

Effect of the Marine Polyketide Plocabulin on Tumor Progression



Abstract
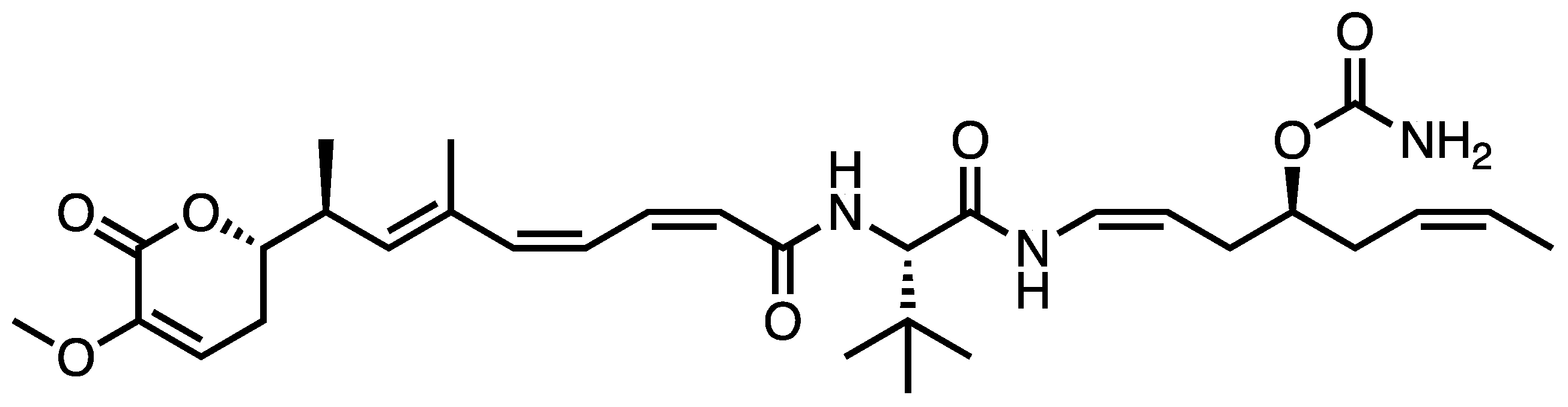
1. Introduction
2. Antitubulin Activity
2.1. In Vitro Studies
2.2. In Vivo Studies
3. Antiangiogenic Activity
4. Clinical Studies
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Martin, T.A.; Ye, L.; Sanders, A.J.; Lane, J.; Jiang, W.G. Cancer Invasion and Metastasis: Molecular and Cellular Perspective. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Kinghorn, A.D.; Carcache De Blanco, E.J.; Lucas, D.M.; Rakotondraibe, H.L.; Orjala, J.; Soejarto, D.D.; Oberlies, N.H.; Pearce, C.J.; Wani, M.C.; Stockwell, B.R.; et al. Discovery of Anticancer Agents of Diverse Natural Origin. Anticancer Res. 2016, 36, 5623–5637. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Díaz Marrero, A.R.; Deniz, I.; Erdoğan, A.; Lukić Bilela, L.; Moulin, C.; Taffin-de-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619. [Google Scholar] [CrossRef]
- Calcabrini, C.; Catanzaro, E.; Bishayee, A.; Turrini, E.; Fimognari, C. Marine Sponge Natural Products with Anticancer Potential: An Updated Review. Mar. Drugs 2017, 15, 310. [Google Scholar] [CrossRef]
- Pereira, R.B.; Evdokimov, N.M.; Lefranc, F.; Valentão, P.; Kornienko, A.; Pereira, D.M.; Andrade, P.B.; Gomes, N.G.M. Marine-Derived Anticancer Agents: Clinical Benefits, Innovative Mechanisms, and New Targets. Mar. Drugs 2019, 17, 329. [Google Scholar] [CrossRef]
- Pera, B.; Barasoain, I.; Pantazopoulou, A.; Canales, A.; Matesanz, R.; Rodriguez-Salarichs, J.; García-Fernandez, L.F.; Moneo, V.; Jiménez-Barbero, J.; Galmarini, C.M.; et al. New Interfacial Microtubule Inhibitors of Marine Origin, PM050489/PM060184, with Potent Antitumor Activity and a Distinct Mechanism. ACS Chem. Biol. 2013, 8, 2084–2094. [Google Scholar] [CrossRef]
- Martín, M.J.; Coello, L.; Fernández, R.; Reyes, F.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez-Sancho, F.; Bueno, S.; et al. Isolation and First Total Synthesis of PM050489 and PM060184, Two New Marine Anticancer Compounds. J. Am. Chem. Soc. 2013, 135, 10164–10171. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef]
- Lafanechère, L. The Microtubule Cytoskeleton: An Old Validated Target for Novel Therapeutic Drugs. Front. Pharmacol. 2022, 13, 969183. [Google Scholar] [CrossRef]
- Vicente, J.J.; Wordeman, L. The Quantification and Regulation of Microtubule Dynamics in the Mitotic Spindle. Curr. Opin. Cell Biol. 2019, 60, 36–43. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Tubulin-Interactive Natural Products as Anticancer Agents. J. Nat. Prod. 2009, 72, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, X.; Cui, H.; Lei, X.; Liu, H.; Wang, Q.; Li, Y. Synthesis of the Analogs of Plocabulin and Their Preliminary Structure-Activity Relationship Study. Bioorg. Med. Chem. Lett. 2021, 51, 128355. [Google Scholar] [CrossRef]
