, acetate). 13C-NMR: (125 MHz, CDCl3): δ 168.9, 168.7, 78.8, 69.0, 42.8, 25.4, 20.3. HRESIMS m/z: calcd for C7H879Br2NaO4 336.8687 [M + Na]+, found: 336.8685 [M + Na]+, 338.8662 [M + Na + 2]+, 340.8643 [M + Na + 4]+.
3.2.6. Acetate 9
Alcohol
7 (20.0 mg, 0.0730 mmol) was acetylated under the same conditions used for
6. After 1 h, TLC analysis (
n-hexane-EtOAc, 6:4) showed the complete conversion of
7 (
RF = 0.4) into a less polar product (
RF = 0.5). Usual work-up followed by filtration through a short pad of silica gel gave crude
9 (24.1 mg). Further purification by analytical HPLC (
n-hexane-EtOAc, 8:2) afforded acetate
9 [
11] (15.1 mg, 66%) as an oil.
9 (2S,3S,4R)-4-bromo-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = +9.1 (c = 0.71, CHCl3). IR (neat) νmax 1780 (s), 1750 (s), 1226 (s), 1187 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 5.47 (1H, d, J = 3.4, H-3), 5.11 (1H, m, H-4), 4.24 (1H, s, H-2), 3.64 (1H, A part of an AB system further coupled, J = 10.1, 5.7, Ha-5), 3.55 (1H, B part of an AB system further coupled, J = 10.1, 9.0, Hb-5), 2.16 (3H, s, acetate). 13C-NMR: (125 MHz, CDCl3): δ 169.5, 169.0, 78.6, 74.7, 38.3, 24.8, 20.4. HRESIMS m/z: calcd for C7H879Br2NaO4 336.8687 [M + Na]+, found: 336.8676 [M + Na]+, 338.8654 [M + Na + 2]+, 340.8634 [M + Na + 4]+.
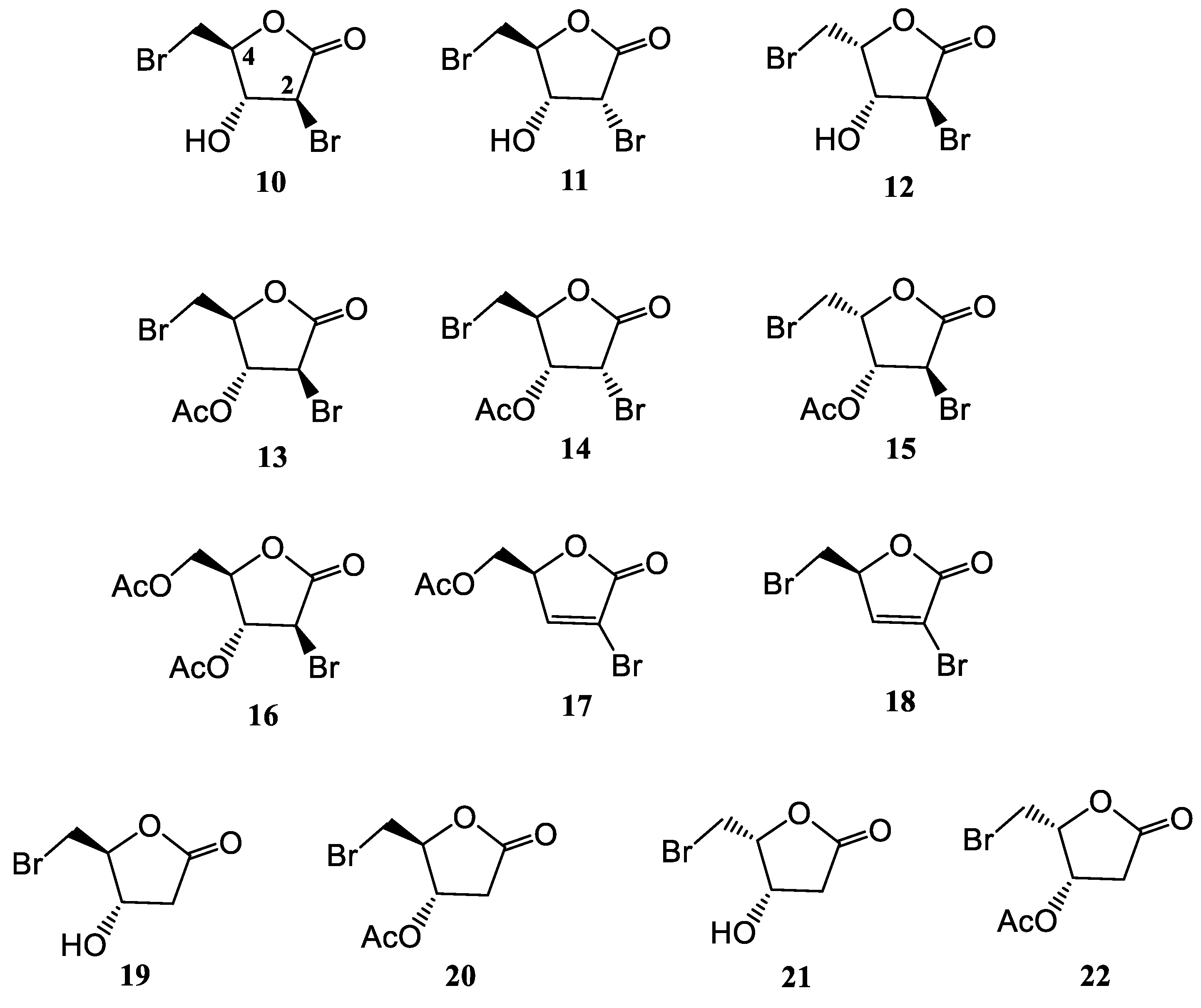
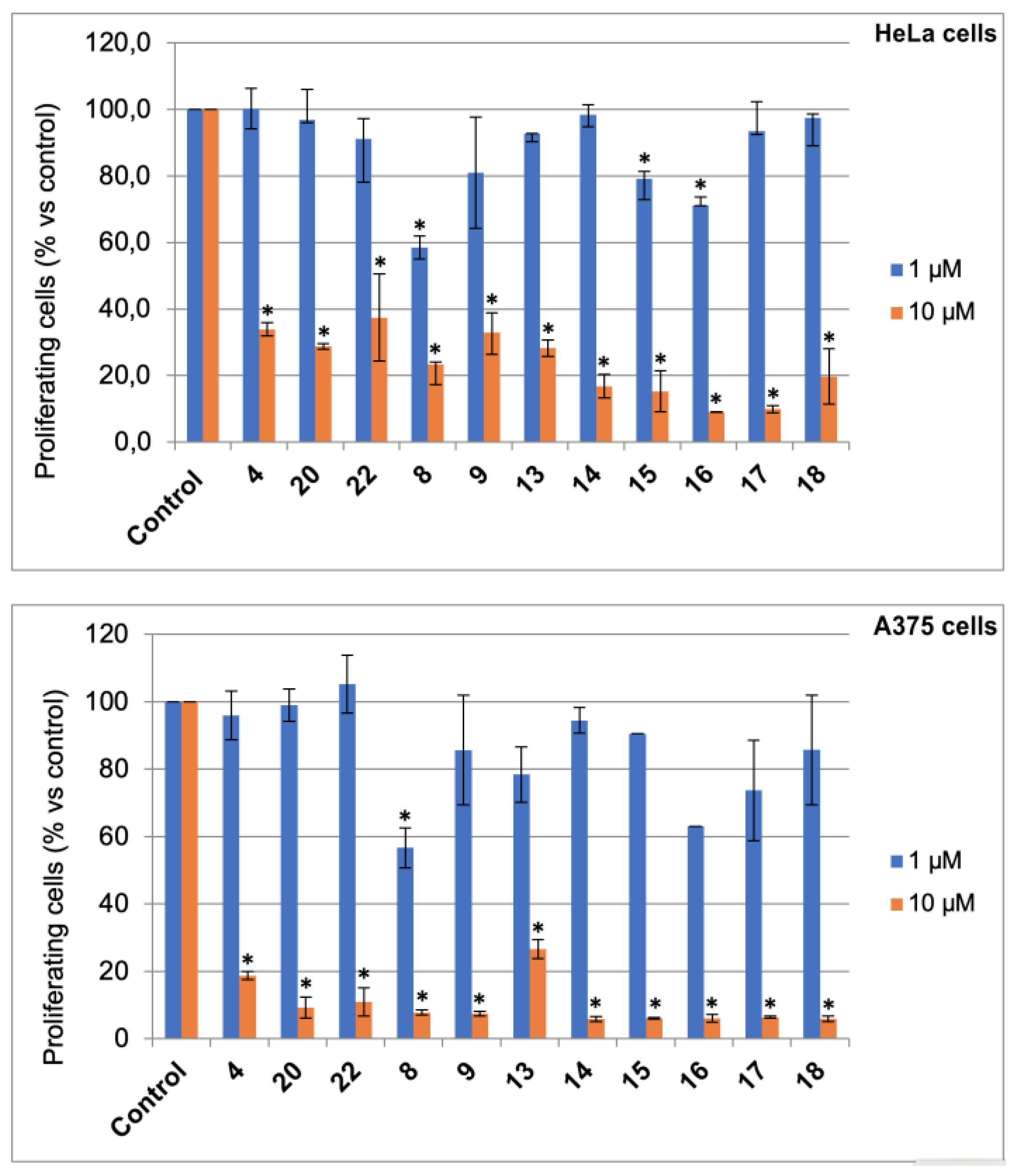
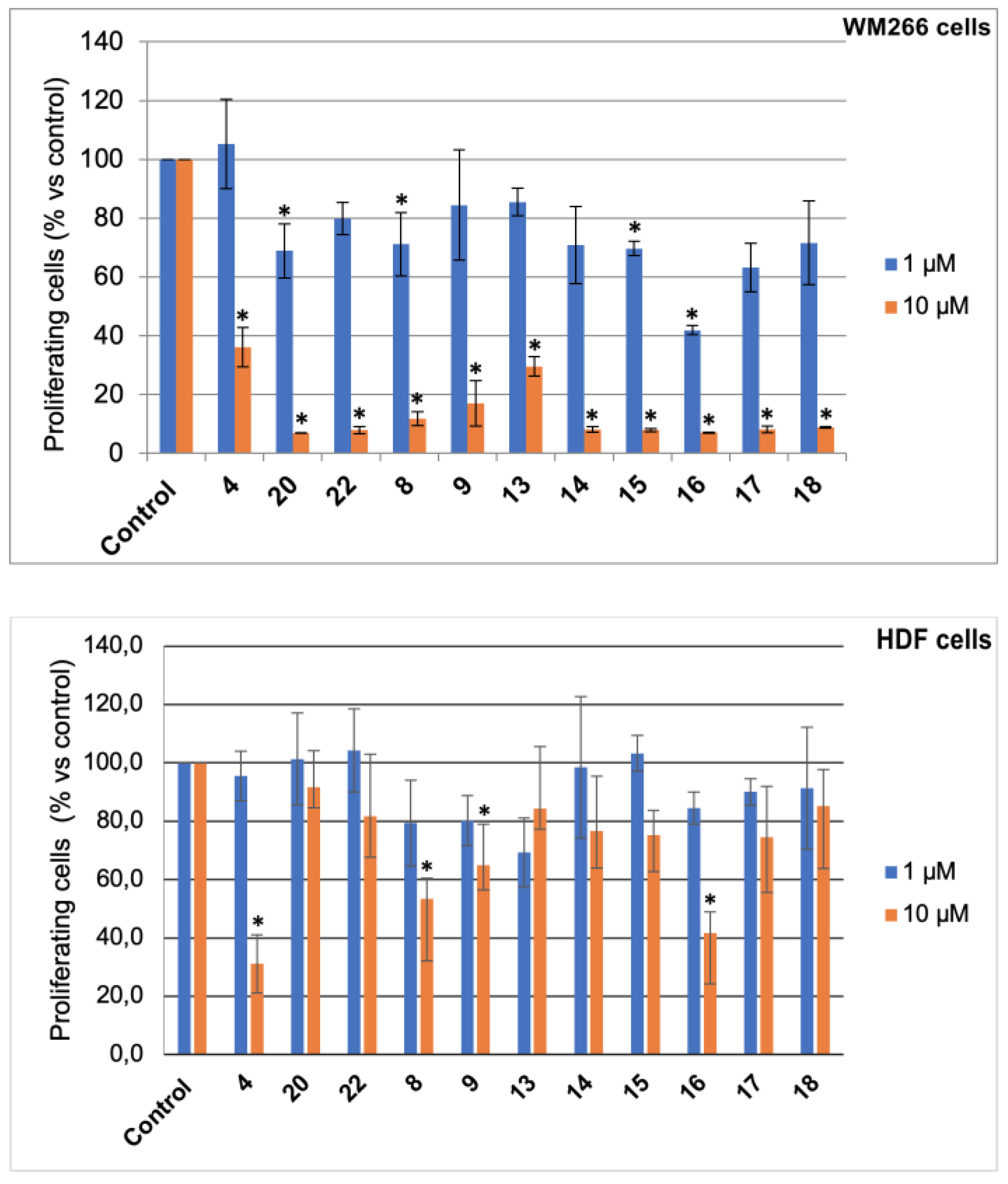
3.3. Bromolactones 10–22
D-ribonolactone was synthesized starting from D-ribose according to a reported procedure [
15] and obtained in pure form by crystallization from EtOH. Its purity was confirmed by
1H and
13C-NMR spectra.
