
Structural Elucidation of a Glucan from Trichaster palmiferus by Its Degraded Products and Preparation of Its Sulfated Derivative as an Anticoagulant

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Extraction, Purification, and Physicochemical Properties of TPG
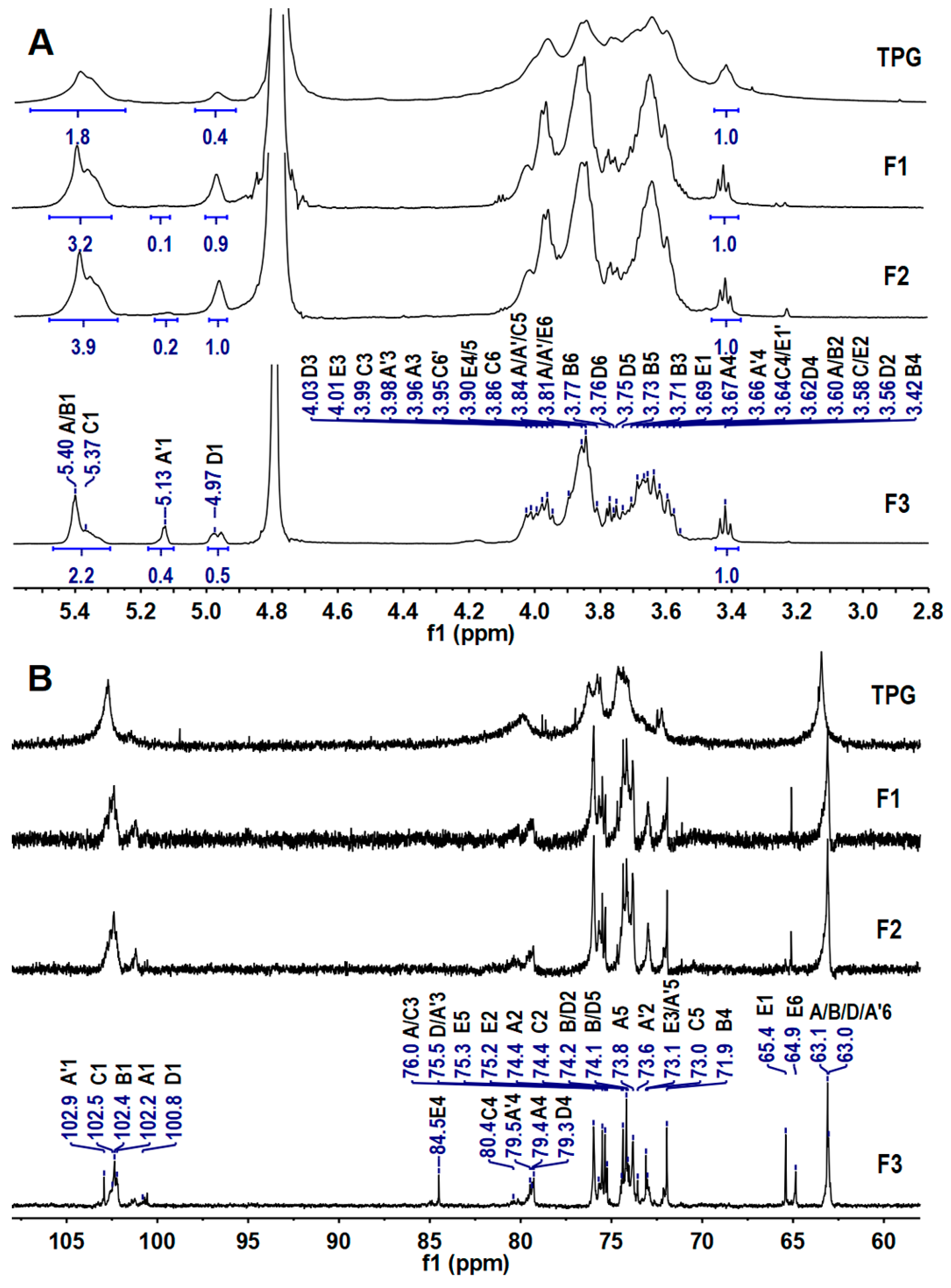
2.2. Elucidation of the Precise Structure of TPG
2.3. Physicochemical Properties of TPGS
2.4. Analysis of Anticoagulant Activity of TPGS
3. Materials and Methods
3.1. Materials
3.2. Extraction, Isolation, and Purification of TPG
3.3. Preparation of Depolymerized Product of TPG
3.4. Sulfated Modification of TPG
3.5. Physicochemical Properties
3.6. Methylation and GC-MS Analysis
3.7. Assays of Anticoagulant Activities
3.8. Measurement of Inhibition of Human Intrinsic Factor Xase
3.9. Determination of Inhibition of Human FIIa and FXa
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Senni, K.; Pereira, J.; Gueniche, F.; Delbarre-Ladrat, C.; Sinquin, C.; Ratiskol, J.; Godeau, G.; Fischer, A.M.; Helley, D.; Colliec-Jouault, S. Marine polysaccharides: A source of bioactive molecules for cell therapy and tissue engineering. Mar. Drugs 2011, 9, 1664–1681. [Google Scholar] [CrossRef]
- Kang, J.; Jia, X.; Wang, N.; Xiao, M.; Song, S.; Wu, S.; Li, Z.; Wang, S.; Cui, S.W.; Guo, Q. Insights into the structure-bioactivity relationships of marine sulfated polysaccharides: A review. Food Hydrocoll. 2022, 123, 107049. [Google Scholar] [CrossRef]
- Yao, W.; Qiu, H.M.; Cheong, K.L.; Zhong, S. Advances in anti-cancer effects and underlying mechanisms of marine algae polysaccharides. Int. J. Biol. Macromol. 2022, 221, 472–485. [Google Scholar] [CrossRef]
- Yang, M.; Zhou, D.; Xiao, H.; Fu, X.; Kong, Q.; Zhu, C. Marine-derived uronic acid-containing polysaccharides: Structures, sources, production, and nutritional functions. Trends Food Sci. Technol. 2022, 122, 1–12. [Google Scholar] [CrossRef]
- Cheong, K.L.; Yu, B.; Chen, J.; Zhong, S. A comprehensive review of the cardioprotective effect of marine algae polysaccharide on the gut microbiota. Foods 2022, 11, 3550. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.I. Echinoderms metabolites: Structure, functions, and biomedical perspectives. Mar. Drugs 2021, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wu, M.; Xiao, C.; Yang, L.; Zhou, L.; Gao, N.; Li, Z.; Chen, J.; Chen, J.; Liu, J.; et al. Discovery of an intrinsic tenase complex inhibitor: Pure nonasaccharide from fucosylated glycosaminoglycan. Proc. Natl. Acad. Sci. USA 2015, 112, 8284–8289. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, Q.; Lv, K.; Ma, H.; Gao, C.; Liu, Y.; Zhang, S.; Zhao, L. Low-molecular-weight fucosylated glycosaminoglycan and its oligosaccharides from sea cucumber as novel anticoagulants: A review. Carbohydr. Polym. 