Abstract
This study aimed to elucidate the structural congeners of natural izenamides A, B, and C (1–3) responsible for cathepsin D (CTSD) inhibition. Structurally modified izenamides were synthesized and biologically evaluated, and their biologically important core structures were identified. We confirmed that the natural statine (Sta) unit (3S,4S)-γ-amino-β-hydroxy acid is a requisite core structure of izenamides for inhibition of CTSD, which is closely related to the pathophysiological roles in numerous human diseases. Interestingly, the statine-incorporated izenamide C variant (7) and 18-epi-izenamide B variant (8) exhibited more potent CTSD-inhibitory activities than natural izenamides.
1. Introduction
Marine cyanobacteria are promising natural sources for drug discovery, as they produce diverse secondary metabolites with various biological relevance. Especially, they can produce numerous modified peptides or depsipeptides with various selectivity profiles and therapeutic potential for protease inhibition [1].
Cathepsin D (CTSD) is a lysosomal aspartic protease that plays critical roles in various physiological and pathological processes [2,3,4,5]. It is involved in metabolic proteolysis, energy metabolism, polypeptide hormone and antigen processing, fibrinolysis, activation of enzyme precursors, apoptotic cell death, and maintenance of intracellular homeostasis [2,6,7,8,9,10,11]. CTSD is widely expressed in various cells of the human body [2,12], with particularly high abundance in the brain. CTSD dysfunction results in the impaired degradation of disease-linked proteins, and is strongly implicated in the pathogenesis of numerous human diseases, especially neurodegenerative disease, such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, neuronal ceroid lipofuscinosis, and progressive disorders of the central nervous system [2,4,13,14]. CTSD is also associated with tumor progression, invasion, and metastasis in human malignancies, especially breast cancers, and is therefore a useful biomarker of breast cancer with poor prognosis [2,14,15,16]. Furthermore, CTSD inhibition can potentially hinder influenza virus replication by modulating host-cell autophagic/apoptotic responses [17]. Given the critical role of CTSD in human diseases, it is considered an attractive molecular target for the treatment of a wide range of diseases. However, novel therapeutic modulators of CTSD have not been sufficiently investigated, highlighting the urgent need for such modulators.
In 2018, izenamides A-C (1–3), which are linear depsipeptides, were first isolated from the marine cyanobacterium 1605-5 by Suenaga et al [18] (Figure 1). Izenamides A (1) and B (2) contain four amino acids (Pro-O-Me, N-Me-Phe, Ala, and Ile); two α-hydroxy acids (two valic acids for 1, and one valic acid and one isoleucic acid for 2); and a γ-amino-β-hydroxy acid moiety known as statine [(3S,4S)-4-amino-3-hydroxy-6-methylheptanoic acid, Sta]. Izenamide C (3) also contains Pro-O-Me, N-Me-Phe, Ile, and valic acid; however, it contains Gly in place of Ala in 1 and 2, as well as isoleucic acid. Unlike izenamides A (1) and B (2), izenamide C (3) lacks a Sta unit in the peptide backbone. Interestingly, izenamides A (1) and B (2) show moderate inhibitory activities against CTSD in vitro, whereas izenamide C (3) does not show CTSD inhibition. From a structural standpoint, modified peptidic CTSD inhibitors, including pepstatin A [19,20], tasiamides [21,22,23], grassystatins [24,25], and phormidepistatin [26], also possess Sta and Sta-like moieties for the inhibition of aspartic proteases, suggesting the role of the Sta unit (in 1 and 2) in CTSD inhibition.
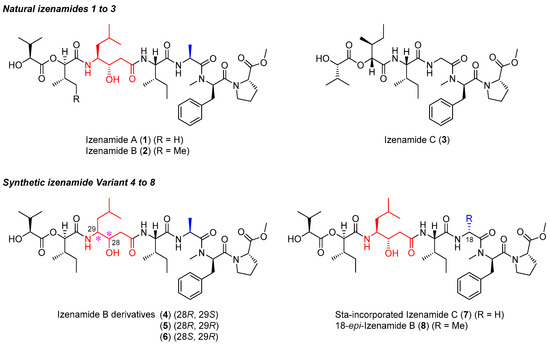
Figure 1.
Structures of natural izenamides (1–3) and synthetic izenamide variants (4–8).
In a previous study, we reported the first total syntheses of izenamides A–C (1–3) and the structural confirmation of 2. Since then, we have been interested in elucidating the pharmacophore of izenamides through systematic structure investigation. We have been particularly interested in the Sta unit, because izenamides C (3) lacks CTSD inhibition [27] without Sta. We anticipated that the core component responsible for the biological activity of izenamides could be identified through structural variation, followed by biological evaluation. In addition, we desired to develop an optimal izenamide variant structure for enhanced CTSD inhibition. The strategy involved initial studies of the Sta unit, including its stereochemical effects on CTSD inhibition by the most potent izenamide, B (2). The study also involved investigating the Ala units of 1 and 2 and their stereochemical effects on CTSD inhibition.
This approach involved the initial preparation of izenamide B variants (4, 5, and 6) consisting of an unnatural diastereomeric Sta unit. A natural Sta-incorporated izenamide C variant (7) was also prepared to confirm the Sta effects of 1 and 2 on CTSD inhibition. To investigate the stereochemical effect of alanine on izenamide B (2), 18-epi-izenamide B (8) consisting of an unnatural d-Ala was prepared (Figure 1). Here, the design and synthesis of izenamide variants and their biological evaluation as CTSD inhibitors are described. In particular, the core structure of izenamides for CTSD inhibition and the optimized structure for improved CTSD-inhibitory activity were identified. In addition, the efficient synthesis of izenamide variants with minimized epimerization of the stereocenters was explored by employing a versatile and diverse synthetic route to prevent 2,5-diketopiperazine (DKP) formation.
2. Results and Discussion
2.1. Synthetic Strategy for Izenamide Variants (4–8)
The synthetic strategy for the novel izenamide variants (4–6) is outlined in Scheme 1, focusing on the diastereomeric modification of the natural Sta and alanine units in izenamide B (2). The envisioned approach involves the convergent assembly of three fragments: tetrapeptides 14–16, diastereomeric Sta precursors 11–13, and acid 9. Tetrapeptides 14–16 were anticipated to be derived from tripeptide 19–21. The diastereomers of Sta precursors 11–13 as key fragments would be conveniently prepared from the known ketone 17 or aldehyde 18. Similarly, izenamide variants (7) and (8) could be synthesized by the amide coupling of tetrapeptide 14 or 15, natural Sta precursor 10, and acid 9. Tetrapeptide 15 was prepared from tripeptide 20. Fragments 9, 10, 14, and 16 were readily prepared as previously described [27].
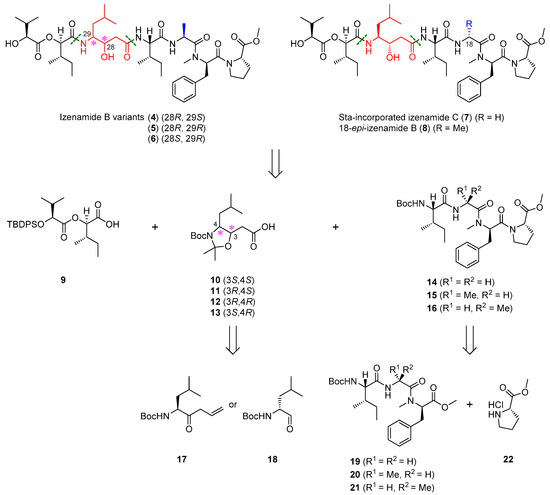
Scheme 1.
Synthetic approach for the preparation of izenamide variants (4–8).
2.2. Synthesis of Diastereomeric Sta Precursors (11–13)
The synthesis of variants 4–6 was initiated by preparing diastereomeric Sta precursors 11–13 to investigate the stereochemical effects of the two stereocenters in the Sta unit on CTSD inhibition. First, the C3-epimeric Sta precursor (3R,4S)-11 was synthesized, as illustrated in Scheme 2. The diastereoselective reduction of ketone 17 [28] with Li(t-BuO)3AlH exclusively afforded alcohol 23 [28], which was subsequently protected with 2,2-dimethoxypropane in the presence of catalytic PPTS. Finally, the desired C3-epimeric Sta precursor (3R,4S)-11 was obtained by the RuO4-mediated oxidation of the terminal alkene of 24.
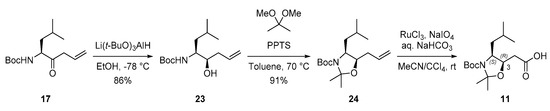
Scheme 2.
Preparation of C3-epimeric Sta precursor (3R,4S)-11.
Following the preparation of intermediate (3R,4S)-11, we focused on the synthesizing unnatural Sta precursors (3R,4R)-12 and (3S,4R)-13, which serve as key fragments for izenamide B variants (5) and (6). Commercially available aldehyde 18 was reacted with the Grignard reagent [29], which led to the nucleophilic addition of allylMgBr to N-Boc-d-leucinal 18 at 0 °C. Diastereomeric mixtures of alcohols 25 and 26 were obtained in 43% and 42% yield, respectively. The structure and stereochemistry of both alcohol 25 and 26 were confirmed by comparison of their spectral data with those of the corresponding enantiomers, including the opposite optical rotation [30]. Acetonide protection of optically pure allylic alcohols 25 and 26 followed by the Ru-mediated oxidation of the terminal alkenes afforded the desired diastereomeric Sta precursors (3R,4R)-12 and (3S,4R)-13, respectively (Scheme 3).