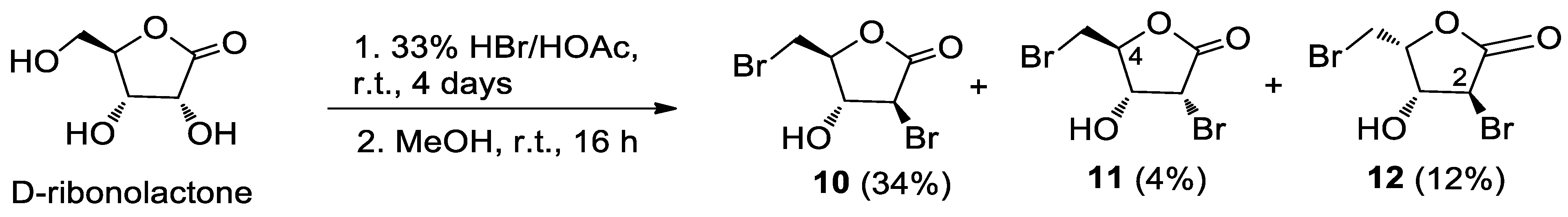
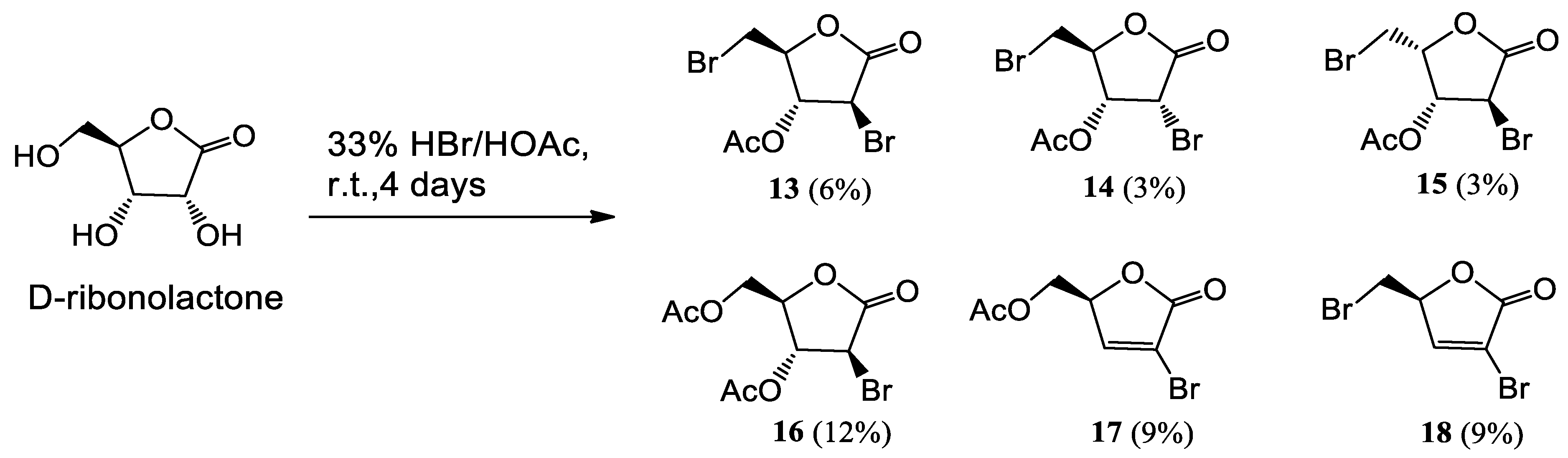
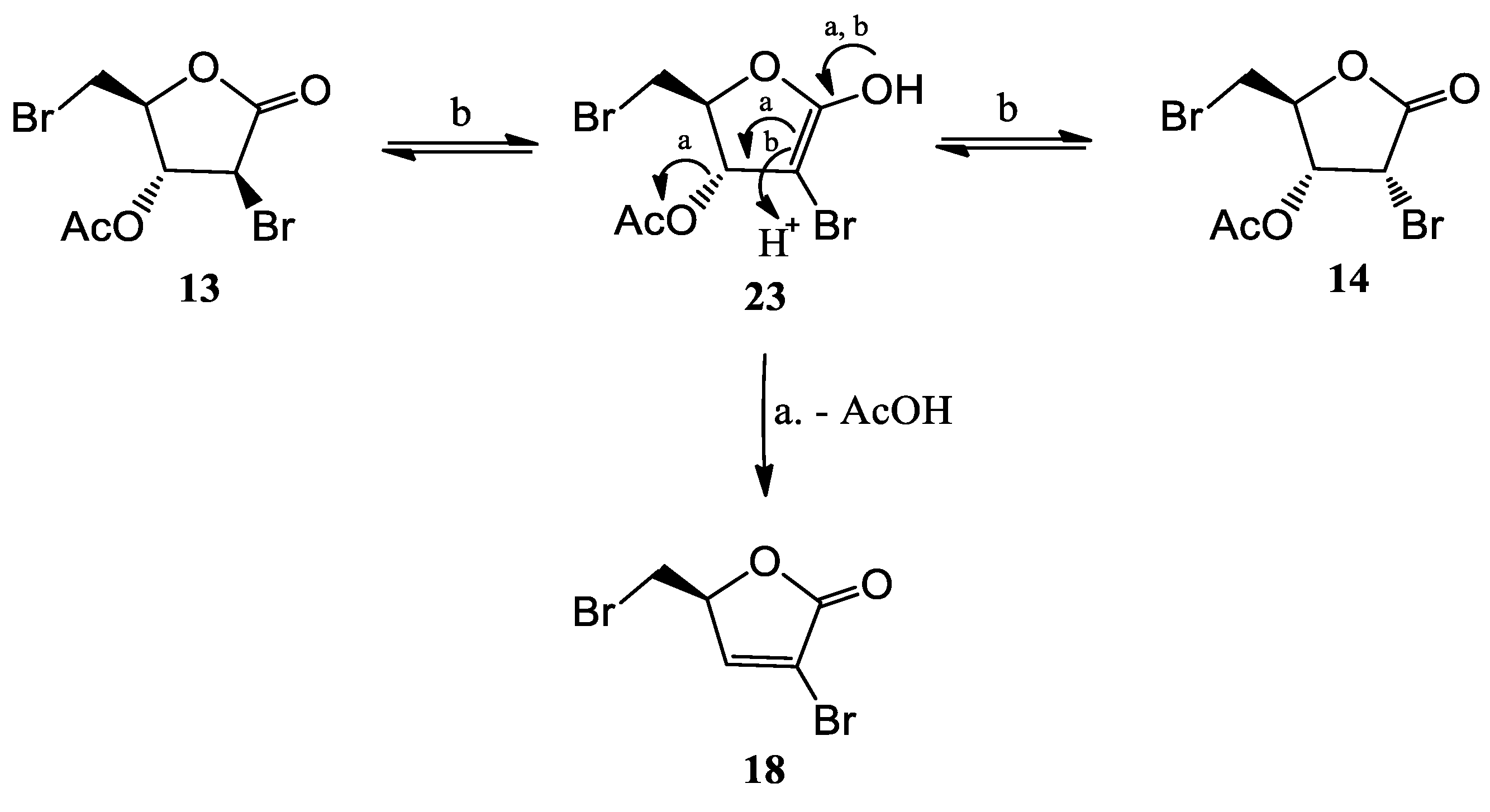
To D-ribonolactone (526 mg, 3.55 mmol), obtained as above, 33% HBr/AcOH (3.0 mL) was added, and the mixture stirred at r.t. for four days. Then, one third of the reaction mixture was partitioned between water and CHCl3 and the aqueous phase was extracted with CHCl3 (3 × 5 mL). The combined organic phases were dried, filtered and taken to dryness. Residual acetic acid was azeotropically removed with n-heptane (2 × 2 mL), to give an oil that was filtered through a short pad of silica gel eluting with EtOAc and then EtOAc/MeOH in (95:5). The combined eluates were taken to dryness furnishing an oily product (167.2 mg). Separation by HPLC on a silica column (250 × 10 mm), using n-hexane-EtOAc (7:3) as the mobile phase gave compounds 13 (22.3 mg, 6%), 14 (11.2 mg, 3%), 15 (11.1 mg, 3%), 16 (41.7 mg, 12%), 17 (24.9 mg, 9%), and 18 (25.0 mg, 9%).
MeOH (5 mL) was added in portions to the remaining original reaction mixture in a water bath. After 16 h, the reaction mixture was taken to dryness under an air stream at 50 °C. The resulting oily semi-solid product was partitioned between