2021, 251, 117034. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhao, L.; Gao, N.; Yin, R.; Li, S.; Sun, H.; Zhou, L.; Zhao, G.; Purcell, S.W.; Zhao, J. From multi-target anticoagulants to DOACs, and intrinsic coagulation factor inhibitors. Blood Rev. 2020, 39, 100615. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, R.; Namburi, R.B.; Ortega-Martinez, O.; Shi, X.; Zaia, J.; Dupont, S.T.; Thorndyke, M.C.; Lindahl, U.; Spillmann, D. Brittlestars contain highly sulfated chondroitin sulfates/dermatan sulfates that promote fibroblast growth factor 2-induced cell signaling. Glycobiology 2014, 24, 195–207. [Google Scholar]
- Zhang, W.; Wang, J.; Jin, W.; Zhang, Q. The antioxidant activities and neuroprotective effect of polysaccharides from the starfish Asterias rollestoni. Carbohydr. Polym. 2013, 95, 9–15. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhao, L.; Cha, Q.; Sun, Y.; Ye, H.; Zeng, X. Structural characterization and immunostimulatory activity of a homogeneous polysaccharide from Sinonovacula constricta. J. Agric. Food Chem. 2015, 63, 7986–7994. [Google Scholar] [CrossRef]
- Meng, Y.; Lyu, F.; Xu, X.; Zhang, L. Recent advances in chain conformation and bioactivities of triple-helix polysaccharides. Biomacromolecules 2020, 21, 1653–1677. [Google Scholar] [CrossRef]
- Otero, P.; Carpena, M.; Garcia-Oliveira, P.; Echave, J.; Soria-Lopez, A.; Garcia-Perez, P.; Fraga-Corral, M.; Cao, H.; Nie, S.; Xiao, J.; et al. Seaweed polysaccharides: Emerging extraction technologies, chemical modifications and bioactive properties. Crit. Rev. Food Sci. Nutr. 2021. [Google Scholar] [CrossRef]
- Vasconcelos, A.F.D.; Dekker, R.F.H.; Barbosa, A.M.; Carbonero, E.R.; Silveira, J.L.M.; Glauser, B.; Pereira, M.S.; Corradi Da Silva, M.D.L. Sulfonation and anticoagulant activity of fungal exocellular β-(1→6)-D-glucan (lasiodiplodan). Carbohydr. Polym. 2013, 92, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Chen, C.; Wang, S.K.; Sun, G.J. Biochemical analysis and hypoglycemic activity of a polysaccharide isolated from the fruit of Lycium barbarum L. Int. J. Biol. Macromol. 2015, 77, 235–242. [Google Scholar] [CrossRef]
- Wang, J.; Nie, S.; Cui, S.W.; Wang, Z.; Phillips, A.O.; Phillips, G.O.; Li, Y.; Xie, M. Structural characterization and immunostimulatory activity of a glucan from natural Cordyceps sinensis. Food Hydrocoll. 2017, 67, 139–147. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, L.; Yan, A.; Feng, L.; Wan, Y. Fractionation, structure and conformation characterization of polysaccharides from Anoectochilus roxburghii. Carbohydr. Polym. 2020, 231, 115688. [Google Scholar] [CrossRef]