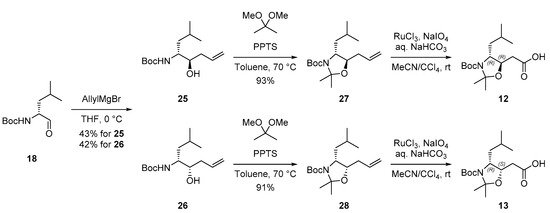
Scheme 3.
Synthesis of Sta precursors (3R,4R)-12 and (3S,4R)-13.
2.3. Completion of Izenamide Variant Synthesis (4–8)
Using diastereomeric Sta precursors 11–13, we assembled fragments for the izenamide B variants (4–6), as shown in Scheme 4. Boc-deprotection of tetrapeptide 16 with TFA and then amide coupling with the diastereomeric Sta precursors 11–13 afforded pentamers 29–31 in yields of 75–77% over two steps. Global deprotection of pentamers 29–31 with TFA and EDC-mediated coupling of the resulting amines with acid 9 produced the corresponding heptamers. Finally, the desired izenamide B variants (4–6) were obtained by desilylation in yields of 61–63% over three steps.
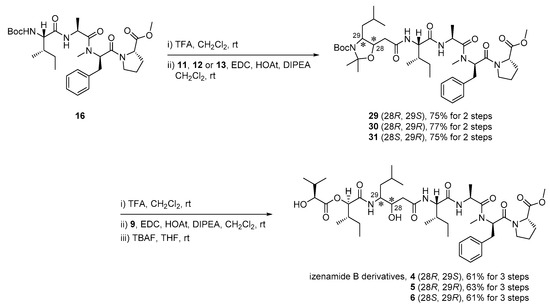
Scheme 4.
Synthesis of izenamide B variants possessing an unnatural Sta unit (4–6).
To investigate the role of the stereochemistry of the Ala unit of 2 in CTSD inhibition, we synthesized Sta-incorporated izenamide C (7) and 18-epi-izenamide B (8). The incorporation of a natural Sta unit into izenamide C (3), which contains a Gly unit instead of an Ala unit, was achieved at the corresponding Sta position in natural izenamide B (2). Additionally, the preparation of 18-epi-izenamide B (8) involved the substitution of unnatural d-Ala unit.
Tripeptide 20 was synthesized by coupling the known acid 32 [27] with N-Me-d-Phe-OMe·TFA salt [27]. The ester hydrolysis of tripeptide 20 and subsequent amidation of the resulting acid with 22 afforded tetrapeptide 15 in a yield of 76% over two steps (Scheme 5).
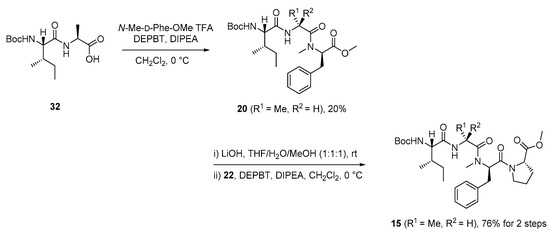
Scheme 5.
Preparation of tetrapeptide 15.
Tetrapeptide 14, acid 9, and natural Sta precursor 10 were prepared following a previously reported synthetic protocol [27]. The synthesis of izenamide variants 7 and 8 (Scheme 6) was completed by Boc-deprotection of tetrapeptides 14 and 15 and subsequent EDC-mediated amide coupling with acid 10. Pentamer intermediates 33 and 34 were obtained in yields of 81% and 76%, respectively, in two steps. The global deprotection of pentamers 33 and 34 and subsequent amide coupling of the resulting amines with acid 9 produced the corresponding heptamers, which were subjected to desilylation to afford Sta-incorporated izenamide C (7) and 18-epi-izenamide B (8) in yields of 62% and 63% over three steps, respectively (All spectral data including 1H, 13C NMR and HRMS of izenamides (4–8) was in Supplementary Materials).
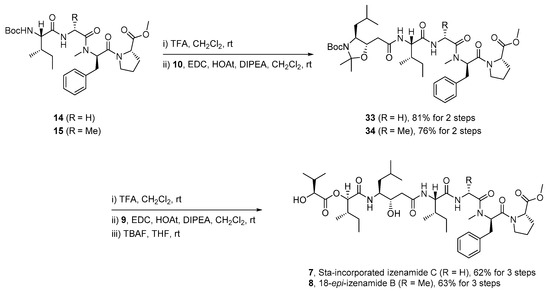
Scheme 6.
Completion of the synthesis of Sta-incorporated izenamide C (7) and 18-epi-izenamide B (8).
2.4. CTSD-Inhibitory Activities of Izenamides (1–3) and Their Variants (4–8)
After the successful synthesis of izenamide variants (4–8), the cathepsin D (CTSD)-inhibitory activities of the five izenamide variants and previously synthesized izenamides A–C (1–3) were evaluated. The CTSD-inhibition assay was performed at Eurofins Panlabs Discovery Services by measuring the percentage inhibition at concentrations of 0.5 and 1.0 μM (Table 1). Synthetic izenamides A (1) and B (2) exhibited similar inhibitory activities against CTSD, while synthetic izenamide C (3), which lacks the natural Sta unit, exhibited no CTSD inhibition, consistent with previous report [18].

Table 1.
Cathepsin D inhibitory activities of synthetic izenamides A–C (1–3) and their variants (4–8).
The natural Sta units of izenamides are essential for binding to CTSD [18]. Co-crystal structural analysis and docking studies predicted that the hydroxy group of the natural Sta in pepstatin A or izenamide A interacts with Asp residues at the active site of CTSD [31]. However, the effects of the stereochemistry of the two stereocenters in the Sta unit on CTSD inhibition remains unexplored. Thus, the stereochemical effects of the Sta unit of the izenamides on their CTSD-inhibitory activity were examined. As shown in Table 1, izenamide B (2) exhibits moderate inhibitory effects against CTSD (8.12% at 0.5 μM and 14.76% at 1.0 μM), whereas the unnatural Sta-incorporated izenamide B variants (4–6) exhibit no CTSD-inhibitory activities. These results confirm that natural (3S,4S)-Sta is essential for CTSD by izenamides.
Notably, the izenamide C variant (7) exhibits more potent CTSD-inhibitory activities (26.99% at 0.5 μM and 36.07% at 1.0 μM) than those of the natural izenamide B (2). The Sta-incorporated izenamide C variant (7) possesses the same peptide sequence as the biologically active 1 and 2, with the exception of alanine being replaced by glycine. The improved CTSD inhibition of the izenamide C variant (7), in contrast to the moderate inhibitory activity of the natural Sta-containing izenamide B (2) against CTSD, indicates that glycine is more appropriate for achieving adequate CTSD-inhibitory activity. These results confirm the crucial role played by the natural Sta unit as a key pharmacophore in the inhibition of CTSD by izenamides.
The sole difference in structure between 2 and 7 was the absence of a methyl substituent on the Gly unit at C18 in the izenamide C variant (7). Therefore, the optimal stereochemistry of the Ala unit in izenamide B (2) for CTSD inhibition was investigated. The 18-epi-izenamide B (8) was prepared, as shown in Scheme 6. Surprisingly, the 18-epi-izenamide B (8) showed the best CTSD-inhibitory effects (40.96% at 1.0 μM) compared to those of 2 and 7 (14.76% and 36.07% at 1.0 μM, respectively).
Overall, this study elucidated the crucial role of the natural Sta unit of izenamides in CTSD inhibition, with the importance of its stereochemistry. The results also suggested that the stereochemistry of the natural Sta unit is critical for its appropriate conformation to interact with Asp residues in the active binding pocket of CTSD, as described previously [18,29]. Additionally, the improved CTSD inhibition observed with the epimeric C18 unit of 8 suggested that it enhances interaction with CTSD.
3. Materials and Methods
3.1. General Information
Unless noted otherwise, all starting materials and reagents were obtained from commercial suppliers and were used without further purification. Tetrahydrofuran was distilled from sodium benzophenone ketyl. Dichloromethane, chloroform and acetonitrile were freshly distilled from calcium hydride. All solvents used for routine isolation of products and chromatography were reagent grade and glass-distilled. Reaction flasks were dried at 100 °C. Air- and moisture-sensitive reactions were performed under argon atmosphere. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Darmstadt, Germany) with the indicated solvents. Thin-layer chromatography was performed using 0.25 mm silica gel plates (Merck, Darmstadt, Germany). High-resolution mass spectra (HRMS) were recorded by JMS-700 (JEOL, Tokyo, Japan) and Q-TOF 6530 MS (Agilent, Santa Clara, CA, USA). Optical rotations were measured with JASCO P-2000 digital polarimeter (JASCO, Easton, MD, USA) at ambient temperature using a 10 mm cylindrical cell. Infrared spectra were recorded on a JASCO FT-IR-4200 spectrometer (JASCO, Easton, MD, USA). 1H and 13C NMR spectra were recorded using BRUKER AVANCE-400 (Bruker, Billerica, MA, USA) and BRUKER AVANCE-800 (Bruker, Billerica, MA, USA). Chemical shifts are expressed in parts per million (ppm, δ) downfield from tetramethylsilane and are referenced to the deuterated solvent (CHCl3 or MeOH). 1H NMR data were reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; dt, doublet of triplet; dq, doublet of quartet; dqu, doublet of quintet; ddd, doublet of doublet of doublet; qd, quartet of doublet, m, multiplet and/or multiple resonances), coupling constant in hertz (Hz) and number of protons.