water and CHCl3 and the aqueous phase was extracted with CHCl3 (3 × 5 mL). The combined organic phases were dried, filtered and taken to dryness. The residual acetic acid was azeotropically removed with n-heptane (2 × 3 mL). Separation of the crude by HPLC on a silica column (250 × 10 mm), using n-hexane-EtOAc (6:4) as the mobile phase, gave the slightly impure alcohol 10 (91.0 mg) and pure 11 (9.4 mg, 4%) and 12 (27.0 mg, 12%). The further purification of 10 by HPLC on an analytical silica column, using n-hexane-EtOAc (75:25) as the mobile phase, furnished pure 10 (82 mg, 34%).
10 (3S,4R,5S)-3-bromo-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = +14.5 (c = 1.21, CHCl3). IR (neat) νmax 3445 (br), 1785 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 4.63 (1H, dd, J = 6.7, 5.8, H-3), 4.54 (1H, ddd, J = 5.8, 5.8, 4.8, H-4), 4.52 (1H, d, J = 6.7, H-2), 3.72 (1H, A part of an AB system further coupled, J = 11.4, 5.7, Ha-5), 3.67 (1H, B part of an AB system further coupled, J = 11.4, 4.8, Hb-5). 13C-NMR: (150 MHz, CDCl3): δ 169.4, 82.3, 78.0, 44.3, 29.9. HRESIMS m/z: calcd for C5H679Br2NaO3 294.8581 [M + Na]+, found: 294.8590 [M + Na]+, 296.8571 [M + Na + 2]+, 298.8549 [M + Na + 4]+.