- Meng, F.; Li, Q.; Qi, Y.; He, C.; Wang, C.; Zhang, Q. Characterization and immunoregulatory activity of two polysaccharides from the root of Ilex asprella. Carbohydr. Polym. 2018, 197, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Deng, J.; Liu, X.; He, P.; He, L.; Zhang, F.; Linhardt, R.J.; Sun, P. Structure and conformation of α-glucan extracted from Agaricus blazei Murill by high-speed shearing homogenization. Int. J. Biol. Macromol. 2018, 113, 558–564. [Google Scholar] [CrossRef]
- Niu, Y.; Yan, W.; Lv, J.; Yao, W.; Yu, L. Characterization of a novel polysaccharide from tetraploid Gynostemma pentaphyllum Makino. J. Agric. Food Chem. 2013, 61, 4882–4889. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Sun, G.; Ren, J.; Zhu, T.; Jia, P.; Qu, W.; Li, Q.; Wu, J.; Ma, H.; Wen, S.; et al. An α-glucan isolated from root of Isatis indigotica, its structure and adjuvant activity. Glycoconj. J. 2014, 31, 317–326. [Google Scholar] [CrossRef]
- Jiang, J.; Kong, F.; Li, N.; Zhang, D.; Yan, C.; Lv, H. Purification, structural characterization and in vitro antioxidant activity of a novel polysaccharide from Boshuzhi. Carbohydr. Polym. 2016, 147, 365–371. [Google Scholar] [CrossRef]
- Shi, X.; Li, O.; Yin, J.; Nie, S. Structure identification of α-glucans from Dictyophora echinovolvata by methylation and 1D/2D NMR spectroscopy. Food Chem. 2019, 271, 338–344. [Google Scholar] [CrossRef]
- Li, B.; Zhang, N.; Wang, D.X.; Jiao, L.; Tan, Y.; Wang, J.; Li, H.; Wu, W.; Jiang, D.C. Structural analysis and antioxidant activities of neutral polysaccharide isolated from Epimedium koreanum Nakai. Carbohydr. Polym. 2018, 196, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, C.; Li, M.; Chen, X.; Ding, K. Structural elucidation of a glucan from Crataegus pinnatifida and its bioactivity on intestinal bacteria strains. Int. J. Biol. Macromol. 2019, 128, 435–443. [Google Scholar] [CrossRef]
- Cheong, K.L.; Xia, L.X.; Liu, Y. Isolation and characterization of polysaccharides from oysters (Crassostrea gigas) with anti-tumor activities using an aqueous two-phase system. Mar. Drugs 2017, 15, 338. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ouyang, K.; Li, J.; Liu, X.; An, Q.; Zhao, M.; Chen, S.; Li, X.; Ye, X.; Zhao, Z.; et al. Sulfated modification, characterization, immunomodulatory activities and mechanism of the polysaccharides from Cyclocarya paliurus on dendritic cells. Int. J. Biol. Macromol. 2020, 159, 108–116. [Google Scholar] [CrossRef]
- Wang, J.; Guo, H.; Zhang, J.; Wang, X.; Zhao, B.; Yao, J.; Wang, Y. Sulfated modification, characterization and structure-antioxidant relationships of Artemisia sphaerocephala polysaccharides. Carbohydr. Polym. 2010, 81, 897–905. [Google Scholar] [CrossRef]