- Jordan, M.A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H.P.; Davis, C.; Littlefield, B.A.; Wilson, L. The Primary Antimitotic Mechanism of Action of the Synthetic Halichondrin E7389 Is Suppression of Microtubule Growth. Mol. Cancer Ther. 2005, 4, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Díez, M.; Guillén-Navarro, M.J.; Pera, B.; Bouchet, B.P.; Martínez-Leal, J.F.; Barasoain, I.; Cuevas, C.; Andreu, J.M.; García-Fernández, L.F.; Díaz, J.F.; et al. PM060184, a New Tubulin Binding Agent with Potent Antitumor Activity Including P-Glycoprotein over-Expressing Tumors. Biochem. Pharmacol. 2014, 88, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; DeLuca, K.; Williams, D.; Jordan, M.A.; Wilson, L. Antiproliferative Mechanism of Action of Cryptophycin-52: Kinetic Stabilization of Microtubule Dynamics by High-Affinity Binding to Microtubule Ends. Proc. Natl. Acad. Sci. USA 1998, 95, 9313–9318. [Google Scholar] [CrossRef]
- Prota, A.E.; Bargsten, K.; Diaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A New Tubulin-Binding Site and Pharmacophore for Microtubule-Destabilizing Anticancer Drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef]
- Navarrete, K.R.; Jiménez, V.A. Interdimeric Curvature in Tubulin-Tubulin Complexes Delineates the Microtubule-Destabilizing Properties of Plocabulin. J. Chem. Inf. Model. 2020, 60, 4076–4084. [Google Scholar] [CrossRef] [PubMed]
- Heredia-Soto, V.; Escudero, J.; Miguel, M.; Ruiz, P.; Gallego, A.; Berjón, A.; Hernández, A.; Martínez-Díez, M.; Zheng, S.; Tang, J.; et al. Antitumoral Effect of Plocabulin in High Grade Serous Ovarian Carcinoma Cell Line Models. Front. Oncol. 2022, 12, 862321. [Google Scholar] [CrossRef]
- Costales-Carrera, A.; Fernández-Barral, A.; Bustamante-Madrid, P.; Guerra, L.; Cantero, R.; Barbáchano, A.; Muñoz, A. Plocabulin Displays Strong Cytotoxic Activity in a Personalized Colon Cancer Patient-Derived 3D Organoid Assay. Mar. Drugs 2019, 17, 648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wozniak, A.; Wellens, J.; Gebreyohannes, Y.K.; Guillén, M.J.; Avilés, P.M.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. Plocabulin, a Novel Tubulin Inhibitor, Has Potent Antitumor Activity in Patient-Derived Xenograft Models of Gastrointestinal Stromal Tumors. Transl. Oncol. 2020, 13, 100832. [Google Scholar] [CrossRef]
- Wang, Y.; Wozniak, A.; Cornillie, J.; Avilés, P.; Debiec-Rychter, M.; Sciot, R.; Schöffski, P. Plocabulin, a Novel Tubulin Inhibitor, Has Potent Antitumour Activity in Patient-Derived Xenograft Models of Soft Tissue Sarcoma. Int. J. Mol. Sci. 2022, 23, 7454. [Google Scholar] [CrossRef]