3.2. Experimental Part
tert-Butyl (4S,5R)-5-allyl-4-isobutyl-2,2-dimethyloxazolidine-3-carboxylate (24). To a solution of ketone 17 (2.20 g, 8.62 mmol) in EtOH (86 mL) was added Li(t-BuO)3AlH (3.29 g, 12.92 mmol) at −78 °C. After stirring for 1 h at the same temperature, the reaction mixture was quenched with Rochelle’s solution and extracted with Et2O. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel to afford alcohol 23 [28] (2.30 g, 86%) as a colorless oil.
To a solution of the alcohol 23 (600 mg, 2.33 mmol) in toluene (8 mL) were added 2,2-dimethoxypropane (3.44 mL, 28.0 mmol) and pyridinium p-toluenesulfonate (58.6 mg, 0.23 mmol) at 70 °C. After stirring for 12 h at the same temperature, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:20) to afford oxazolidine 24 (623 mg, 91%) as a colorless oil. [α= +1.74 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3) δ 5.80–5.76 (m, 1H), 5.14 (s, 0.5H), 5.12 (s, 0.5H), 5.08 (d, J = 9.4 Hz, 1H), 4.03 (d, J = 5.2 Hz, 1H), 3.99 (d, J = 5.0 Hz, 0.5H), 3.82 (d, J = 5.1 Hz, 0.5 H), 2.35 (dt, J = 14.1, 6.9 Hz, 1H), 2.27 (dt, J = 13.2, 6.4 Hz, 1H), 1.65-1.60 (m, 1H), 1.53 (d, J = 28.5 Hz, 3H), 1.50 (d, J = 13.9 Hz, 3H), 1.45 (s, 9H), 1.43–1.36 (m, 1H), 1.33–1.21 (m, 1H), 0.94–0.89 (m, 6H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 151.9, 134.3, 117.0, 92.7, 79.6, 76.6, 57.1, 39. 7, 33.4, 28.6 (3C), 27.4, 24.6, 23.8, 23.5, 22.9, minor rotamer δ 152.5, 134.2, 117.0, 92.2, 79.6, 76.2, 57.1, 39.4, 33.3, 28.4 (3C), 28.2, 25.2, 24.8, 23.1, 23.0; IR (thin film, neat) νmax 3448, 3079, 2956, 2870, 1698, 1502, 1455, 1366, 1253, 1176, 1114, 754 cm−1; LR-MS (FAB+) m/z 298 (M + H)+; HR-MS (FAB+) calcd for C17H32NO3 (M + H)+ 298.2382; found 298.2374.
To a solution of oxazolidine 24 (600 mg, 1.91 mmol) in a mixture of CCl4, CH3CN, and H2O (1:1:2, 20 mL) were added NaHCO3 (1.18 g, 14.1 mmol), NaIO4 (2.59 g, 12.1 mmol) and RuCl3 (42 mg, 0.20 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel to afford (3R,4S)-statine derivative 11, which was used for the next step without further purification.
tert-Butyl (4R,5R)-5-allyl-4-isobutyl-2,2-dimethyloxazolidine-3-carboxylate (27). To a solution of aldehyde 18 (1.70 g, 7.90 mmol) in THF (27 mL) was added AllylMgBr solution (1.0 M in Et2O, 16.0 mL, 16.0 mmol) at 0 °C. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:10 to 1:5) to afford alcohol 25 (874 mg, 43%) as a colorless oil and 26 (854 mg, 42%) as white solid [28,30].
tert-Butyl ((4R,5R)-5-hydroxy-2-methyloct-7-en-4-yl)carbamate (25). [α= +23.15 (c 3.30, MeOH) (ent-25, lit. [α= −29.70 (c 3.30, MeOH) [30]); 1H NMR (400 MHz, CDCl3) δ 5.82 (m, 1H), 5.16-5.10 (m, 2H), 4.66 (m, 1H), 3.65-3.53 (m, 2H), 2.32-2.15 (m, 3H), 1.64 (m, 1H), 1.47 (m, 1H), 1.42 (s, 9H), 1.29 (m, 1H), 0.91 (d, J = 1.8 Hz, 3H), 0.90 (d, J = 1.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.2, 134.6, 118.3, 79.1, 72.7, 52.0, 41.8, 39.1, 28.4 (3C), 24.8, 23.2, 22.1; LR-MS (ESI+) m/z 158 (M − Boc + 2H)+; HR-MS (ESI+) calcd for C9H20NO (M − Boc + 2H)+ 158.1539; found 158.1536.
tert-Butyl ((4R,5S)-5-hydroxy-2-methyloct-7-en-4-yl)carbamate (26). [α= +12.11 (c 0.36, MeOH) (ent-26, lit. [α= −13.90 (c 0.36, MeOH) [30]); 1H NMR (400 MHz, CDCl3) δ 5.89-5.80 (m, 1H), 5.15-5.11 (m, 2H), 4.65-4.56 (brs, 1H), 3.65 (brs, 2H), 2.20 (m, 2H), 1.66 (m, 1H), 1.44 (s, 9H), 1.33-1.26 (m, 2H), 0.93 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 156.6, 135.1, 118.2, 80.1, 74.1, 53.5, 38.8, 38.2, 28.6 (3C), 25.0, 23.9, 21.8; LR-MS (ESI+) m/z 158 (M − Boc + 2H)+; HR-MS (ESI+) calcd for C9H20NO (M − Boc + 2H)+ 158.1539; found 158.1534.
To a solution of alcohol 25 (530 mg, 2.06 mmol) in toluene (7 mL) were added 2,2-dimethoxypropane (3.06 mL, 25.0 mmol) and pyridinium p-toluenesulfonate (53 mg, 0.21 mmol) at 70 °C. After stirring for 12 h at the same temperature, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:20) to afford oxazolidine 27 (580 mg, 93%) as a colorless oil. [α= −8.03 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3) δ 5.77 (ddt, J = 17.2, 10.2, 7.0 Hz, 1H), 5.13 (s, 0.5H), 5.09 (s, 0,5H), 5.08 (d, J = 10.4 Hz, 1H), 3.86 (t, J = 6.4 Hz, 1H), 3.76 (s, 0.5H), 3.63 (s, 0.5H), 2.40–2.35 (m, 1H), 2.30–2.27 (m, 1H), 1.70–1.61 (m, 1H), 1.60–1.53 (m, 3H), 1.50–1.42 (m, 14H), 0.90 (d, J = 6.4 Hz, 6H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 151.8, 134.2, 117.8, 93.8, 80.2, 60.1, 42.8, 40.4, 28.8, 28.5 (3C), 28.4, 27.8, 25.5, 24.0, 21.3, minor rotamer δ 156.2, 134.6, 117.8, 79.1, 72.7, 52.0, 41.9, 39.1, 28.5, 28.4 (3C), 25.5, 24.8, 23.2, 22.2, 21.3; IR (thin film, neat) νmax 2958, 2871, 1701, 1468, 1388, 1366, 1255, 1176, 1082, 856 cm−1; LR-MS (FAB+) m/z 298 (M + H)+; HR-MS (FAB+) calcd for C17H32NO3 (M + H)+ 298.2382; found 298.2397.
To a solution of oxazolidine 27 (500 mg, 1.68 mmol) in a mixture of CCl4, CH3CN, and H2O (1:1:2, 16 mL) were added NaHCO3 (1.0 g, 11.9 mmol), NaIO4 (2.2 g, 10.3 mmol), and RuCl3 (35 mg, 0.17 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel to afford (3R,4R)-statine derivative 12, which was used for the next step without further purification.
tert-Butyl (4R,5S)-5-allyl-4-isobutyl-2,2-dimethyloxazolidine-3-carboxylate (28). To a solution of alcohol 26 (500 mg, 1.94 mmol) in toluene (7 mL) were added 2,2-dimethoxypropane (2.94 mL, 24.0 mmol) and pyridinium p-toluenesulfonate (50 mg, 0.20 mmol) at 70 °C. After stirring for 12 h at the same temperature, the reaction mixture was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:20) to afford oxazolidine 28 (536 mg, 91%) as a colorless oil. [α= −6.12 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3) δ 5.80–5.72 (m, 1H), 5.10 (s, 0.5H), 5.08 (s, 0.5H), 5.04 (d, J = 9.8 Hz, 1H), 3.99 (d, J = 5.2 Hz, 1H), 3.96 (d, J = 5.1 Hz, 0.5H), 3.79 (d, J = 5.2 Hz, 0.5H), 2.31 (dt, J = 14.3, 7.0 Hz, 1H), 2.25-2.21 (m, 1H), 1.60–1.56 (m, 1H), 1.50 (d, J = 28.0 Hz, 3H), 1.46 (d, J = 13.2 Hz, 3H), 1.41 (s, 9H), 1.38–1.31 (m, 1H), 1.30–1.18 (m, 1H), 0.91–0.85 (m, 6H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 151.8, 134.3, 117.0, 92.6, 79.5, 76.5, 57.1, 39.6, 33.3, 28.5 (3C), 27.3, 24.6, 23.7, 23.4, 22.8, minor rotamer δ 152.4, 134.9, 117.7, 92.1, 79.5, 76.1, 57.1, 39.3, 33.3, 28.4 (3C), 28.2, 25.1, 24.7, 23.1, 23.0; IR (thin film, neat) νmax 2957, 2870, 1698, 1456, 1366, 1254, 1178, 1114, 864 cm−1; LR-MS (FAB+) m/z 298 (M + H+); HR-MS (FAB+) calcd for C17H32NO3 (M + H+) 298.2382; found 298.2386.