- Martinichen-herrero, J.C.; Carbonero, E.R.; Sassaki, G.L. Anticoagulant and antithrombotic activities of a chemically sulfated galactoglucomannan obtained from the lichen Cladonia ibitipocae. Int. J. Biol. Macromol. 2005, 35, 97–102. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, Y.; Wang, D.; Wu, N.; Wang, K.; Zhang, Y. Preparation, chemical structure and α-glucosidase inhibitory activity of sulfated polysaccharide from Grifola frondosa. J. Funct. Foods 2022, 98, 105289. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Z.; Shen, M.; Nie, S.; Gong, B.; Li, H.; Zhao, Q.; Li, W.; Xie, M. Sulfated modification, characterization and antioxidant activities of polysaccharide from Cyclocarya paliurus. Food Hydrocoll. 2016, 53, 7–15. [Google Scholar] [CrossRef]
- Li, T.; Ma, H.; Li, H.; Tang, H.; Huang, J.; Wei, S.; Yuan, Q.; Shi, X.; Gao, C.; Mi, S.; et al. Physicochemical properties and anticoagulant activity of purified heteropolysaccharides from Laminaria japonica. Molecules 2022, 27, 3027. [Google Scholar] [CrossRef]
- Yuan, Q.; Li, H.; Wang, Q.; Sun, S.; Fang, Z.; Tang, H.; Shi, X.; Wen, J.; Huang, L.; Bai, M.; et al. Deaminative-cleaved S. monotuberculatus fucosylated glycosaminoglycan: Structural elucidation and anticoagulant activity. Carbohydr. Polym. 2022, 298, 120072. [Google Scholar] [CrossRef]
- Xiao, C.; Zhao, L.; Gao, N.; Wu, M.; Zhao, J. Nonasaccharide inhibits intrinsic factor Xase complex by binding to factor IXa and disrupting factor IXa-factor VIIIa interactions. Thromb. Haemost. 2019, 119, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Feng, Y.; He, W.; Wang, L.; Wang, R.; Dong, L.; Wang, C. Post-screening characterisation and in vivo evaluation of an anti-inflammatory polysaccharide fraction from Eucommia ulmoides. Carbohydr. Polym. 2017, 169, 304–314. [Google Scholar] [CrossRef]
- Yang, J.; Luo, K.; Li, D.; Yu, S.; Cai, J.; Chen, L.; Du, Y. Preparation, characterization and in vitro anticoagulant activity of highly sulfated chitosan. Int. J. Biol. Macromol. 2013, 52, 25–31. [Google Scholar] [CrossRef]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric method for determination of sugars and related substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Dodgson, K.S.; Price, R.G. A note on the determination of the ester sulphate content of sulphated polysaccharides. Biochem. J. 1962, 84, 106–110. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, X.; Ma, M.; Long, T.; Xiao, C.; Zhang, J.; Liu, J.; Zhao, L. Immunoenhancing glucuronoxylomannan from Tremella aurantialba Bandoni et Zang and its low-molecular-weight fractions by radical depolymerization: Properties, structures and effects on macrophages. Carbohydr. Polym. 2020, 238, 116184. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lai, S.; Huang, R.; Wu, M.; Gao, N.; Xu, L.; Qin, H.; Peng, W.; Zhao, J. Structure and anticoagulant activity of fucosylated glycosaminoglycan degraded by deaminative cleavage. Carbohydr. Polym. 2013, 98, 1514–1523. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Retention Time (min) a | Methylated Sugars | Mass Fragments (m/z) | Type of Linkage | Area (%) |
