
New Cladiellin-Type Diterpenoids from the South China Sea Soft Coral Cladiella krempfi: Structures and Molecular Docking Analysis in EGFRs



Abstract
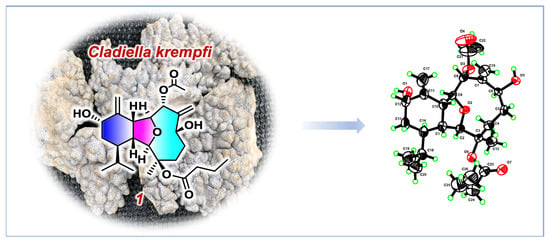
:
1. Introduction
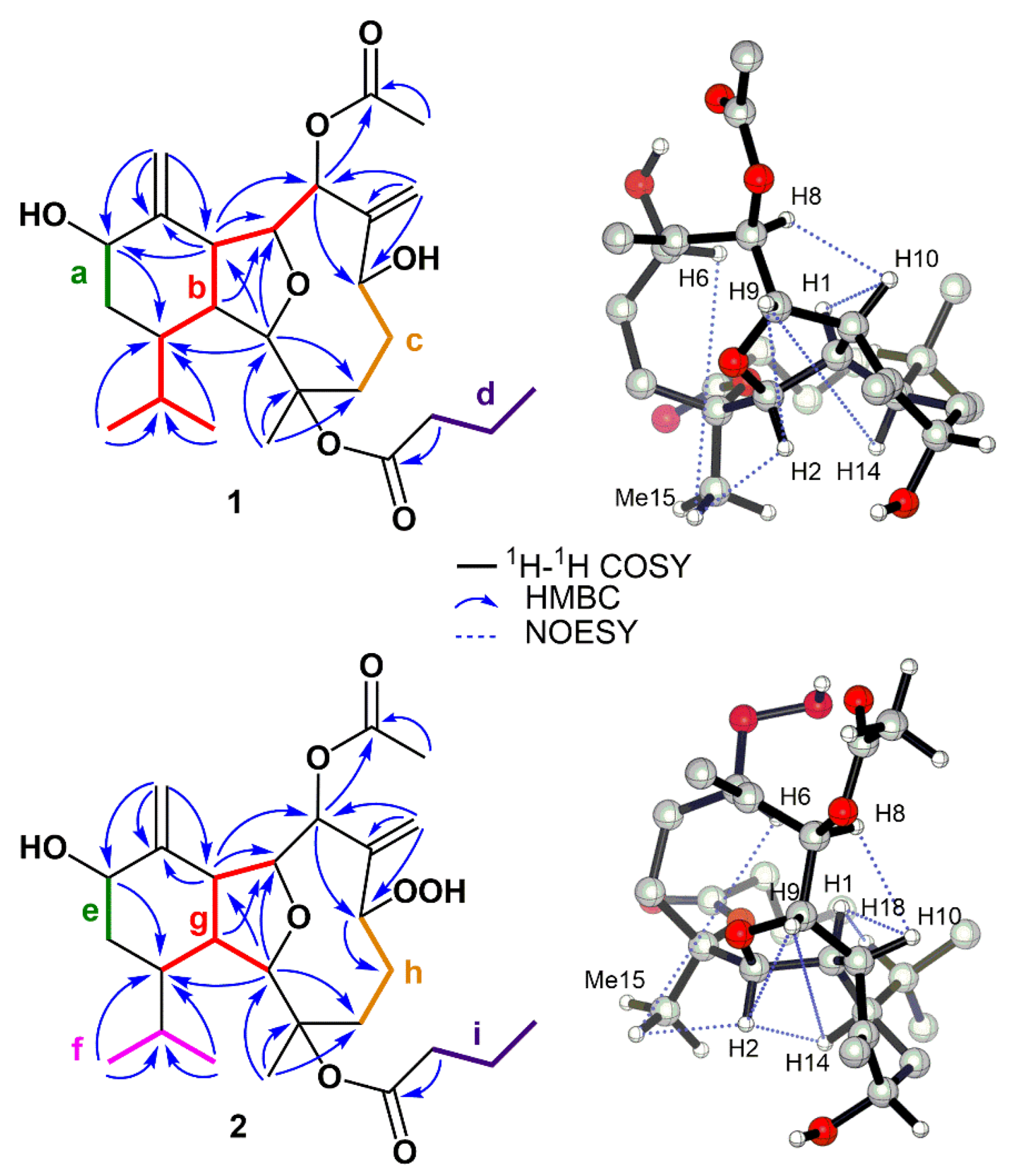
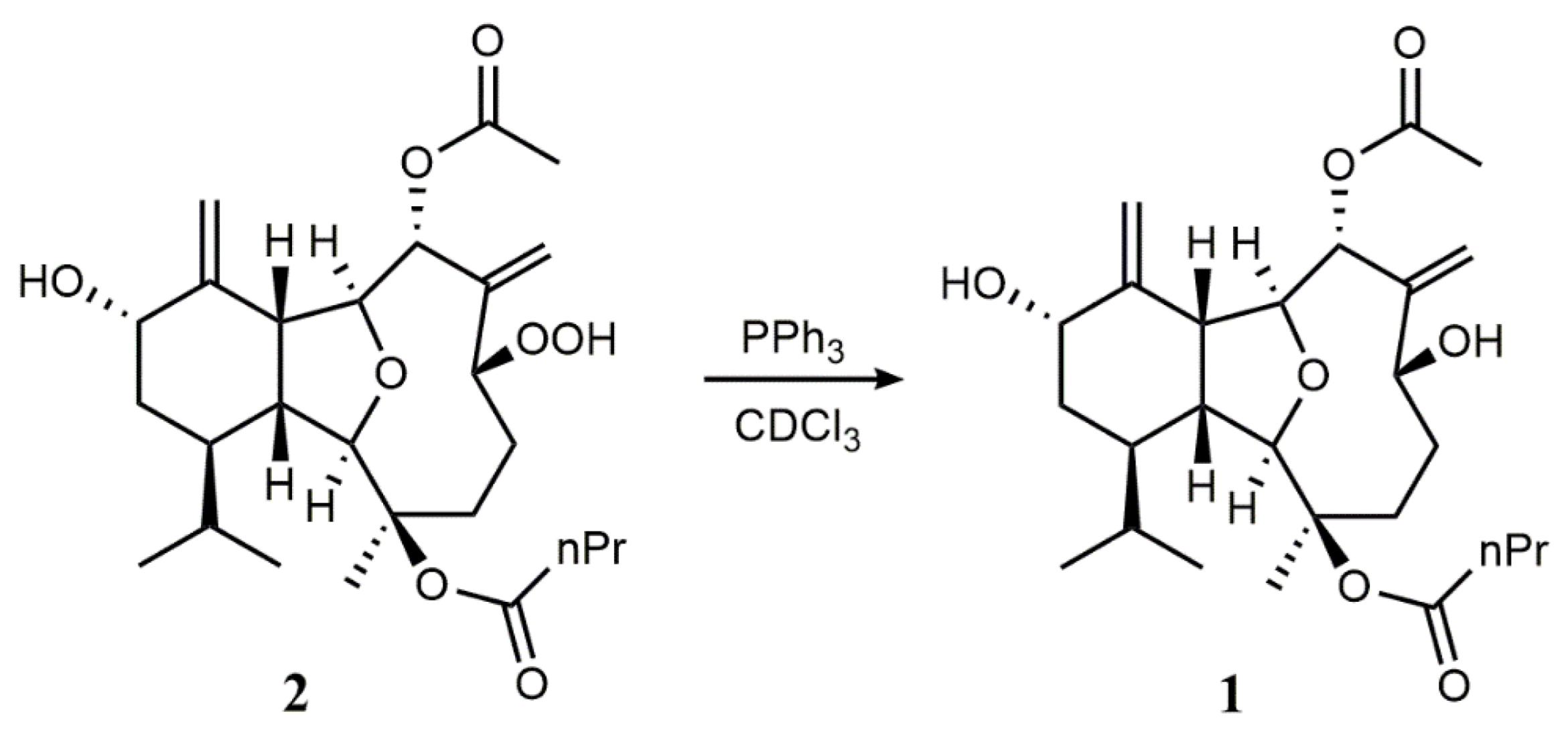