To a solution of oxazolidine 28 (400 mg, 1.34 mmol) in a mixture of CCl4, CH3CN, and H2O (1:1:2, 16 mL) were added NaHCO3 (900 mg, 10.7 mmol), NaIO4 (1.9 g, 8.9 mmol), and RuCl3 (31 mg, 0.15 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel to afford (3S,4R)-statine derivative 13, which was used for the next step without further purification.
(28R,29S)-Statine derivative-l-Ile-l-Ala-NMe-d-Phe-l-Pro-OMe, Pentamer (29). To a solution of tetrapeptide 16 (140 mg, 0.24 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.24 mmol) in CH2Cl2 (2.4 mL) was added (3R,4S)-statine 11 (100 mg, 0.32 mmol), DIPEA (0.21 mL, 1.20 mmol), HOAt (43 mg, 0.32 mmol), and EDC·HCl (93 mg, 0.49 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to give pentamer 29 (141 mg, 75% for 2 steps) as white solid. [α= +19.52 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.22-7.18 (m, 4H), 7.16 (t, J = 7.2 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.30 (d, J = 6.5 Hz, 1H), 5.61 (dd, J = 9.5, 6.4 Hz, 1H), 4.82–4.78 (m, 1H), 4.52 (d, J = 8.8 Hz, 1H), 4.42 (t, J = 7.4 Hz, 1H), 4.23 (dd, J = 7.5, 5.2 Hz, 1H), 3.84 (ddd, J = 9.7, 5.6, 2.4 Hz, 1H), 3.71 (s, 3H), 3.64–3.61 (m, 1H), 3.39 (ddd, J = 10.6, 6.8, 4.2 Hz, 1H), 3.21–3.16 (m, 2H), 3.00 (s, 3H), 2.93 (dd, J = 14.4, 9.6 Hz, 1H), 2.37 (d, J = 12.9 Hz, 1H), 2.32–2.29 (m, 1H), 2.23–2.19 (m, 1H), 1.99–1.95 (m, 1H), 1.93–1.89 (m, 1H), 1.88–1.77 (m, 3H), 1.72–1.63 (m, 2H), 1.49–1.42 (m, 3H), 1.41 (s, 9H), 1.39–1.36 (m, 2H), 1.32–1.28 (m, 1H), 1.08 (ddd, J = 13.4, 9.6, 7.3 Hz, 1H), 0.94 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H), 0.80 (d, J = 6.9 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.7, 172.3, 172.3, 170.2, 168.0, 156.2, 136.6, 129.5 (2C), 128.2 (2C), 126.7, 94.2, 79.6, 72.4, 59.4, 58.4, 55.8, 53.2, 52.2, 47.1, 44.8, 40.7, 39.3, 36.6, 34.7, 30.5, 28.8, 28.4 (5C), 25.4, 24.8 (2C), 23.8, 21.6, 17.8, 15.8, 11.7; IR (thin film, neat) νmax 3308, 2958, 1749, 1633, 1453, 1365, 1265, 1174, 749 cm−1; LR-MS (FAB+) m/z 732 (M − C(CH3)2 + 3H)+; HR-MS (FAB+) calcd for C38H62N5O9 (M − C(CH3)2 + 3H)+ 732.4548; found 732.4568.
(28R,29R)-Statine derivative-l-Ile-l-Ala-NMe-d-Phe-l-Pro-OMe, Pentamer (30). To a solution of tetrapeptide 16 (150 mg, 0.26 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.26 mmol) in CH2Cl2 (2.4 mL) were added (3R,4R)-statine derivative 12 (123 mg, 0.39 mmol), DIPEA (0.23 mL, 1.30 mmol), HOAt (46 mg, 0.34 mmol), and EDC·HCl (100 mg, 0.52 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to give pentamer 30 (155 mg, 77% for 2 steps) as white solid. [α= +70.45 (c 0.20, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.21–7.17 (m, 4H), 7.16–7.14 (m, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.07 (d, J = 8.3 Hz, 1H), 5.60 (dd, J = 9.2, 6.6 Hz, 1H), 4.85–4.80 (m, 1H), 4.69 (d, J = 9.8 Hz, 1H), 4.43 (t, J = 7.5 Hz, 1H), 4.26 (dd, J = 8.3, 5.0 Hz, 1H), 3.96 (d, J = 10.3 Hz, 1H), 3.71 (s, 3H), 3.63–3.60 (m, 1H), 3.40–3.38 (m, 1H), 3.22-3.17 (m, 2H), 3.01 (s, 3H), 2.92 (dd, J = 14.3, 9.4 Hz, 1H), 2.38 (dd, J = 13.8, 10.5 Hz, 1H), 2.31 (dd, J = 13.9, 2.1 Hz, 1H), 2.21–2.20 (m, 1H), 1.99–1.96 (m, 1H), 1.92–1.90 (m, 1H), 1.86–1.80 (m, 3H), 1.68–1.64 (m, 2H), 1.53–1.49 (m, 1H), 1.45–1.44 (m, 3H), 1.43 (s, 9H), 1.40–1.39 (m, 1H), 1.33–1.32 (m, 1H), 1.10–1.07 (m, 1H), 0.93 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H), 0.81 (d, J = 6.9 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.8, 172.5, 172.3, 170.1, 167.9, 156.0, 136.6, 129.5 (2C), 128.2 (2C), 126.7, 95.1, 79.2, 70.9, 59.4, 58.2, 55.8, 52.4, 52.2, 47.0, 44.8, 41.9, 41.5, 36.7, 34.7, 30.5, 29.7, 28.8, 28.4 (3C), 28.4, 25.4, 24.8, 24.7, 23.1, 22.3, 17.9, 15.7, 11.7; IR (thin film, neat) νmax 3306, 2927, 1749, 1635, 1454, 1365, 1266, 1174, 700 cm−1; LR-MS (FAB+) m/z 732 (M − C(CH3)2 + 3H)+; HR-MS (FAB+) calcd for C38H62N5O9 (M − C(CH3)2 + 3H)+ 732.4548; found 732.4546.
(28S,29R)-Statine derivative-l-Ile-l-Ala-NMe-d-Phe-l-Pro-OMe, Pentamer (31). To a solution of tetrapeptide 16 (142 mg, 0.25 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.25 mmol) in CH2Cl2 (2.4 mL) were added (3S,4R)-statine derivative 13 (117 mg, 0.37 mmol), DIPEA (0.21 mL, 1.20 mmol), HOAt (44 mg, 0.32 mmol), and EDC·HCl (95 mg, 0.50 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to afford pentamer 31 (143 mg, 75% for 2 steps) as white solid. [α= +72.55 (c 0.20, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.23–7.19 (m, 4H), 7.16 (t, J = 7.2 Hz, 1H), 6.74 (s, 1H), 6.49 (dd, J = 102.1, 7.2 Hz, 1H), 5.66 (m, 1H), 4.77–4.72 (m, 1H), 4.53 (d, J = 8.0 Hz, 1H), 4.45–4.42 (m, 1H), 4.22–4.18 (m, 1H), 3.84–3.80 (m, 1H), 3.72 (s, 3H), 3.64–3.59 (m, 1H), 3.49–3.43 (m, 1H), 3.24–3.18 (m, 2H), 2.99 (s, 3H), 2.97–2.93 (m, 1H), 2.38–2.32 (m, 2H), 2.22–2.19 (m, 1H), 1.93–1.90 (m, 1H), 1.85–1.81 (m, 3H), 1.65–1.61 (m, 2H), 1.54–1.49 (m, 2H), 1.45 (s, 3H), 1.42 (s, 9H), 1.29–1.26 (m, 2H), 1.12–1.10 (m, 1H), 0.91 (d, J = 6.7 Hz, 3H), 0.88 (d, J = 6.5 Hz, 3H), 0.87 (d, J = 7.2 Hz, 3H), 0.85 (t, J = 7.6 Hz, 3H), 0.83 (d, J = 6.9 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.4, 172.1, 170.0, 168.1, 168.1, 156.7, 136.7, 129.5 (2C), 128.3 (2C), 126.7, 92.7, 80.0, 72.1, 59.3, 57.8, 55.6, 53.3, 52.2, 47.0, 45.1, 39.4, 38.9, 37.3, 34.7, 30.3, 29.7, 28.8, 28.4 (4C), 25.3, 24.8, 23.6, 21.5, 17.6, 15.4, 14.1, 11.4; IR (thin film, neat) νmax 2926, 1635, 1366, 1175, 781 cm−1; LR-MS (FAB+) m/z 732 (M − C(CH3)2 + 3H)+; HR-MS (FAB+) calcd for C38H62N5O9 (M − C(CH3)2 + 3H)+ 732.4548; found 732.4526.