|---|---|---|---|---|
| 1.00 | 2,3,4,6-Me4-Glcp | 43, 71, 87, 101, 117, 129, 145, 161, 205 | Glcp-(1→ | 17.2 |
| 1.25 | 2,3,6-Me3-Glcp | 43, 87, 99, 101, 113, 117, 129, 131, 161, 173, 233 | →4)-Glcp-(1→ | 69.66 |
| 1.40 | 2,6-Me2-Glcp | 43, 87, 97, 117, 159, 185 | →3,4)-Glcp-(→ | 0.61 |
| 1.53 | 2,3-Me2-Glcp | 43, 71, 85, 87, 99, 101, 117, 127, 159, 161, 201 | →4,6)-Glcp-(→ | 12.53 |
| Residues | H/C | Chemical Shifts (δ, ppm) | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| A | H | 5.40 | 3.61 | 3.97 | 3.66 a | 3.85 | 3.81/3.91 |
| C | 102.36 | 74.41 | 75.98 | 79.42/80.30 | 73.90 | 63.11 | |
| B | H | 5.39 | 3.60 | 3.71 | 3.42 | 3.73 | 3.77/3.85 |
| C | 102.22 | 74.07 | 75.47 | 71.89 | 74.15 | 63.11 | |
| C | H | 5.37 | 3.58 | 4.00 | 3.67 | 3.87 | 3.86/3.98 |
| C | 102.54 | 74.35 | 75.94 | 79.96 | 73.01 | 69.95 | |
| A’ | H | 5.13 | 3.61 | 3.98 | 3.63 | 3.85 | 3.81/3.91 |
| C | 102.87 | 73.60 | 75.82 | 79.85 | 73.10 | 63.11 | |
| D | H | 4.97 | 3.59 | 4.03 | 3.63 | 3.75 | 3.76/3.84 |
| C | 100.95 | 74.00 | 75.85 | 79.35 | 74.14 | 63.19 | |
| E | H | 3.64/3.69 | 3.58 | 4.01 | 3.89 | 3.76 | 3.68/3.80 |
| C | 65.22 | 75.31 | 73.2 | 84.49 | 75.32 | 64.62 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, H.; Yuan, Q.; Tang, H.; Tan, H.; Li, T.; Wei, S.; Huang, J.; Yao, Y.; Hu, Y.; Zhong, S.; et al. Structural Elucidation of a Glucan from Trichaster palmiferus by Its Degraded Products and Preparation of Its Sulfated Derivative as an Anticoagulant. Mar. Drugs 2023, 21, 148. https://doi.org/10.3390/md21030148
Ma H, Yuan Q, Tang H, Tan H, Li T, Wei S, Huang J, Yao Y, Hu Y, Zhong S, et al. Structural Elucidation of a Glucan from Trichaster palmiferus by Its Degraded Products and Preparation of Its Sulfated Derivative as an Anticoagulant. Marine Drugs. 2023; 21(3):148. https://doi.org/10.3390/md21030148
Chicago/Turabian StyleMa, Haiqiong, Qingxia Yuan, Hao Tang, Hongjie Tan, Tingting Li, Shiying Wei, Jinwen Huang, Yue Yao, Yaping Hu, Shengping Zhong, and et al. 2023. "Structural Elucidation of a Glucan from Trichaster palmiferus by Its Degraded Products and Preparation of Its Sulfated Derivative as an Anticoagulant" Marine Drugs 21, no. 3: 148. https://doi.org/10.3390/md21030148
APA StyleMa, H., Yuan, Q., Tang, H., Tan, H., Li, T., Wei, S., Huang, J., Yao, Y., Hu, Y., Zhong, S., Liu, Y., Gao, C., & Zhao, L. (2023). Structural Elucidation of a Glucan from Trichaster palmiferus by Its Degraded Products and Preparation of Its Sulfated Derivative as an Anticoagulant. Marine Drugs, 21(3), 148. https://doi.org/10.3390/md21030148