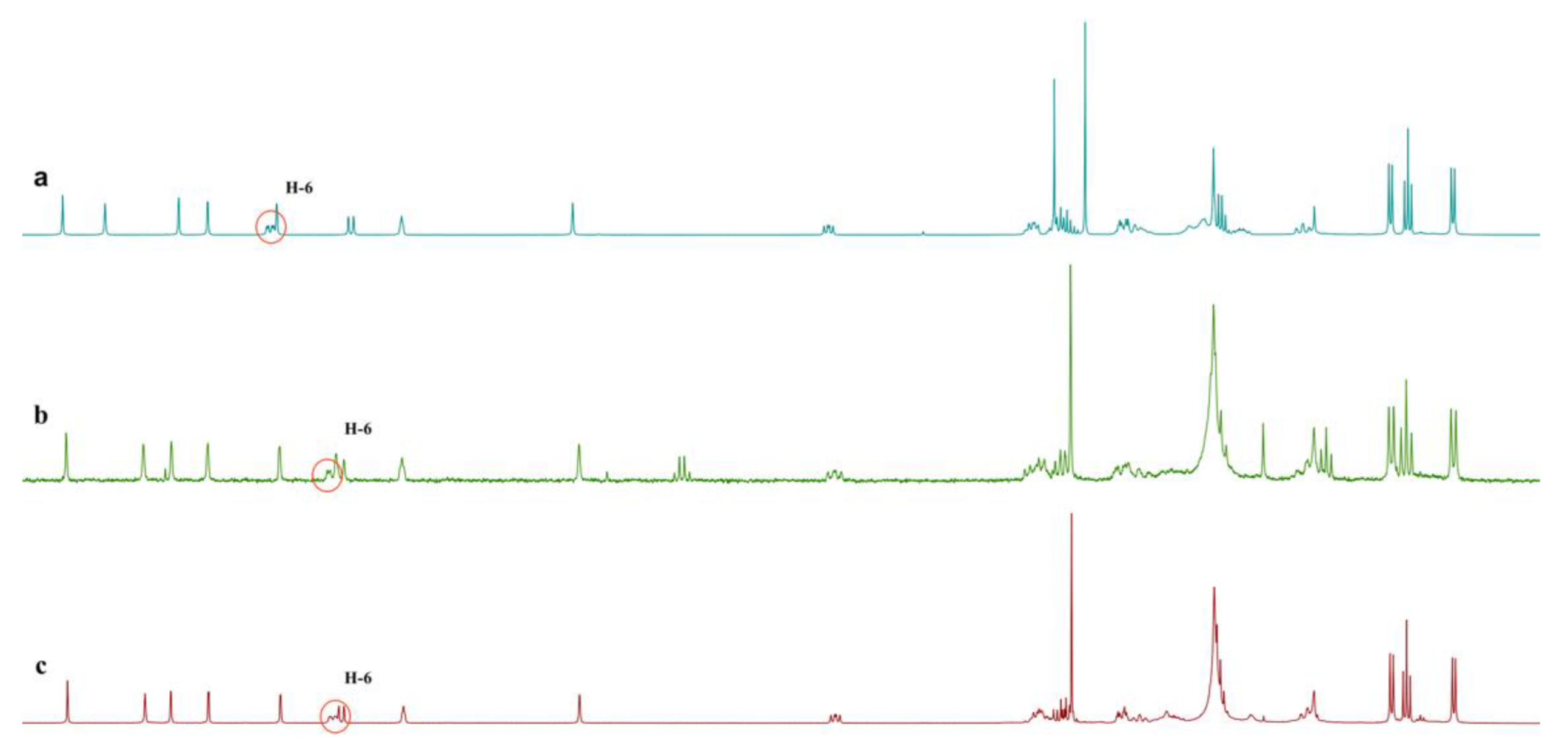
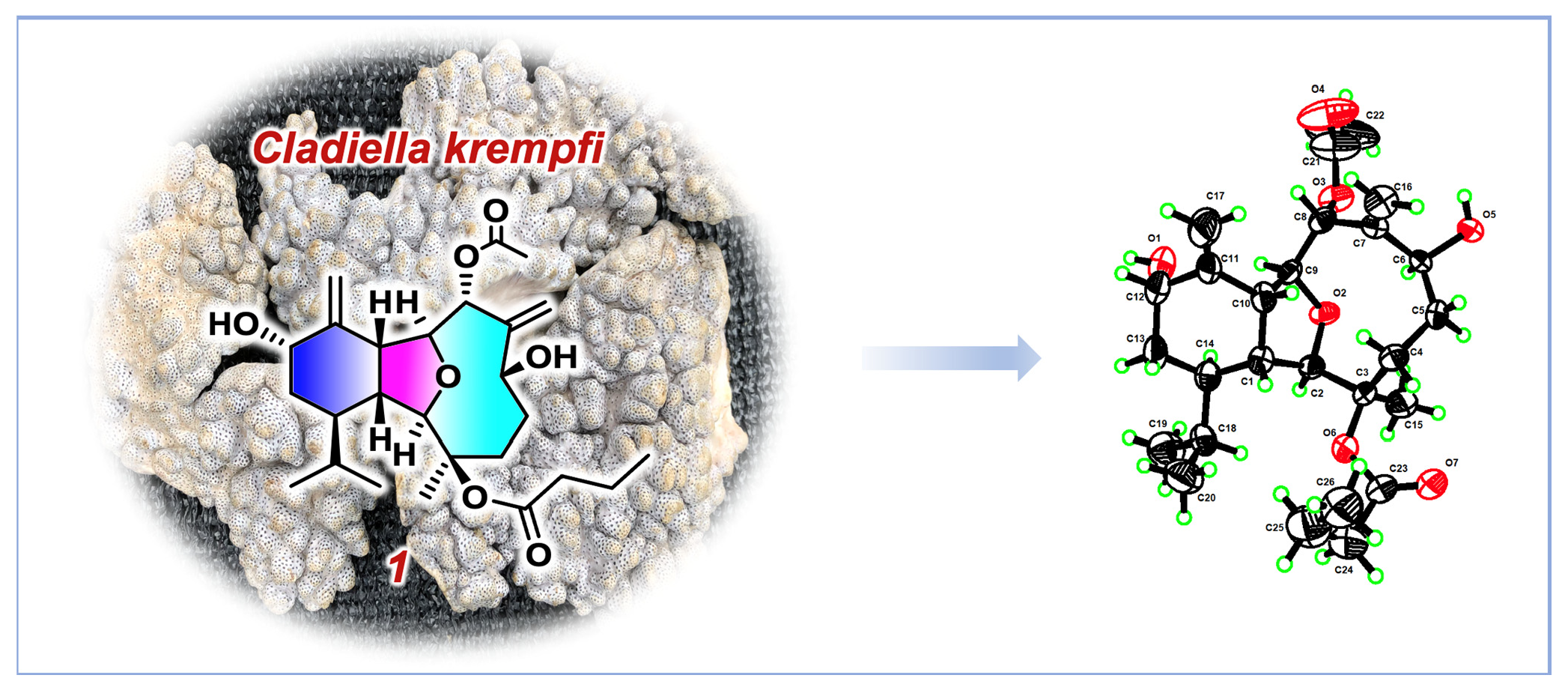