11 (3R,4R,5S)-3-bromo-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = +16.2 (c = 0.23, CHCl3). IR (neat) νmax 3450 (br), 1784 (s) cm−1. 1H-NMR (700 MHz, CDCl3): δ 4.71 (1H, d, J = 6.0, H-2), 4.59 (1H, ddd, J = 5.4, 4.1, 4.1, H-4), 4.36 (1H, ddd, J = 6.8, 5.7, 5.7, H-3), 3.76 (1H, A part of an AB system further coupled, J = 11.9, 4.2, Ha-5), 3.67 (1H, B part of an AB system further coupled, J = 11.9, 3.9, Hb-5), 2.51 (1H, d, J = 6.8, OH). 13C-NMR: (175 MHz, CDCl3): δ 169.0, 81.7, 70.2, 45.9, 29.9. HRESIMS m/z: calcd for C5H679Br2NaO3 294.8581 [M + Na]+, found: 294.8579 [M + Na]+, 296.8561 [M + Na + 2]+, 298.8542 [M + Na + 4]+.
12 (3S,4R,5R)-3-bromo-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = +3.2 (c = 0.27, CHCl3). IR (neat) νmax 3432 (br), 1778 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 4.99 (1H, dt, J = 7.2, 7.2, 3.4, H-4), 4.66 (1H, dd, J = 3.4, 1.1, H-3), 4.25 (1H, bd, J = 1.1, H-2), 3.68 (2H, d, J = 7.2, H2-5). 13C-NMR: (125 MHz, CDCl3): δ 171.4, 80.7, 73.9, 41.1, 25.5. HRESIMS m/z: calcd for C5H679Br2NaO3 294.8581 [M + Na]+, found: 294.8563 [M + Na]+, 296.8543 [M + Na + 2]+, 298.8526 [M + Na + 4]+.
13 (2S,3R,4S)-4-bromo-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: IR (neat) νmax 1796 (s), 1748 (s), 1217 cm−1. 1H-NMR (700 MHz, CDCl3): δ 5.49 (1H, dd, J = 3.2, 3.2, H-3), 4.68 (1H, ddd, J = 6.2, 6.2, 3.1, H-4), 4.43 (1H, d, J = 3.4, H-2), 3.74 (2H, AB system further coupled; A part: J = 11.5, 5.5; B part: J = 11.5, 4.7, H2-5), 2.17 (3H, s, acetate). 13C-NMR: (175 MHz, CDCl3): δ 169.5, 169.2, 82.4, 77.8, 38.5, 30.0, 20.6. HRESIMS m/z: calcd for C7H879Br2NaO4 336.8687 [M + Na]+, found: 336.8690 [M + Na]+, 338.8670 [M + Na + 2]+, 340.8649 [M + Na + 4]+.
14 (2S,3R,4R)-4-bromo-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = +30.1 (c = 0.073, CHCl3). IR (neat) νmax 1797 (s), 1748 (s), 1237 (s) cm−1. 1H-NMR (700 MHz, CDCl3): δ 5.16 (1H, dd, J = 6.2, 6.2, H-3), 4.84 (1H, d, J = 6.5, H-2), 4.75 (1H, ddd, J = 5.7, 3.8, 3.8, H-4), 3.75 (1H, A part of an AB system further coupled, J = 12.0, 3.9, Ha-5), 3.65 (1H, B part of an AB system further coupled, J = 12.0, 3.6, Hb-5), 2.20 (3H, s, acetate). 13C-NMR: (175 MHz, CDCl3): δ 169.3, 168.7, 79.0, 70.9, 40.8, 30.0, 20.4. HRESIMS m/z: calcd for C7H879Br2NaO4 336.8687 [M + Na]+, found: 336.8692 [M + Na]+, 338.8671 [M + Na + 2]+, 340.8653 [M + Na + 4]+.
15 (2R,3R,4S)-4-bromo-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = −22.4 (c = 0.23, CHCl3). IR (neat) νmax 1796 (s), 1753 (s), 1212 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 5.48 (1H, d, J = 3.5, H-3), 5.11 (1H, ddd, J = 9.0, 5.7, 3.5, H-4), 4.24 (1H, s, H-2), 3.64 (1H, A part of an AB system further coupled, J = 10.2, 5.6, Ha-5), 3.55 (1H, B part of an AB system further coupled, J = 10.2, 9.0, Hb-5), 2.17 (3H, s, acetate). 13C-NMR: (125 MHz, CDCl3): δ 169.5, 169.0, 78.6, 74.7, 38.3, 24.8, 20.4. HRESIMS m/z: calcd for C7H879Br2NaO4 336.8687 [M + Na]+, found: 336.8671 [M + Na]+, 338.8652 [M + Na + 2]+, 340.8631 [M + Na + 4]+.
16 ((2R,3R,4S)-3-acetoxy-4-bromo-5-oxotetrahydrofuran-2-yl)methyl acetate: [α]D20 = −4.9 (c = 0.72, CHCl3). IR (neat) νmax 1803 (s), 1741 (s), 1220 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 5.39 (1H, dd, J = 3.2, 3.2, H-3), 4.64 (1H, m, H-4), 4.47 (1H, d, J = 3.5, H-2), 4.41 (2H, AB system further coupled; A part: J = 12.4, 3.9; B part: J = 12.4, 5.1, H2-5), 2.14 (3H, s, acetate), 2.12 (3H, s, acetate). 13C-NMR: (125 MHz, CDCl3): δ 170.2, 169.6, 169.5, 81.5, 76.9, 62.5, 38.7, 20.7, 20.5. HRESIMS m/z: calcd for C9H1179BrNaO6 316.9637 [M + Na]+, found: 316.9643 [M + Na]+, 318.9621 [M + Na + 2]+.