- Goda, K.; Bacso, Z.; Szabo, G. Multidrug Resistance Through the Spectacle of P-Glycoprotein. Curr. Cancer Drug Targets 2009, 9, 281–297. [Google Scholar] [CrossRef]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of Hypoxia and Necrosis in Multicellular Tumor Spheroids Is Associated with Resistance to Chemotherapy Treatment. Oncotarget 2017, 8, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Marshall, L.J.; Triunfol, M.; Seidle, T. Patient-Derived Xenograft vs. Organoids: A Preliminary Analysis of Cancer Research Output, Funding and Human Health Impact in 2014–2019. Animals 2020, 10, 1923. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P. The Story of Imatinib in GIST—A Journey through the Development of a Targeted Therapy. Oncol. Res. Treat. 2018, 41, 472–477. [Google Scholar] [CrossRef]
- Huang, W.-K.; Wu, C.-E.; Wang, S.-Y.; Chang, C.-F.; Chou, W.-C.; Chen, J.-S.; Yeh, C.-N. Systemic Therapy for Gastrointestinal Stromal Tumor: Current Standards and Emerging Challenges. Curr. Treat. Options Oncol. 2022, 23, 1303–1319. [Google Scholar] [CrossRef]
- Edmonson, J.H.; Marks, R.S.; Buckner, J.C.; Mahoney, M.R. Contrast of Response to Dacarbazine, Mitomycin, Doxorubicin, and Cisplatin (DMAP) plus GM-CSF between Patients with Advanced Malignant Gastrointestinal Stromal Tumors and Patients with Other Advanced Leiomyosarcomas. Cancer Investig. 2002, 20, 605–612. [Google Scholar] [CrossRef]
- Wiltink, L.M.; Haas, R.L.M.; Gelderblom, H.; van de Sande, M.A.J. Treatment Strategies for Metastatic Soft Tissue Sarcomas. Cancers 2021, 13, 1722. [Google Scholar] [CrossRef]
- Seidi, K.; Jahanban-Esfahlan, R.; Zarghami, N. Tumor Rim Cells: From Resistance to Vascular Targeting Agents to Complete Tumor Ablation. Tumour Biol. 2017, 39, 1010428317691001. [Google Scholar] [CrossRef] [PubMed]
- Rieker, R.J.; Weitz, J.; Lehner, B.; Egerer, G.; Mueller, A.; Kasper, B.; Schirmacher, P.; Joos, S.; Mechtersheimer, G. Genomic Profiling Reveals Subsets of Dedifferentiated Liposarcoma to Follow Separate Molecular Pathways. Virchows Arch. 2010, 456, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Pantazopoulou, A.; Galmarini, C.M.; Peñalva, M.A. Molecular Basis of Resistance to the Microtubule-Depolymerizing Antitumor Compound Plocabulin. Sci. Rep. 2018, 8, 8616. [Google Scholar] [CrossRef] [PubMed]
- Bocci, G.; Nicolaou, K.C.; Kerbel, R.S. Protracted Low-Dose Effects on Human Endothelial Cell Proliferation and Survival in Vitro Reveal a Selective Antiangiogenic Window for Various Chemotherapeutic Drugs. Cancer Res. 2002, 62, 6938–6943. [Google Scholar]
- Maj, E.; Papiernik, D.; Wietrzyk, J. Antiangiogenic Cancer Treatment: The Great Discovery and Greater Complexity (Review). Int. J. Oncol. 2016, 49, 1773–1784. [Google Scholar] [CrossRef]
- Schwartz, E.L. Anti-Vascular Actions of Microtubule-Binding Drugs. Clin. Cancer Res. 2009, 15, 2594–2601. [Google Scholar] [CrossRef]