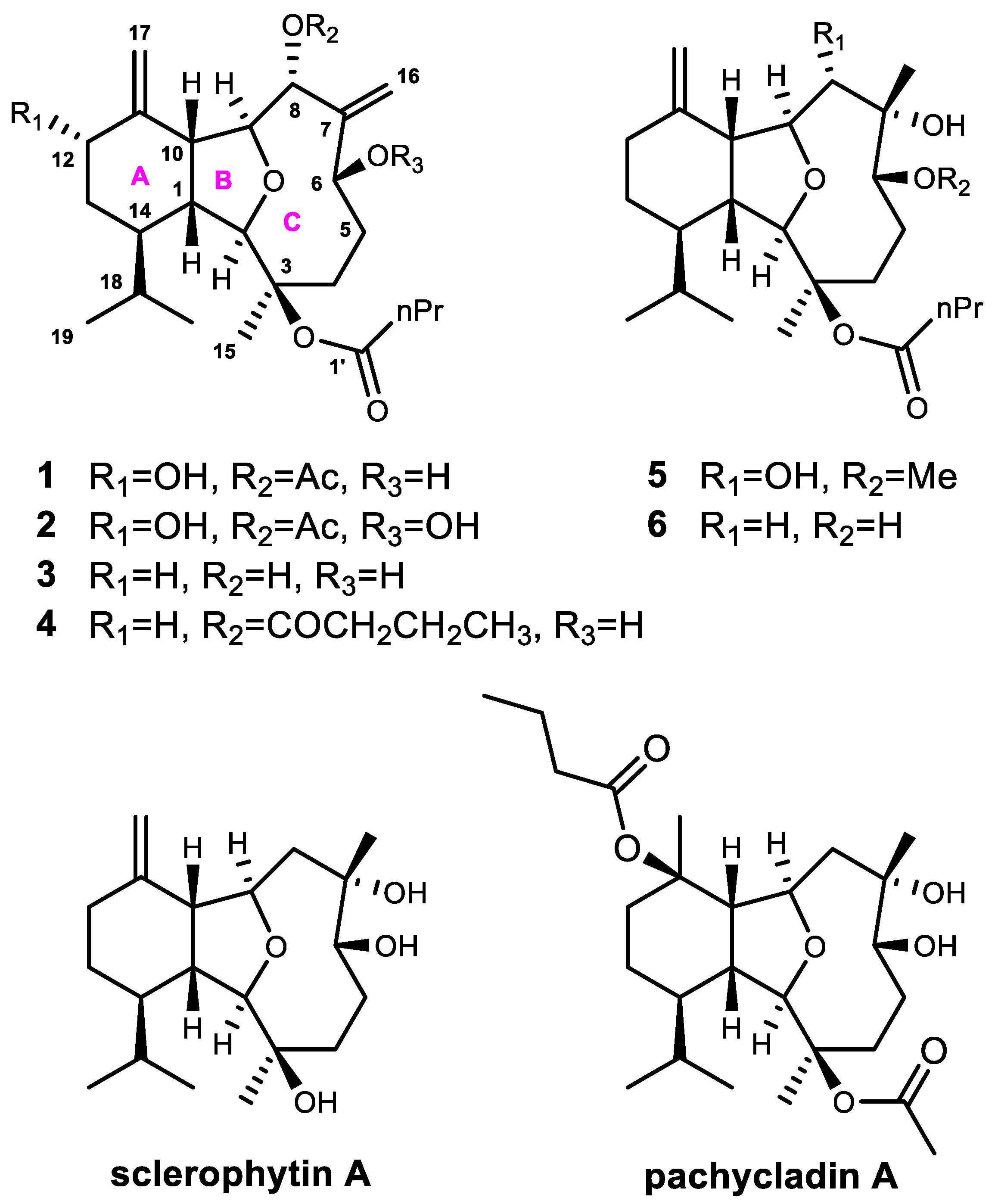
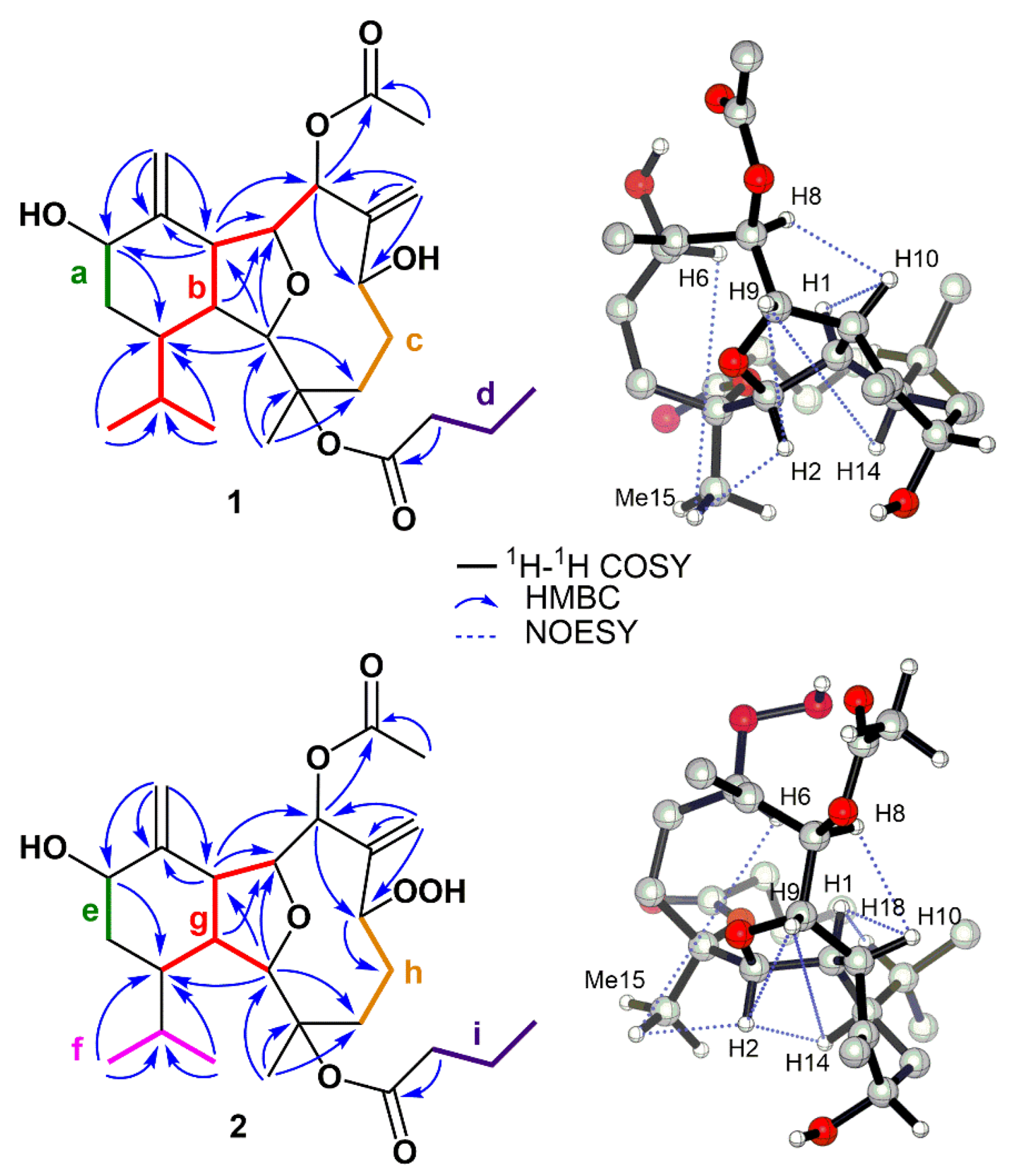
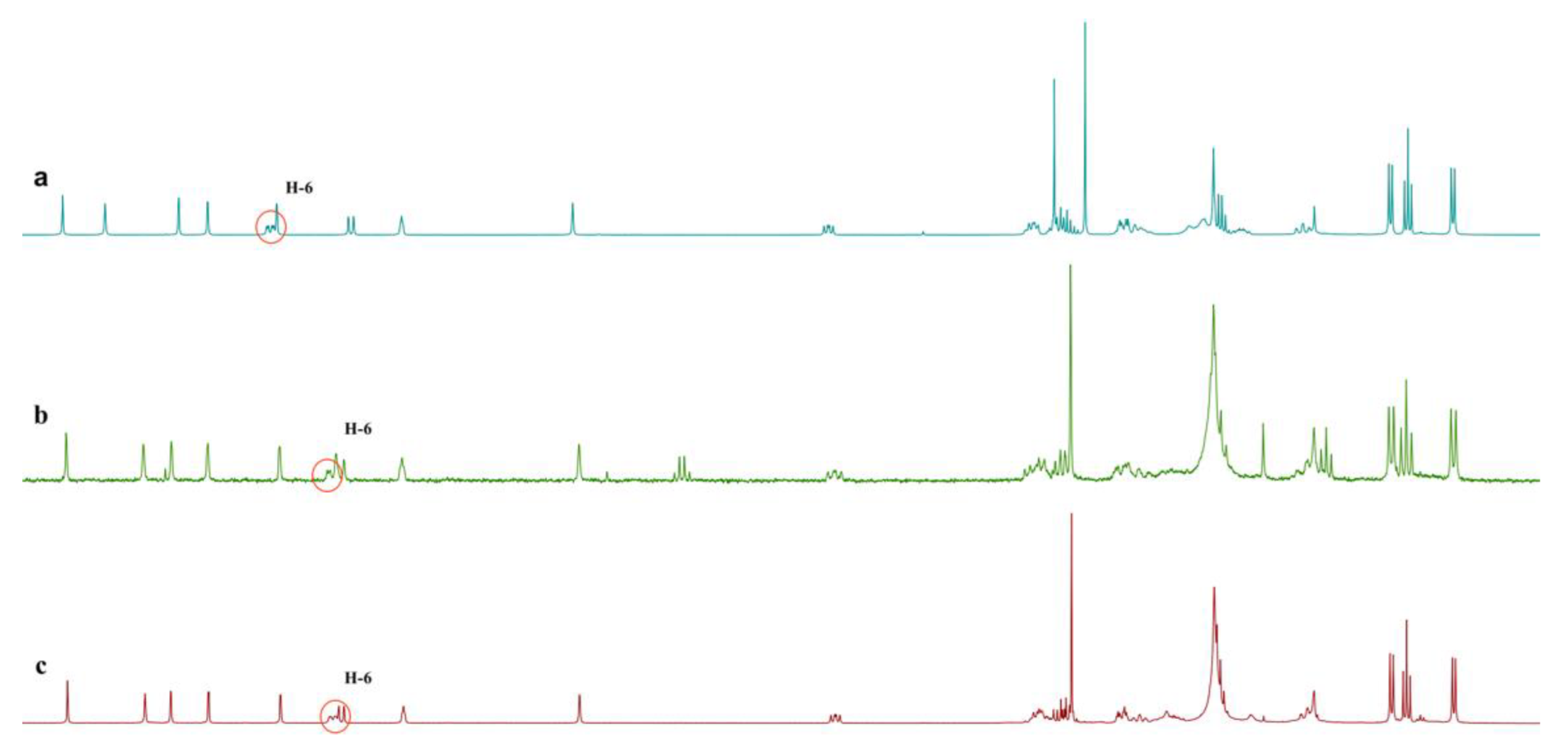