(28R, 29S)-Izenamide B variant (4). To a solution of pentamer 29 (41 mg, 0.05 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.05 mmol) in CH2Cl2 (1.0 mL) were added acid 9 (27 mg, 0.05 mmol), DIPEA (0.05 mL, 0.30 mmol), HOAt (9 mg, 0.07 mmol) and EDC·HCl (20 mg, 0.10 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel. To a solution of the above heptamer (0.05 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 0.20 mL, 0.20 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (acetone/n-hexane = 1:3) to afford (28R, 29S)-izenamide B variant 4 (27 mg, 61% for 3 steps) as white solid. [α= +3.07 (c 1.00, MeOH); 1H NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.23–7.21 (m, 4H), 7.16–7.15 (m, 1H), 5.70 (dd, J = 10.0, 5.9 Hz, 1H), 4.95 (d, J = 4.1 Hz, 1H), 4.69 (q, J = 7.0 Hz, 1H), 4.42-4.40 (m, 1H), 4.20 (d, J = 6.6 Hz, 1H), 4.11 (d, J = 4.3 Hz, 1H), 3.90 (ddd, J = 11.5, 7.0, 2.9 Hz, 1H), 3,79 (ddd, J = 9.9, 7.1, 2.9 Hz, 1H), 3.71 (s, 3H), 3.47–3.44 (m, 1H), 3.38–3.35 (m, 1H), 3.11 (dd, J = 14.3, 5.8 Hz, 1H), 3.06 (s, 3H), 2.93 (dd, J = 14.3, 10.0 Hz, 1H), 2.44 (dd, J = 14.3, 2.9 Hz, 1H), 2.35 (dd, J = 14.3, 9.6 Hz, 1H), 2.27–2.24 (m, 1H), 2.11–2.07 (m, 1H), 1.98–1.95 (m, 1H), 1.92–1.90 (m, 1H), 1.86–1.85 (m, 1H), 1.82–1.80 (m, 1H), 1.57–1.51 (m, 2H), 1.46–1.43 (m, 4H), 1.33–1.29 (m, 4H), 1.17–1.14 (m, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 7.4 Hz, 3H), 0.93 (d, J = 6.5 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 7.0 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H), 0.82 (d, J = 7.0 Hz, 3H); 13C NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 175.3, 174.7, 174.6, 173.9, 173.2, 172.0, 170.3, 138.3, 130.8 (2C), 129.3 (2C), 127.6, 77.9, 76.5, 73.1, 61.0, 59.2, 57.2, 53.1, 52.6, 46.4, 41.3, 40.2, 38.6, 38.0, 35.6, 33.3, 31.1, 30.4, 30.0, 27.3, 26.2, 26.0, 25.7, 24.3, 21.6, 19.4, 17.0, 16.9, 16.0, 14.7, 11.9, 11.7; IR (thin film, neat) νmax 3646, 3307, 2925, 2858, 2360, 1746, 1643, 1456, 1054, 1016, 850 cm−1; LR-MS (FAB+) m/z 846 (M + H)+; HR-MS (FAB+) calcd for C44H72N5O11 (M + H)+ 846.5228; found 846.5220.
(28R, 29R)-Izenamide B variant (5). To a solution of pentamer 30 (50 mg, 0.06 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.06 mmol) in CH2Cl2 (1.0 mL) were added acid 9 (33 mg, 0.06 mmol), DIPEA (0.06 mL, 0.30 mmol), HOAt (11 mg, 0.08 mmol), and EDC·HCl (25 mg, 0.13 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel. To a solution of the above heptamer (0.06 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 0.20 mL, 0.20 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (acetone/n-hexane = 1:3) to afford (28R, 29R)-izenamide B variant 5 (35 mg, 63% for 3 steps) as a white solid. [α= −40.20 (c 0.25, MeOH); 1H NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.24–7.21 (m, 4H), 7.16–7.15 (m, 1H), 5.68 (dd, J = 9.9, 6.0 Hz, 1H), 4.89 (m, 1H), 4.66 (q, J = 7.0 Hz, 1H), 4.42-4.40 (m, 1H), 4.20 (d, J = 6.8 Hz, 1H), 4.05 (d, J = 2.7 Hz, 1H), 3.99-3.96 (m, 2H), 3.71 (s, 3H), 3.47-3.44 (m, 1H), 3.40-3.38 (m, 1H), 3.11 (dd, J = 14.3, 5.9 Hz, 1H), 3.06 (s, 3H), 2.93 (dd, J = 14.3, 10.0 Hz, 1H), 2.35-2.29 (m, 2H), 2.27-2.24 (m, 1H), 1.97-1.93 (m, 1H), 1.87-1.85 (m, 3H), 1.82-1.78 (m, 1H), 1.63-1.59 (m, 1H), 1.58-1.54 (m, 1H), 1.52-1.42 (m, 3H), 1.35-1.31 (m, 5H), 1.18-1.14(m, 1H), 0.95 (d, J = 7.5 Hz, 3H), 0.93 (d, J = 6.7 Hz, 6H), 0.91 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H), 0.87 (t, J = 7.5 Hz, 3H), 0.83 (d, J = 7.0 Hz, 3H), 0.80 (d, J = 6.9 Hz, 3H); 13C NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 176.9, 174.6, 174.0, 173.9, 173.5, 173.2, 170.3, 138.3, 130.8 (2C), 129.3 (2C), 127.6, 74.8, 71.4, 60.9, 59.0, 57.2, 52.6, 52.0, 48.5, 46.5, 42.0, 41.9, 39.3 (2C), 38.2, 35.6 (2C), 31.1, 30.0, 27.4, 26.2, 25.9, 25.7, 23.7, 22.3, 16.8, 16.0, 13.5 (2C), 12.2 (2C), 11.6; IR (thin film, neat) νmax 3311, 2922, 2853, 1633, 1573, 1461, 1261, 1138, 802, 676 cm−1; LR-MS (FAB+) m/z 846 (M + H)+; HR-MS (FAB+) calcd for C44H72N5O11 (M + H)+ 846.5228; found 846.5224.
(28S,29R)-Izenamide B variant (6). To a solution of pentamer 31 (45 mg, 0.06 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.06 mmol) in CH2Cl2 (1.0 mL) were added acid 9 (30 mg, 0.06 mmol), DIPEA (0.05 mL, 0.30 mmol), HOAt (10 mg, 0.08 mmol), and EDC·HCl (22 mg, 0.11 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel. To a solution of the above heptamer (0.06 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 0.20 mL, 0.20 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (acetone/n-hexane = 1:3) to afford (28S,29R)-izenamide B variant 6 (30 mg, 61% for 3 steps) as a white solid. [α= +7.53 (c 1.00, MeOH); 1H NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.24–7.20 (m, 4H), 7.16–7.14 (m, 1H), 5.68 (dd, J = 9.9, 6.0 Hz, 1H), 5.00 (d, J = 4.0 Hz, 0.5H), 4.86 (overlapped, 0.5H), 4.66-4.63 (m, 1H), 4.41 (dd, J = 8.1, 6.9 Hz, 1H), 4.20 (dd, J = 7.2, 5.3 Hz, 1H), 4.17 (d, J = 4.1 Hz, 0.5H), 4.01 (d, J = 2.8 Hz, 0.5H), 3.94–3.92 (m, 1H), 3.86 (ddd, J = 9.4, 6.1, 3.5 Hz, 0.5H), 3.77 (ddd, J = 9.7, 6.8, 3.0 Hz, 0.5H), 3.71 (s, 3H), 3.48–3.45 (m, 1H), 3.42–3.37 (m, 1H), 3.11 (dd, J = 14.3, 5.9 Hz, 1H), 3.05 (d, J = 1.5 Hz, 3H), 2.93 (dd, J = 14.2, 10.0 Hz, 1H), 2.39–2.34 (m, 1H), 2.33–2.31 (m, 0.5H), 2.28–2.24 (m, 1.5H), 2.13–2.10 (m, 0.5H), 2.04–1.99 (m, 0.5H), 1.94–1.91 (m, 1.5H), 1.88–1.83 (m, 2.5H), 1.79–1.72 (m, 1.5H), 1.67–1.57 (m, 1.5H), 1.50-1.41(m, 5H), 1.33–1.26 (m, 2H), 1.16-1.11 (m, 1H), 1.02 (d, J = 6.9 Hz, 1.5H), 0.96 (t, J = 6.6 Hz, 3H), 0.94-0.92 (m, 4.5H), 0.90 (dd, J = 6.9, 2.5 Hz, 3H), 0.88 (d, J = 6.7 Hz, 3H), 0.87-0.85 (m, 4.5H), 0.83 (dd, J = 7.0, 1.8 Hz, 3H), 0.80 (d, J = 6.9 Hz, 1.5H); 13C NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 176.9, 175.3, 174.5, 174.0, 173.2, 172.1, 170.3, 138.3, 130.8 (2C), 129.3 (2C), 127.6, 78.0, 76.7, 74.9, 72.5, 60.9, 58.8, 57.2, 52.6, 46.5, 41.1, 40.0, 39.3, 38.3, 35.6, 33.2, 31.0, 30.0, 27.4, 26.2, 25.9, 25.7, 24.2, 21.7, 19.5, 16.8, 16.7, 15.9, 14.8, 12.2, 12.0, 11.5; IR (thin film, neat) νmax 3326, 2961, 1747, 1639, 1530, 1455, 1176, 1033, 700 cm−1; LR-MS (FAB+) m/z 846 (M + H)+; HR-MS (FAB+) calcd for C44H72N5O11 (M + H)+ 846.5228; found 846.5230.