- Hotchkiss, K.A.; Ashton, A.W.; Mahmood, R.; Russell, R.G.; Sparano, J.A.; Schwartz, E.L. Inhibition of Endothelial Cell Function in Vitro and Angiogenesis in Vivo by Docetaxel (Taxotere): Association with Impaired Repositioning of the Microtubule Organizing Center. Mol. Cancer Ther. 2002, 1, 1191–1200. [Google Scholar]
- Kamat, A.A.; Kim, T.J.; Landen, C.N.; Lu, C.; Han, L.Y.; Lin, Y.G.; Merritt, W.M.; Thaker, P.H.; Gershenson, D.M.; Bischoff, F.Z.; et al. Metronomic Chemotherapy Enhances the Efficacy of Antivascular Therapy in Ovarian Cancer. Cancer Res. 2007, 67, 281–288. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Martin, M.; Bouchet, B.P.; Guillen-Navarro, M.J.; Martínez-Diez, M.; Martinez-Leal, J.F.; Akhmanova, A.; Aviles, P. Plocabulin, a Novel Tubulin-Binding Agent, Inhibits Angiogenesis by Modulation of Microtubule Dynamics in Endothelial Cells. BMC Cancer 2018, 18, 164. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Azzi, S.; Hebda, J.; Gavard, J. Vascular Permeability and Drug Delivery in Cancers. Front. Oncol. 2013, 3, 211. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, R.; Jacobs, F.; Benvenuti, C.; Gaudio, M.; Franceschini, R.; Tancredi, R.; Pedrazzoli, P.; Santoro, A.; Zambelli, A. From Seaside to Bedside: Current Evidence and Future Perspectives in the Treatment of Breast Cancer Using Marine Compounds. Front. Pharmacol. 2022, 13, 909566. [Google Scholar] [CrossRef]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching Cancer Pharmacology with Drugs of Marine Origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef]
- Elez, E.; Gomez-Roca, C.; Soto Matos-Pita, A.; Argiles, G.; Valentin, T.; Coronado, C.; Iglesias, J.; Macarulla, T.; Betrian, S.; Fudio, S.; et al. First-in-Human Phase I Study of the Microtubule Inhibitor Plocabulin in Patients with Advanced Solid Tumors. Investig. New Drugs 2019, 37, 674–683. [Google Scholar] [CrossRef]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-Induced Peripheral Neuropathy: A Current Review. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Seretny, M.; Currie, G.L.; Sena, E.S.; Ramnarine, S.; Grant, R.; MacLeod, M.R.; Colvin, L.A.; Fallon, M. Incidence, Prevalence, and Predictors of Chemotherapy-Induced Peripheral Neuropathy: A Systematic Review and Meta-Analysis. Pain 2014, 155, 2461–2470. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Bagues, A.; Tu, L.; Liu, J.Y.H.; Lu, Z.; Rudd, J.A.; Nurgali, K.; Abalo, R. Mechanisms of Chemotherapy-Induced Neurotoxicity. Front. Pharmacol. 2022, 13, 750507. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Experimental Models | Plocabulin: Concentrations/Doses and Time of Treatment | IC50 or GI50 (nM) | Association with Other Anticancer Drugs | Mechanisms of Action | References | |
|---|---|---|---|---|---|---|