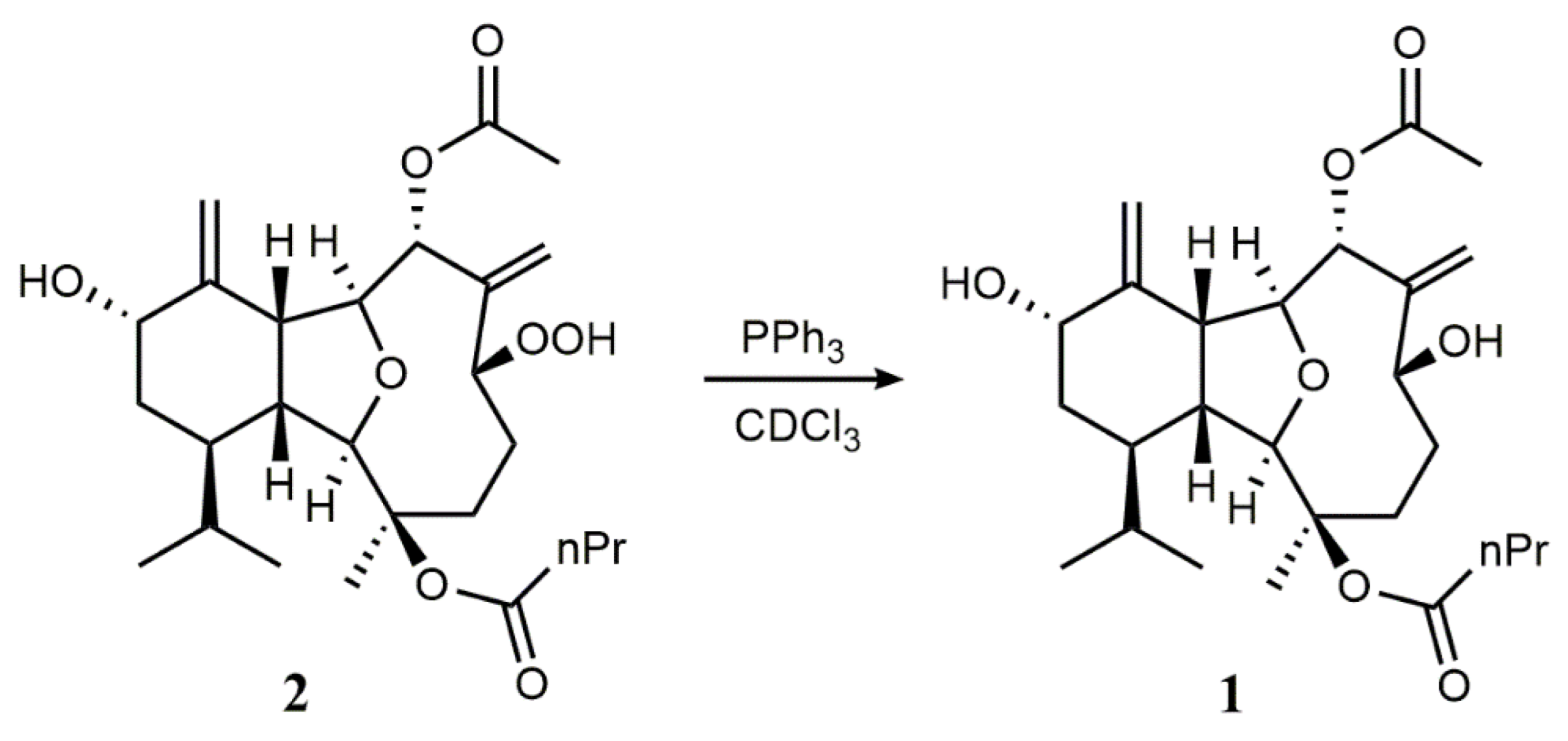
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