17 (S)-(4-bromo-5-oxo-2,5-dihydrofuran-2-yl)methyl acetate: [α]D20 = −65.9 (c = 0.42, CHCl3). IR (neat) νmax 1776 (s), 1744 (s), 1227 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 7.48 (1H, d, J = 1.9, H-3), 5.16 (1H, ddd, J = 4.7, 4.2, 1.9, H-4), 4.35 (2H, AB system further coupled: A part J = 12.2, 4.2; B part J = 12.2, 4.7, H2-5), 2.08 (3H, s, acetate). 13C-NMR: (125 MHz, CDCl3): δ 170.4, 167.5, 148.6, 115.0, 80.1, 62.1, 20.5. HRESIMS m/z: calcd for C7H779BrNaO4 256.9425 [M + Na]+, found: 256.9428 [M + Na]+, 258.9410 [M + Na + 2]+.
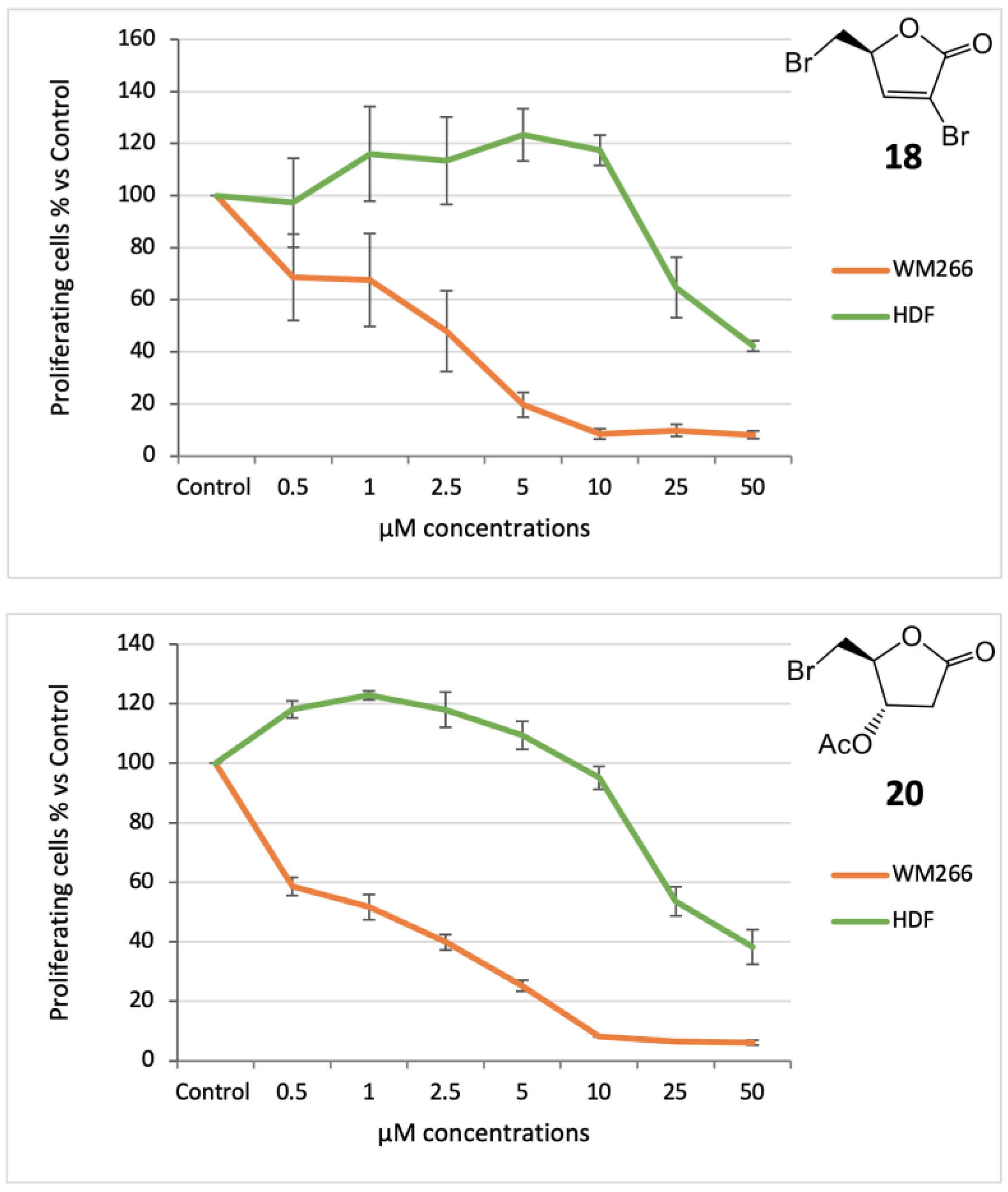
18 (S)-3-bromo-5-(bromomethyl)furan-2(5H)-one: [α]
D20 = −39.3 (c = 0.17, CHCl
3). IR (neat) ν
max 1780 (s), 1609, 1151, 1033, 988 cm
−1.
1H-NMR [
20] (500 MHz, CDCl
3): δ 7.59 (1H, d,
J = 1.8, H-3), 5.20 (1H, ddd,
J = 7.0, 4.5, 1.8, H-4), 3.67 (1H, A part of an AB system further coupled,
J = 10.8, 4.5, Ha-5), 3.49 (1H, B part of an AB system further coupled,
J = 10.8, 7.0, Hb-5).
13C-NMR: (125 MHz, CDCl
3): δ 167.2, 150.1, 115.6, 80.0, 29.6. HRESIMS
m/
z: calcd for C
5H
479Br
2NaO
2 276.8476 [M + Na]
+, found: 276.8471 [M + Na]
+, 278.8452 [M + Na + 2]
+, 280.8430 [M + Na + 4]
+.