| Human prostate (PC3, 22RV1), pancreas (PANC-1, MiaPaCa-2), ovary (IGROV-1, A2780), lung (NCI-H460, NCI-H23, A549), liver (SK-HEP-1, HEPG2), leukemia (MOLT4, K562), kidney (RXF393, CAKI-1), stomach (HS746T, HGC-27), colon (LoVo, HT29, HCT-116), and breast (MDA-MB-231, MCF-7, BT-474) cancer cell lines | Range of concentrations tested not indicated 72 h | GI50 PC3, 0.114 22RV1, 0.0636 PANC-1, 0.0997 MiaPaCa-2, 0.145 IGROV-1, 0.0429 A2780, 0.152 NCI-H460, 0.101 NCI-H23, 0.129 A549, 0.0892 SK-HEP-1, 0.752 HEPG2, 2.76 MOLT4, 0.102 K562, 0.151 RXF393, 0.0420 CAKI-1, 0.525 HS746T, 2.10 HGC-27, 0.0659 LoVo, 0.146 HT29, 0.0403 HCT-116, 4.68 MDA-MB-231, 0.0909 MCF-7, 4.07 BT-474, 0.054 | ↑ cellular microtubules disruption ↓ mitosis ↓ cellular proliferation | [9] | ||
| Human ovarian cancer cell lines cultured in 2D or in 3D spheroids (PEA1, PEA2, PEO1, PEO4, PEO6, PEO14, PEO23, PEO16, OVCAR-3, 59M, OV866(2), TOV3041G) | Up to 10 nM 72 h | IC50 2D PEA1, 0.07 PEA2, 0.23 PEO1, 0.03 PEO4, 0.05 PEO6, 0.37 PEO14, >10 PEO23, 0.35 PEO16, 0.30 OVCAR-3, 0.03 OV866(2), 0.08 TOV3041G, 0.07 59M, 1.15 | IC50 3D >10 >10 >10 0.16 0.24 >10 >10 0.05 >10 >10 >10 >10 | No synergistic or additive effects with cisplatin, gemcitabine, doxorubicin, trabectedin | ↑ depolymerizing effect on microtubules ↓ invasion (PEA1, PEA2, PEO14 and OV866(2) in 2D) ↓ migration (PEA2, PEO14 and OV866(2) in 2D and OV866(2) also in 3D spheroids) | [21] |
| Colorectal cancer patient-derived tumor organoids | Up to 5 nM 96 h | IC50 Patient#3, 1.1 Patient#4, 0.9 Patient#29, 0.7 | ↓ cell viability of colorectal cancer organoids | [22] | ||
| Human ovarian cancer (IGROV-1, IGROV/ET, A2780, A2780/Dox), human colon cancer (LoVo, LoVo/Dox) cell linesXenografted (MDA-MB-231, HCT-116, HGC-27, H-460, 22RV1 and Caki-1) female athymic nu/nu mice | Range of concentrations tested not indicated 72 h 16 mg/kg i.v. (0, 7, 14 day) | GI50 IGROV-1, 0.4 IGROV-1/ET, 4.0 A2780, 2.5 A2780/Dox, 17 LoVo, 0.1 Lovo/Dox, 5.0 | ↓ tubulin polymerization, alterations in the dynamic instability of microtubules, and blockage of the cell cycle in both interphase and mitosis ↓ microtubules’ shortening and growing to a similar extent ↓ tumor growth | [17] | ||
| 3 patient-derived xenografted nude mice of GIST characterized by different GIST mutations (UZLX-GIST3KIT 11 harbored KIT mutation in exon 11; UZLX-GIST9FKIT 11+17 harbored mutations in exons 11 and 17; UZLX-GIST2BFKIT 9 harbored mutation in exon 9) | 16 mg/kg i.v.once a week for 22 days | ↓ tumor growth ↑ tumor necrosis ↓ tumor vasculature | [23] | |||
| 7 patient-derived xenografted nude mice of sarcoma (dedifferentiated liposarcoma, leiomyosarcoma, undifferentiated sarcoma, intimal sarcoma, and CIC-rearranged sarcoma) | 16 mg/kg i.v. once a week for 22 days | ↓ tumor growth Tumor stabilization in dedifferentiated liposarcoma and intimal sarcoma Tumor regression in leiomyosarcoma, CIC-rearranged sarcoma, and undifferentiated sarcoma models ↑ tumor necrosis ↓ tumor vasculature | [24] | |||