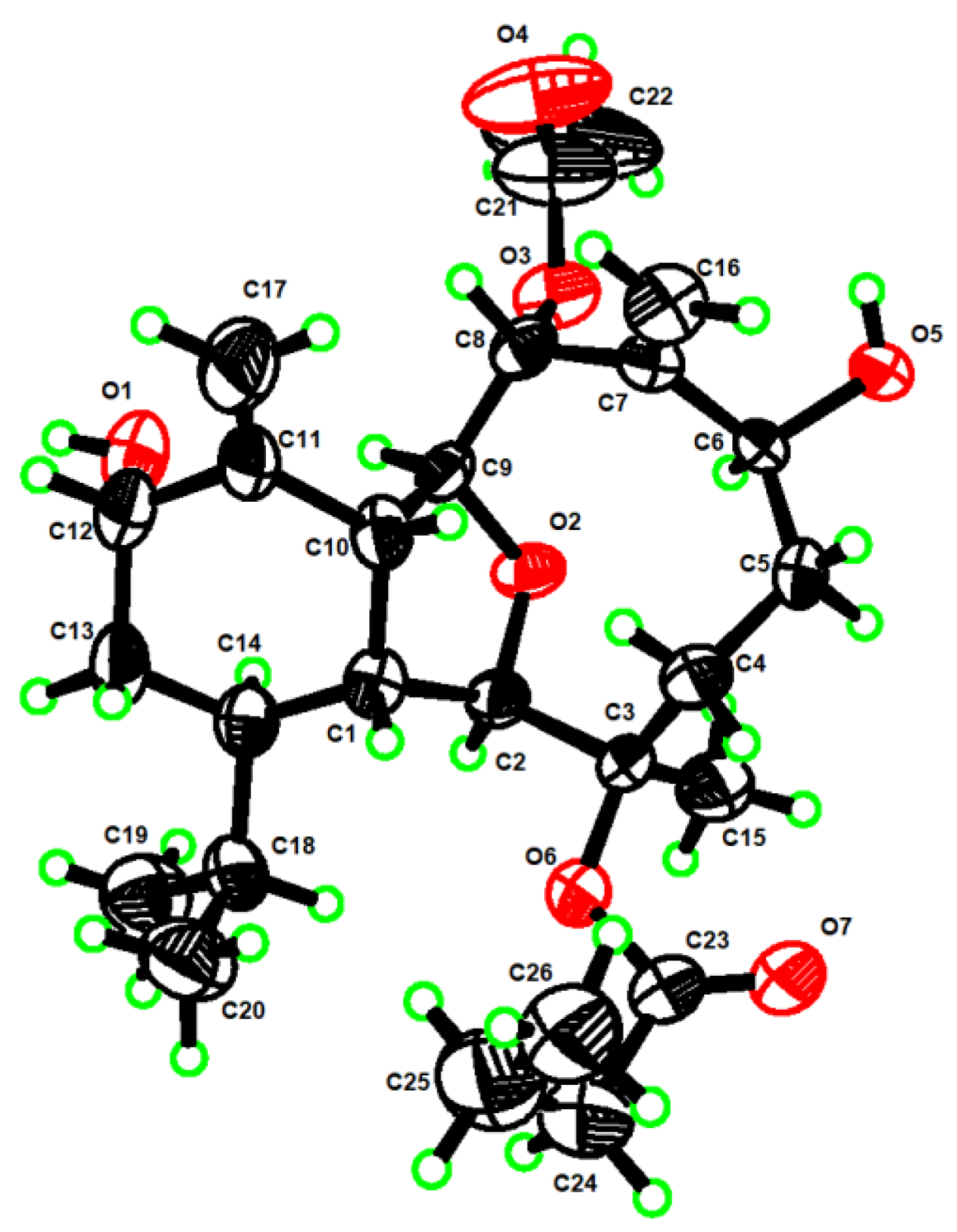
4. X-ray Crystal Structure Analysis of 1
5. Bioassay Procedures