Boc-l-Ile-d-Ala-NMe-d-Phe-OMe (20). To a solution of Boc-NMe-d-Phe-OMe [27] (660 mg, 2.30 mmol) in CH2Cl2 (9.0 mL) was added TFA (1.5 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (2.30 mmol) in CH2Cl2 (11 mL) were added Boc-l-Ile-l-Ala-OH 32 (816 mg, 2.70 mmol), DIPEA (1.2 mL, 6.90 mmol) and DEPBT (1.30 g, 4.34 mmol) at 0 °C. After stirring for 12 h at the same temperature, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to afford Boc-l-Ile-d-Ala-NMe-d-Phe-OMe tripeptide 20 (218 mg, 20% for 2 steps) as white solid. [α= +48.23 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.25 (t, J = 7.5 Hz, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.3 Hz, 2H), 6.65 (d, J = 5.6 Hz, 1H), 5.05 (dd, J = 10.9, 5.3 Hz, 1H), 5.00 (d, J = 8.0 Hz, 1H), 4.73 (quint, J = 6.8 Hz, 1H), 3.99-3.94 (m, 1H), 3.71 (s, 3H), 3.36 (dd, J = 14.7, 5.3 Hz, 1H), 3.05 (dd, J = 14.7, 10.9 Hz, 1H), 2.82 (s, 3H), 1.84-1.80 (m, 1H), 1.42 (s, 9H), 1.39-1.33 (m, 1H), 1.25 (d, J = 6.8 Hz, 3H), 1.08-1.02 (m, 1H), 0.87 (d, J = 7.0 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.5, 170.8, 170.3, 136.7, 128.8 (2C), 128.6 (2C), 126.9, 79.8, 59.5, 59.0, 52.4, 45.5, 37.9, 34.3, 33.4, 28.3 (3C), 28.3, 24.7, 18.2, 15.5, 11.7; IR (thin film, neat) νmax 3308, 2967, 1743, 1714, 1638, 1497, 1366, 1172, 1017, 750 cm-1; LR-MS (ESI+) m/z 478 (M + H)+; HR-MS (ESI+) calcd for C25H40N3O6 (M + H)+ 478.2912; found 478.2915.
Boc-l-Ile-d-Ala-NMe-d-Phe-l-Pro-OMe (15). To a solution Boc-l-Ile-d-Ala-NMe-d-Phe-OMe 20 (210 mg, 0.44 mmol) in a mixture of THF, MeOH and H2O (1:1:1, 4 mL) was added LiOH·H2O (34 mg, 0.81 mmol) at room temperature. After stirring for 2 h, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above acid (0.44 mmol) in CH2Cl2 (4 mL) were added l-Pro-OMe·HCl 22 (81 mg, 0.49 mmol), DIPEA (0.23 mL, 1.3 mmol), and DEPBT (615 mg, 2.06 mmol) at 0 °C. After stirring for 12 h at the same temperature, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane =1:1) to give Boc-l-Ile-d-Ala-NMe-d-Phe-l-Pro-OMe tetrapeptide 15 (170 mg, 76% for 2 steps) as white solid. [α= +37.07 (c 1.00, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.23-7.21 (m, 3H), 7.18-7.14 (m, 2H), 6.63 (dd, J = 14.4, 6.7 Hz, 1H), 5.53 (dd, J = 8.0, 6.9 Hz, 1H), 5.02 (d, J = 8.2 Hz, 1H), 4.83-4.80 (m, 1H), 4.40 (dd, J = 8.5, 5.5 Hz, 1H), 3.96-3.90 (m, 1H), 3.68 (s, 3H), 3.59-3.51 (m, 1H), 3.39-3.34 (m, 1H), 3.29 (dt, J = 10.5, 6.1 Hz, 1H), 3.27 (dd, J = 13.7, 8.5 Hz, 1H), 3.02 (s, 3H), 2.77 (dd, J = 13.6, 6.5 Hz, 1H), 2.16-2.09 (m, 1H), 1.91-1.86 (m, 1H), 1.85-1.76 (m, 3H), 1.42 (s, 9H), 1.29 (d, J = 6.8 Hz, 3H), 1.11-0.99 (m, 1H), 0.88 (d, J = 6.7 Hz, 3H), 0.87 (t, J = 6.6 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.5, 172.1, 170.3, 167.8, 155.6, 137.0, 129.5 (2C), 128.3 (2C), 126.6, 79.8, 59.2, 58.8, 56.1, 52.2, 46.6, 45.6, 37.8, 34.7, 30.5, 28.8, 28.3 (3C), 25.0, 18.5, 17.5, 15.5, 11.6; IR (thin film, neat) νmax 3298, 2969, 1748, 1712, 1637, 1497, 1365, 1173, 1044, 870 cm-1; LR-MS (ESI+) m/z 576 (M + H)+; HR-MS (ESI+) calcd for C30H47N4O7 (M + H)+ 576.3471; found 576.3473.
(28R,29S)-Statine derivative-l-Ile-Gly-NMe-d-Phe-l-Pro-OMe, Pentamer (33). To a solution of Boc-NMe-l-Ile-Gly-NMe-d-Phe-OMe 14 (140 mg, 0.25 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.25 mmol) in CH2Cl2 (2.4 mL) were added (3S,4S)-natural statine 10 (118 mg, 0.37 mmol), DIPEA (0.2 mL, 1.14 mmol), HOAt (44 mg, 0.32 mmol), and EDC·HCl (95 mg, 0.50 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to afford pentamer 33 153 mg (81% for 2 steps) as white solid. [α= +54.65 (c 0.20, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.24–7.21 (m, 2H), 7.20–7.19 (m, 2H), 7.16 (t, J = 7.1 Hz, 1H), 6.72–6.68 (m, 2H), 5.55 (t, J = 7.6 Hz, 1H), 4.40–4.37 (m, 2H), 4.22–4.20 (m, 1H), 4.06 (dd, J = 17.6, 4.4 Hz, 1H), 3.85 (dd, J = 18.9, 4.0 Hz, 1H), 3.70 (s, 3H), 3.64–3.58 (m, 1H), 3.37–3.31 (m, 2H), 3.25 (dd, J = 13.7, 8.0 Hz, 1H), 2.95 (s, 3H), 2.80 (dd, J = 13.7, 7.1 Hz, 1H), 2.61–2.55 (m, 1H), 2.46 (d, J = 14.4 Hz, 1H), 2.14–2.11 (m, 1H), 1.95–1.89 (m, 2H), 1.87–1.78 (m, 3H), 1.75-1.65 (m, 3H), 1.65–1.56 (m, 5H), 1.55–1.52 (m, 1H), 1.46 (s, 9H), 1.12–1.07 (m, 1H), 0.90 (d, J = 6.9 Hz, 3H), 0.90–0.89 (m, 6H), 0.88 (t, J = 7.5 Hz, 3H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.5, 170.8, 170.0, 167.9, 167.7, 151.6, 136.9, 129.4 (2C), 128.4 (2C), 126.7, 94.6, 79.9, 68.1, 60.9, 58.9, 57.5, 56.2, 52.2, 46.8, 41.2, 37.4, 35.0, 29.6, 28.8, 28.5 (6C), 25.4, 25.0, 24.7, 24.0 (2C), 21.3, 15.6, 11.5; IR (thin film, neat) νmax 3309, 2958, 1747, 1634, 1388, 1254, 1175, 1088, 701 cm−1; LR-MS (ESI+) m/z 758 (M + H)+; HR-MS (ESI+) calcd for C40H64N5O9 (M + H)+ 758.4704; found 758.4713.
(28R,29S)-Statine derivative-l-Ile-d-Ala-NMe-d-Phe-l-Pro-OMe, Pentamer (34), To a solution of tetrapeptide 15 (140 mg, 0.24 mmol) in CH2Cl2 (1.6 mL) was added TFA (0.4 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.24 mmol) in CH2Cl2 (2.4 mL) were added (3S,4S)-natural statine 10 (100 mg, 0.32 mmol), DIPEA (0.2 mL, 1.14 mmol), HOAt (43 mg, 0.32 mmol), and EDC·HCl (93 mg, 0.49 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (EtOAc/n-hexane = 1:1) to give pentamer 34 (143 mg, 76% for 2 steps) as white solid. [α= +23.40 (c 0.20, CHCl3); 1H NMR (800 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 7.24-7.21 (m, 4H), 7.18-7.16 (m, 1H), 6.69 (d, J = 6.2 Hz, 1H), 6.42 (d, J = 6.4 Hz, 1H), 5.52 (dd, J = 8.4, 6.5 Hz, 1H), 4.79 (dq, J = 6.9, 6.8 Hz, 1H), 4.41 (dd, J = 8.6, 5.3 Hz, 1H), 4.32–4.28 (m, 1H), 3.96 (d, J = 9.7 Hz, 1H), 3.70 (s, 3H), 3.60–3.53 (m, 1H), 3.35 (dt, J = 10.3, 6.2 Hz, 1H), 3.32–3.30 (m, 1H), 3.29 (dd, J = 13.6, 8.7 Hz, 1H), 3.05 (s, 3H), 2.78 (dd, J = 13.6, 6.4 Hz, 1H), 2.47 (dd, J = 14.8, 10.3 Hz, 1H), 2.34 (d, J = 15.2 Hz, 1H), 2.15–2.11 (m, 1H), 1.96–1.76 (m, 11H), 1.65–1.60 (m, 1H), 1.55–1.50 (m, 1H), 1.48–1.46 (m, 1H), 1.43 (s, 9H), 1.32 (d, J = 6.7 Hz, 3H), 1.15–1.07 (m, 1H), 0.90 (t, J = 7.0 Hz, 3H), 0.90 (d, J = 6.8 Hz, 9H); 13C NMR (200 MHz, CDCl3, a mixture of rotamers) Major rotamer δ 172.6 (2C), 172.2, 169.6, 167.8, 156.3, 137.1, 129.6 (2C), 128.4 (2C), 126.6, 93.4, 79.3, 70.5, 58.7, 57.3, 56.3, 52.3, 52.2, 46.6, 45.9, 41.6, 40.1, 37.5, 34.7, 30.9, 30.6, 28.8, 28.4 (4C), 25.1, 25.0, 24.8, 23.1, 22.2, 18.3, 15.4, 11.5; IR (thin film, neat) νmax 3298, 2928, 1647, 1453, 1261, 1199, 800 cm−1; LR-MS (FAB+) m/z 732 (M − C(CH3)2 + 3H)+; HR-MS (FAB+) calcd for C38H62N5O9 (M − C(CH3)2 + 3H)+ 732.4548; found 732.4561.