| Phase | Population | Intervention | Key Outcome(s) | Status and/or Key Results | Reference or Clinical Trial Identification Number |
|---|---|---|---|---|---|
| I | Forty-four patients with advanced solid tumors (11 colorectal carcinoma, 5 breast carcinoma, 5 cervix carcinoma, 5 NSCLC, others a) | Plocabulin (i.v.), starting dose: 1.3 mg/m2 administered on D1, D8 and D15 every four weeks | DLTs, MTD, RD | MTD = 14.5 mg/m2, 2/2 patients with DLTs (grade 3 peripheral sensory neuropathy) | [47] |
| I | Sixty patients with advanced solid tumors | Plocabulin (i.v.), starting dose: 4 mg/m2 administered on D1–3 and D15–17 every 28 days | MTD, RD | Completed. Results not yet available. | NCT01299636 |
| I | Fifty-seven patients with advanced solid tumors (18 NSCLC, 13 endometrial or cervical cancer, 13 epithelial cancer, 4 breast cancer, others b) | Plocabulin (6–10.5 mg/m2) and gemcitabine (800 or 1000 mg/m2) | DLTs, MTD, RD | 9% of patients with DLTs (44 patients evaluated), all-cause mortality: 14.5% (55 patients evaluated), serious adverse events: 47.3% (55 patients evaluated) | NCT02533674 |
| II | Twenty-two women with advanced, hormone receptor positive, HER2 negative breast cancer | Plocabulin 9.3 mg/m2 on D1 and D8 every three weeks | PFS rate at 4 months (primary endpoint), OS | PFS rate: 11.1% (18 patients evaluated), OS: 6.6 months (median value; 18 patients evaluated), serious adverse events: 19% (21 patients evaluated) | Eudra CT 2015-002395-24 |
| II | Thirty-two patients with advanced colorectal cancer | Plocabulin 9.3 mg/m2 on D1 and D8 every three weeks | PFS rate at 3 months (primary endpoint), OS | PFS rate: 20.7% (29 patients evaluated), OS: not reached (29 patients evaluated), all-cause mortality: 53% (30 patients evaluated), serious adverse events: 20% (30 patients evaluated) | NCT03427268 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turrini, E.; Maffei, F.; Fimognari, C. Effect of the Marine Polyketide Plocabulin on Tumor Progression. Mar. Drugs 2023, 21, 38. https://doi.org/10.3390/md21010038
Turrini E, Maffei F, Fimognari C. Effect of the Marine Polyketide Plocabulin on Tumor Progression. Marine Drugs. 2023; 21(1):38. https://doi.org/10.3390/md21010038
Chicago/Turabian StyleTurrini, Eleonora, Francesca Maffei, and Carmela Fimognari. 2023. "Effect of the Marine Polyketide Plocabulin on Tumor Progression" Marine Drugs 21, no. 1: 38. https://doi.org/10.3390/md21010038
APA StyleTurrini, E., Maffei, F., & Fimognari, C. (2023). Effect of the Marine Polyketide Plocabulin on Tumor Progression. Marine Drugs, 21(1), 38. https://doi.org/10.3390/md21010038