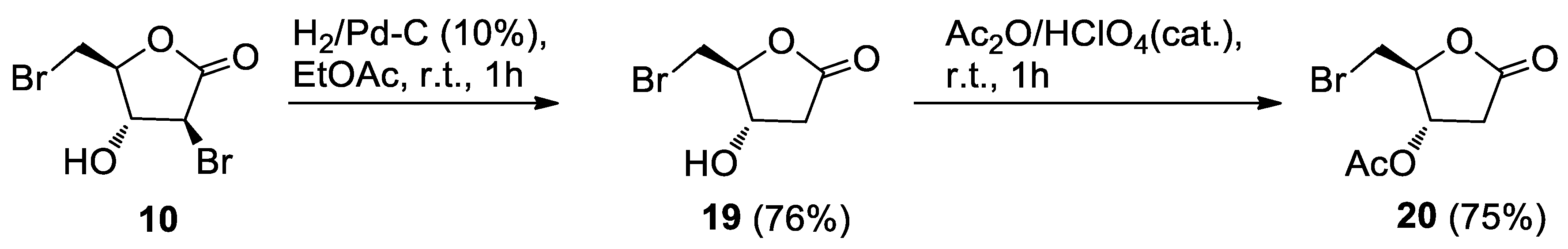
3.3.1. Alcohol 19
Pd/C (10% w/w, 4.5 mg) and Et3N (15 μL) were added to the alcohol 10 (22.5 mg, 0.0822 mmol) in EtOAc (3 mL), and the mixture was stirred under a hydrogen atmosphere. The same equipment used for the hydrogenation of 5 was used. After 1 h, the reaction mixture was worked-up as for 5. Further filtration on a short pad of silica gel, eluting with EtOAc, gave crude 19. HPLC purification on an analytical silica column (n-hexane-EtOAc, 3:7) afforded the pure alcohol 19 (12.8 mg, 76%) as an oil.
19 (4S,5S)-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: [α]D20 = −16.4 (c = 0.23, CHCl3). IR (neat) νmax 3421 (br), 1776 (s), 1172 (s) cm−1. 1H-NMR (500 MHz, CDCl3): δ 4.61, 4.56 (1H each. m’s, H-3 and H-4), 3.61 (1H, A part of an AB system further coupled, J = 11.3, 3.9, Ha-5), 3.53 (1H, B part of an AB system further coupled, J = 11.3, 5.9, Hb-5), 2.99 (1H, dd, J = 18.3, 7.4, Ha-2), 2.58 (1H, dd, J = 18.3, 3.7, Hb-2). 13C-NMR: (150 MHz, CDCl3): δ 174.0, 84.8, 70.6, 37.8, 31.2. HRESIMS m/z: calcd for C5H779BrNaO3 216.9476 [M + Na]+, found: 216.9485 [M + Na]+, 218.9463 [M + Na + 2]+.
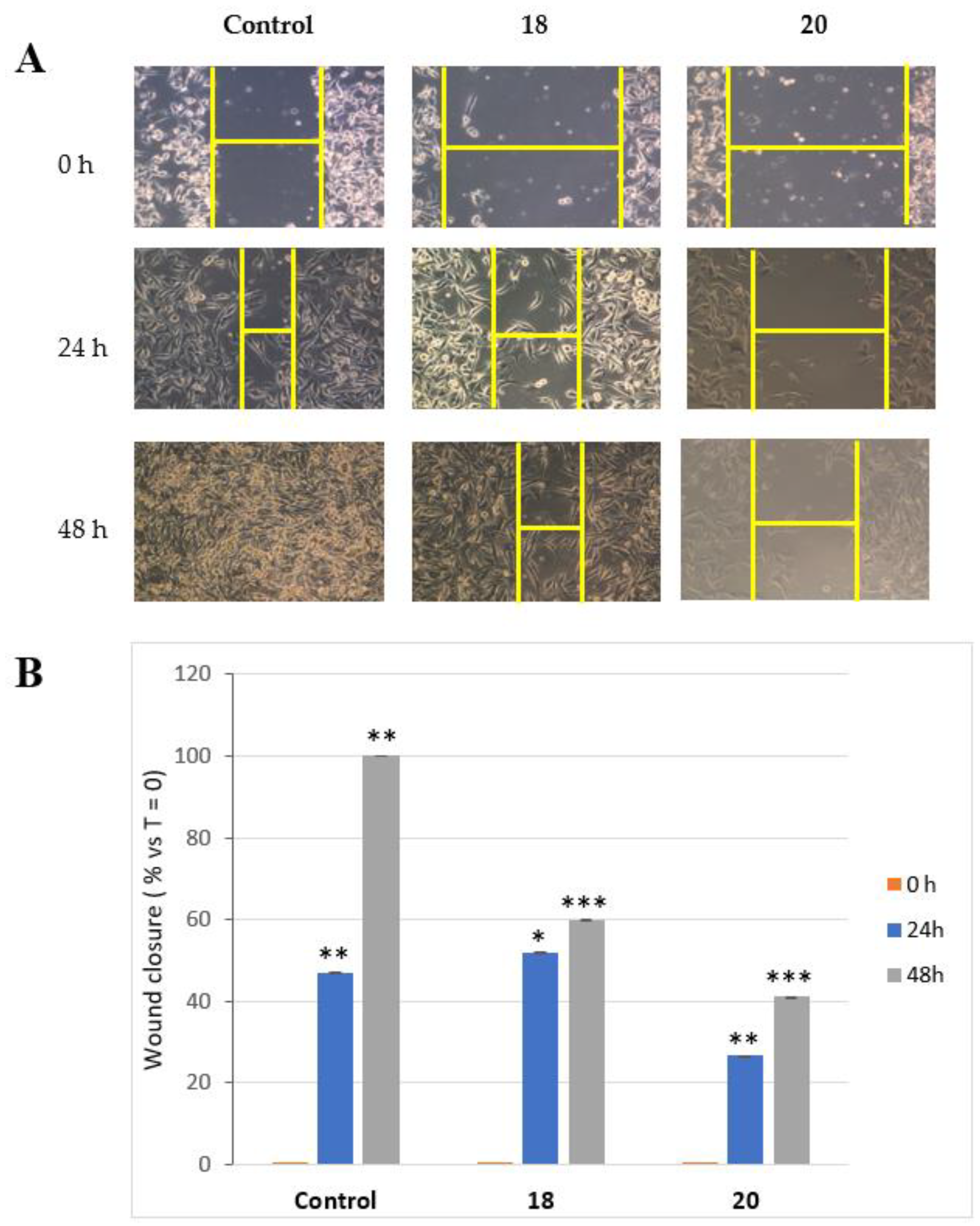
3.3.2. Acetate 20
A 65% HClO4 solution (two drops) was added to the alcohol 19 (7.0, 0.0359 mmol) in acetic anhydride (0.5 mL). After 1 h, the mixture was worked-up as usual. Purification by HPLC on an analytical silica column (n-hexane-EtOAc, 3:7) gave pure acetate 20 (6.4 mg, 75%), as a clear oil.