Sta-incorporated-izenamide C (7). To a solution of pentamer 33 (56 mg, 0.07 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.2 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.07 mmol) in CH2Cl2 (1.0 mL) were added acid 9 (38 mg, 0.07 mmol), DIPEA (0.063 mL, 0.36 mmol), HOAt (13 mg, 0.10 mmol), and EDC·HCl (28 mg, 0.15 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel. To a solution of heptamer (0.07 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 0.20 mL, 0.20 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched by 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatograph on silica gel (acetone/n-hexane = 1:3) to afford the Sta-introduced izenamide C 7 (38 mg, 62% for 3 steps) as white solid. [α= −28.30 (c 0.20, MeOH); 1H NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.29–7.24 (m, 1H), 7.22–7.20 (m, 3H), 7.16–7.14 (m, 1H), 5.56 (t, J = 7.5 Hz, 1H), 4.96 (d, J = 4.2 Hz, 1H), 4.36 (dd, J = 8.4, 5.6 Hz, 1H), 4.29 (d, J = 6.7 Hz, 1H), 4.12 (d, J = 16.8 Hz, 1H), 4.12 (d, J = 4.4 Hz, 1H), 4.04–3.98 (m, 2H), 3.89 (d, J = 16.9 Hz, 1H), 3.70 (s, 3H), 3.43 (dt, J = 10.4, 6.9 Hz, 1H), 3.40–3.37 (m, 1H), 3.20 (dd, J = 13.7, 8.0 Hz, 1H), 3.02 (s, 3H), 2.83 (dd, J = 13.7, 7.1 Hz, 1H), 2.44–2.35 (m, 2H), 2.21–2.16 (m, 1H), 2.12–2.08 (m, 1H), 1.95–1.88 (m, 3H), 1.87–1.80 (m, 2H), 1.60–1.52 (m, 3H), 1.49-1.43 (m, 1H), 1.34–1.27 (m, 2H), 1.21–1.16 (m, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H), 0.95 (t, J = 7.4 Hz, 3H), 0.94 (d, J = 7.0 Hz, 3H), 0.92 (d, J = 6.4 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.90 (t, J = 7.5 Hz, 3H), 0.89 (d, J = 6.5 Hz, 3H); 13C NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 176.4, 174.9, 174.7, 174.7, 172.9, 171.1, 171.0, 139.4, 131.3 (2C), 130.2 (2C), 128.4, 79.1, 77.2, 72.2, 61.4, 60.1, 58.6, 53.4, 53.1, 49.1, 42.7, 42.3, 42.2, 39.2, 38.8, 36.5, 34.1, 31.4, 30.7, 28.0, 26.8, 26.8, 26.6, 24.6, 23.0, 20.1, 17.8, 16.8, 15.5, 12.7, 12.5; IR (thin film, neat) νmax 3329, 2965, 1744, 1647, 1456, 1033, 810 cm−1; LR-MS (ESI+) m/z 758 (M + H)+; HR-MS (ESI+) calcd for C43H70N5 O11(M + H)+ 832.5066; found 832.5079.
18-epi-izenamide B (8). To a solution of pentamer 34 (48 mg, 0.06 mmol) in CH2Cl2 (0.8 mL) was added TFA (0.8 mL) dropwise at room temperature. After stirring for 1 h, the reaction mixture was concentrated in vacuo. The residue was used in the next step without further purification. To a solution of the above amine salt (0.06 mmol) in CH2Cl2 (1.0 mL) were added acid 9 (32 mg, 0.06 mmol), DIPEA (0.05 mL, 0.30 mmol), HOAt (11 mg, 0.08 mmol), and EDC·HCl (24 mg, 0.13 mmol) at room temperature. After stirring for 12 h, the reaction mixture was quenched with 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was filtered through a short column of silica gel. To a solution of heptamer (0.06 mmol) in THF (1.0 mL) was added TBAF (1 M in THF, 0.20 mL, 0.20 mmol) at room temperature. After stirring for 6 h, the reaction mixture was quenched 1N HCl and extracted with CH2Cl2. The combined organic layer was dried over MgSO4 and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (acetone/n-hexane = 1:3) to give 18-epi-izenamide B 8 (33 mg, 63% for 3 steps) as white solid. [α= −52.70 (c 0.10, MeOH); 1H NMR (800 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 7.24–7.20 (m, 4H), 7.17–7.14 (m, 1H), 5.53 (dd, J = 9.0, 5.8 Hz, 1H), 4.97 (d, J = 4.1 Hz, 1H), 4.83 (q, J = 7.0 Hz, 1H), 4.35 (dd, J = 8.6, 5.1 Hz, 1H), 4.27 (d, J = 6.7 Hz, 1H), 4.13 (d, J = 4.5 Hz, 1H), 4.03–3.99 (m, 2H), 3.70 (s, 3H), 3.42–3.38 (m, 1H), 3.36–3.33 (m, 1H), 3.25 (dd, J = 13.5, 9.0 Hz, 1H), 3.11 (s, 3H), 2.79 (dd, J = 13.5, 5.7 Hz, 1H), 2.42 (qd, J = 14.6, 6.8 Hz, 2H), 2.19–2.13 (m, 1H), 2.12–2.07 (m, 1H), 1.97–1.93 (m, 1H), 1.91–1.80 (m, 4H), 1.61–1.55 (m, 2H), 1.52 (ddd, J = 13.6, 7.5, 3.5 Hz, 1H), 1.46 (dq, J = 6.5, 6.4 Hz, 1H), 1.32 (d, J = 6.9 Hz, 3H), 1.32–1.25 (m, 2H), 1.18 (ddd, J = 13.6, 9.5, 7.4 Hz, 1H), 1.01 (d, J = 6.9 Hz, 3H), 0.97 (d, J = 6.8 Hz, 3H), 0.95 (t, J = 7.5 Hz, 3H), 0.92 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.90 (t, J = 7.5 Hz, 3H), 0.88 (d, J = 6.5 Hz, 3H); 13C NMR (200 MHz, CD3OD, a mixture of rotamers) Major rotamer δ 176.4, 175.2, 174.9, 174.7, 173.9, 172.7, 170.9, 139.5, 131.3 (2C), 130.2 (2C), 128.4, 79.0, 77.3, 72.3, 61.2, 59.9, 58.7, 53.5, 53.0, 48.9, 48.0, 42.3, 42.1, 39.2, 39.1, 36.4, 34.1, 32.2, 30.7, 28.1, 26.9, 26.6, 26.6, 24.6, 23.0, 20.1, 18.4, 17.9, 16.8, 15.5, 12.7, 12.6; IR (thin film, neat) νmax 3680, 2966, 2844, 1637, 1346, 1055, 1033, 691 cm−1; LR-MS (ESI+) m/z 846 (M + H)+; HR-MS (ESI+) calcd for C44H72N5O11 (M + H)+ 846.5223; found 846.5230.
3.3. Cathepsin D (CTSD) Human Pepsin Aspartic Peptidase Enzymatic Assay
Human liver cathepsin D was used for the enzymatic essay. Test compound or vehicle was preincubated with 190 mU/mL enzyme in modified acetate buffer pH 4.0 for 15 minutes at 37 °C. The reaction was initiated by addition of 20 μM MOCAc-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(Dnp)-d-Arg NH2 for another 10-minute incubation period. Determination of the amount of MOCAc formed was read spectrofluorimetrically at 340 nm/400. Compounds were screened at 0.5 and 1.0 μM. Cathepsin-D-Human-Pepsin-Aspartic-Peptidase-Enzymatic-Assay-Panlabs (eurofinsdiscovery.com, accessed on 22 February 2022).
4. Conclusions
In this study, a range of structural variants of izenamide depsipeptides were designed and synthesized to elucidate core structures responsible for CTSD inhibition. The divergent syntheses of four diastereomers of the Sta unit of izenamide B and evaluation of CTSD inhibition by the izenamide variants were conducted. The results confirmed the essential role of the natural (3S,4S)-Sta unit of izenamides A (1) and B (2) in CTSD inhibition. The incorporation of the natural Sta unit into izenamide C (3) led us to identify an izenamide C variant (7) that exhibited more potent CTSD inhibition than izenamide B (2). Moreover, the most potent 18-epi-izenamide B (8), possessing an unnatural d-Ala in izenamide B (2), was identified based on the substituent effects of the alanine unit. These results highlighted the important role of the Ala or Gly unit, in addition to the natural Sta unit of izenamides, in CTSD inhibition. Overall, these findings provide valuable information for the further development of novel depsipeptide-based CTSD inhibitors.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md21050281/s1, the 1H and 13C NMR spectra of 24, 27, 28, 29, 30, 31, 4, 5, 6, 20, 15, 33, 34, 7 and 8.
Author Contributions
Conceptualization and methodology, H.S.K. and H.K.; analysis and validation, H.S.K., H.K., T.K., C.L.; data curation, H.S.K., H.K., C.L. and S.L.; writing—original draft preparation, H.S.K. and H.K.; writing—review and editing, S.-H.K., and Y.-G.S.; supervision, Y.-G.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF-2022R1C1C2003502).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The original data presented in the study are included in the article/Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shah, S.A.A.; Akhter, N.; Auckloo, B.N.; Khan, I.; Lu, Y.; Wang, K.; Wu, B.; Guo, Y.W. Structural diversity, biological properties and applications of natural products from cyanobacteria. a review. Mar. Drugs 2017, 15, 354. [Google Scholar] [CrossRef]
- Benes, P.; Vetvicka, V.; Fusek, M. Cathepsin D—Many functions of one aspartic protease. Crit. Rev. Oncol. Hematol. 2008, 68, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Maurer, A.; Nieke, S.; Kalbacher, H. Cathepsin D: A cellular roadmap, Biochem. Biophys. Res. Commun. 2008, 376, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The role of cathepsin d in the pathogenesis of human neurodegenerative disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1697. [Google Scholar] [CrossRef]
- Simon, D.I.; Ezratty, A.M.; Loscalzo, J. The fibrin (ogen) olytic properties of cathepsin D. Biochem. 1994, 33, 6555–6563. [Google Scholar] [CrossRef]
- Koike, M.; Shibata, M.; Ohsawa, Y.; Nakanishi, H.; Koga, T.; Kametaka, S.; Waguri, S.; Momoi, T.; Kominami, E.; Peters, C. Involvement of two different cell death pathways in retinal atrophy of cathepsin D-deficient mice. Mol. Cell Neurosci. 2003, 22, 146–161. [Google Scholar] [CrossRef]
- Saftig, P.; Hetman, M.; Schmahl, W.; Weber, K.; Heine, L.; Mossmann, H.; Köster, A.; Hess, B.; Evers, M.; von Figura, K. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995, 14, 3599–3608. [Google Scholar] [CrossRef]
- Baechle, D.; Flad, T.; Cansier, A.; Steffen, H.; Schittek, B.; Tolson, J.; Herrmann, T.; Dihazi, H.; Beck, A.; Mueller, G.A. Cathepsin D is present in human eccrine sweat and involved in the postsecretory processing of the antimicrobial peptide DCD-1L. J. Biol. Chem. 2006, 281, 5406–5415. [Google Scholar] [CrossRef]
- Hakala, J.K.; Oksjoki, R.; Laine, P.; Du, H.; Grabowski, G.A.; Kovanen, P.T.; Pentikäinen, M.O. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1430–1436. [Google Scholar] [CrossRef]
- Bańkowska, A.; Gacko, M.; Chyczewska, E.; Worowska, A. Biological and diagnostic role of cathepsin D. Rocz. Akad. Med. Bialymst. 1997, 42, 79–85. [Google Scholar] [PubMed]
- Reid, W.A.; Valler, M.J.; Kay, J. Immunolocalization of cathepsin D in normal and neoplastic human tissues. J. Clin. Pathol. 1986, 39, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Bunk, J.; Huarcaya, S.P.; Drobny, A.; Dobert, J.P.; Walther, L.; Rose-John, S.; Arnold, P.; Zunke, F. Cathepsin D variants associated with neurodegenerative diseases show dysregulated functionality and modified α-synuclein degradation properties. Front. Cell Dev. Biol. 2021, 9, 581805. [Google Scholar] [CrossRef] [PubMed]
- Drobny, A.; Huarcaya, S.P.; Dobert, J.; Kluge, A.; Bunk, J.; Schlothauer, T.; Zunke, F. The role of lysosomal cathepsins in neurodegeneration: Mechanistic insights, diagnostic potential and therapeutic approaches. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119243. [Google Scholar] [CrossRef] [PubMed]
- Dubey, V.; Luqman, S. Cathepsin D as a promising target for the discovery of novel anticancer agents. Curr. Cancer Drug Targets 2017, 17, 404–422. [Google Scholar] [CrossRef]
- Ashraf, Y.; Mansouri, H.; Laurent-Matha, V.; Alcaraz, L.B.; Roger, P.; Guiu, S.; Derocq, D.; Robin, G.; Michaud, H.; Delpech, H.; et al. Immunotherapy of triple-negative breast cancer with cathepsin D-targeting antibodies. J. Immunother. Cancer 2019, 7, 29. [Google Scholar] [CrossRef]
- Matarrese, P.; Nencioni, L.; Checconi, P.; Ciarlo, L.; Gambardella, L.; Ascione, B.; Sgarbanti, R.; Garaci, E.; Malorni, W.; Palamara, A.T. Pepstatin A alters host cell autophagic machinery and leads to a decrease in influenza A virus production. J. Cell Physiol. 2011, 226, 3368–3377. [Google Scholar] [CrossRef]
- Kanamori, Y.; Iwasaki, A.; Sumimoto, S.; Matsubara, T.; Sato, T.; Suenaga, K. Izenamides A and B, statine-containing depsipeptides, and an analogue from a marine cyanobacterium. J. Nat. Prod. 2018, 81, 1673–1681. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Morishima, H.; Matsuzaki, M.; Hamada, M.; Takeuchi, T. Pepstatin, a new pepsin inhibitor produced by Actinomycetes. J. Antibiot. 1970, 23, 259–262. [Google Scholar] [CrossRef]
- Morishima, H.; Takita, T.; Aoyagi, T.; Takeuchi, T.; Umezawa, H. The structure of pepstatin. J. Antibiot. 1970, 23, 263–265. [Google Scholar] [CrossRef]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium. Symploca sp. J. Nat. Prod. 2003, 66, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, W.; Zong, C.; Wang, P.; Li, Y. Total synthesis and stereochemical reassignment of tasiamide B. J. Pept. Sci. 2010, 16, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Al-Awadhi, F.H.; Ratnayake, R.; Paul, V.J.; Luesch, H. Tasiamide F, a potent inhibitor of cathepsins D and E from a marine cyanobacterium. Bioorg. Med. Chem. 2016, 24, 3276–3282. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Eksioglu, E.A.; Liu, C.; Paul, V.J.; Luesch, H. Grassystatins A-C from marine cyanobacteria, potent cathepsin E inhibitors that reduce antigen presentation. J. Med. Chem. 2009, 52, 5732–5747. [Google Scholar] [CrossRef]
- Al-Awadhi, F.H.; Law, B.K.; Paul, V.J.; Luesch, H. Grassystatins D-F, potent aspartic protease inhibitors from marine cyanobacteria as potential antimetastatic agents targeting invasive breast cancer. J. Nat. Prod. 2017, 80, 2969–2986. [Google Scholar] [CrossRef]
- Sullivan, P.; Krunic, A.; Davis, L.J.; Kim, H.S.; Burdette, J.E.; Orjala, J. Phormidepistatin from the cyanobacterium UIC 10484: Assessing the phylogenetic distribution of the statine pharmacophore. J. Nat. Prod. 2021, 84, 2256–2264. [Google Scholar] [CrossRef]
- Lim, C. Total syntheses of cathepsin D inhibitory izenamides A, B, and C and structural confirmation of izenamide B. Molecules 2019, 24, 3424. [Google Scholar] [CrossRef]
- Huang, W.; Ma, J.; Yuan, M.; Xu, L.; Wei, B. A facile approach to trans-4,5-pyrrolidine lactam and application in the synthesis of nemonapride and streptopyrrolidine. Tetrahedron 2011, 67, 7829–7837. [Google Scholar] [CrossRef]
- Bartolo, N.D.; Read, J.A.; Valentín, E.M.; Woerpel, K.A. Reactions of allylmagnesium reagents with carbonyl compounds and compounds with C=N double bonds: Their diastereoselectivities generally cannot be analyzed using the Felkin-Anh and chelation-control models. Chem. Rev. 2020, 120, 1513–1619. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Bischoff, A.; Cappiello, J. Asymmetric total synthesis of the gastroprotective microbial agent AI-77-B. Eur. J. Org. Chem. 2003, 5, 821–832. [Google Scholar] [CrossRef]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Hosur, M.V.; Sowder, R.C.; Cachau, R.E.; Collins, J.; Silva, A.M.; Erickson, J.W. Crystal structures of native and inhibited forms of human cathepsin D: Implications for lysosomal targeting and drug design. Proc. Natl. Acad. Sci. USA 1993, 90, 6796–6800. [Google Scholar] [CrossRef] [PubMed]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).