20 (2S,3S)-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: [α]D20 = −19.5 (c = 0.43, CHCl3). IR (neat) νmax 1790 (s), 1741 (s), 1236 (s), 1162, 1041 cm−1. 1H-NMR (600 MHz, CDCl3): δ 5.25 (1H, d, J = 8.0, H-3), 4.73 (1H, bs, H-4), 3.74 (1H, A part of an AB system further coupled, J = 11.5, Ha-5), 3.65 (1H, B part of an AB system further coupled, J = 11.5, 3.8, Hb-5), 3.13 (1H, dd, J = 19.0, 8.0, Ha-2), 2.62 (1H, d, J = 19.0, Hb-2), 2.11 (3H, s, acetate). 13C-NMR: (150 MHz, CDCl3): δ 173.4, 170.4, 82.6, 72.6, 35.0, 32.2, 20.7. HRESIMS m/z: calcd for C7H979BrNaO4 258.9582 [M + Na]+, found: 258.9583 [M + Na]+, 260.9564 [M + Na + 2]+.
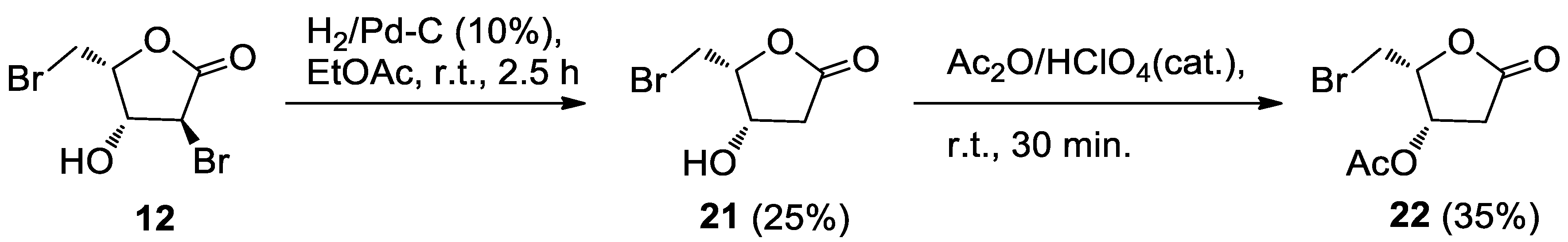
3.3.3. Alcohol 21
Pd/C (10% w/w, 3.0 mg) and Et3N (10 μL) were added to the alcohol 12 (11.9 mg, 0.0434 mmol) in EtOAc (6 mL) and the mixture was stirred under a hydrogen atmosphere. The same equipment employed for the hydrogenation of 5 was used. After 2.5 h, the reaction mixture was worked-up as usual. Further filtration through a short pad of silica gel eluting with EtOAc gave crude 21. HPLC purification on an analytical silica column (n-hexane-EtOAc, 2:8) afforded pure 21 (3.4 mg, 25%) as an oil.
21 (4S,5R)-5-(bromomethyl)-4-hydroxydihydrofuran-2(3H)-one: IR (neat) νmax 3419 (br), 1768 (s) cm−1. 1H-NMR (700 MHz, CDCl3): δ 4.73 (1H, bdd, J = 5.6, 3.8, H-3), 4.61 (1H, m, H-4), 3.69–3.62 (2H, m, H2-5), 2.84 (1H, dd, J = 17.9, 5.6, Ha-2) 2.65 (1H, d, J = 17.9, Hb-2), 2.4–2.0 (1H. broad, OH). 13C-NMR: (175 MHz, CDCl3): δ 174.3, 81.8, 67.7, 38.5, 26.3. HRESIMS m/z: calcd for C5H779BrNaO3 216.9476 [M + Na]+, found: 216.9472 [M + Na]+, 218.9451 [M + Na + 2]+.
3.3.4. Acetate 22
A 65% HClO4 solution (two drops) was added to alcohol 21 (2.6 mg, 0.0133 mmol) in acetic anhydride (0.5 mL). After 30 min, the mixture was worked-up as usual. Filtration through a short pad of silica gel gave crude 22 as an oil. Further purification by HPLC on an analytical silica column (n-hexane-EtOAc, 4:6) gave pure acetate 22 (1.1 mg, 35%), as an oil.
22 (2R,3S)-2-(bromomethyl)-5-oxotetrahydrofuran-3-yl acetate: IR (neat) νmax 1795 (s), 1735 (s), 1237(s) cm−1. 1H-NMR (700 MHz, CDCl3): δ 5.58 (1H, ddd, J = 5.8, 4.4, 1.1, H-3), 4.76 (1H, ddd, J = 8.3, 5.9, 4.3, H-4), 3.59 (2H, AB system further coupled, A part: J = 10.3, 5.9; B part: J = 10.3, 8.3), 2.93 (1H, dd, J = 18.3, 6.0, Ha-2), 2.67 (1H, dd, J = 18.3, 1.1, Hb-2), 2.13 (3H, s, acetate). 13C-NMR: (175 MHz, CDCl3): δ 173.0, 169.6, 80.3, 69.4, 36.8, 25.7, 20.7. HRESIMS m/z: calcd for C7H979BrNaO4 258.9582 [M + Na]+, found: 258.9569 [M + Na]+, 260.9550 [M + Na + 2]+.

 ,
, 


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}