5.1. EGFR Activity Assays
5.2. Anti-Tumor Assays
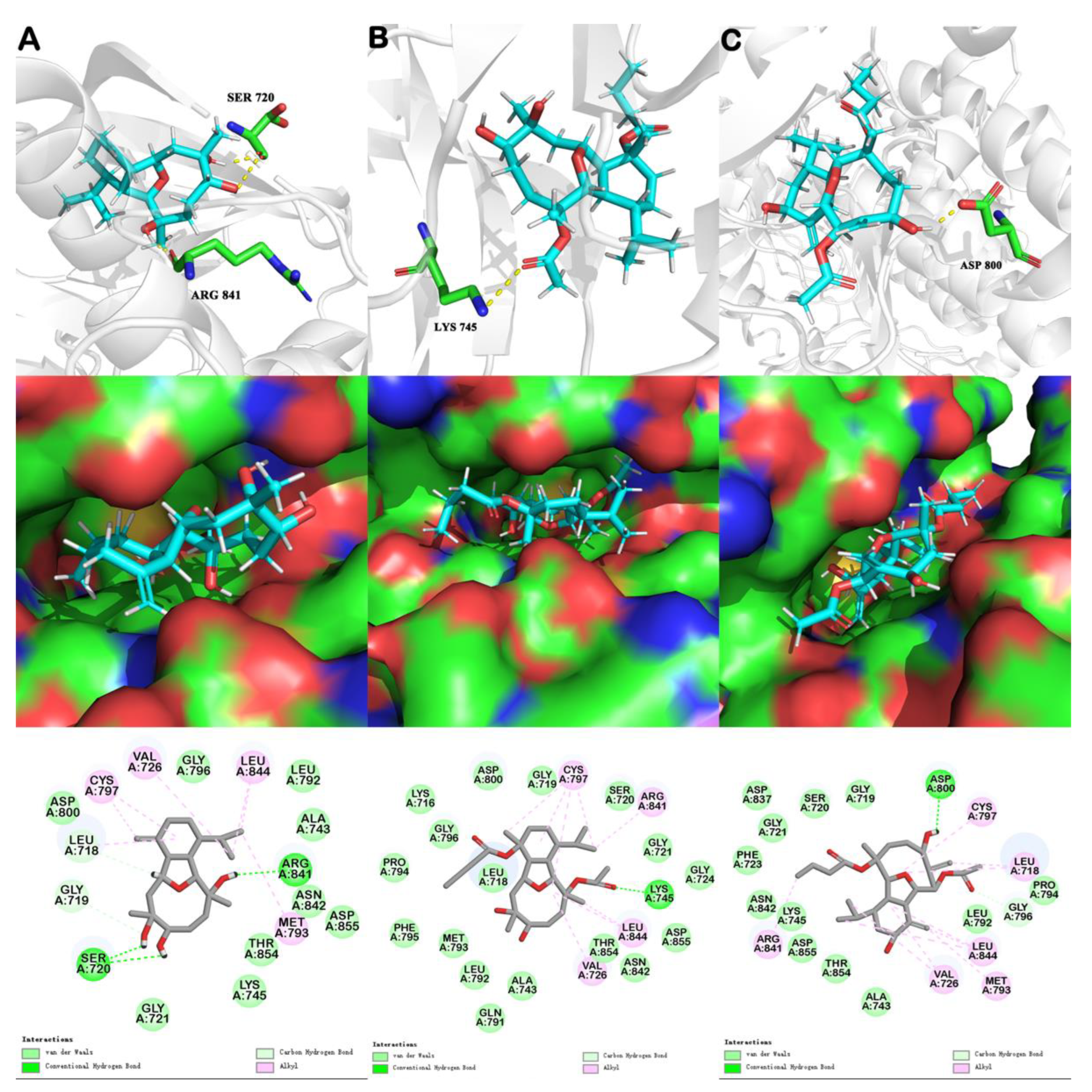
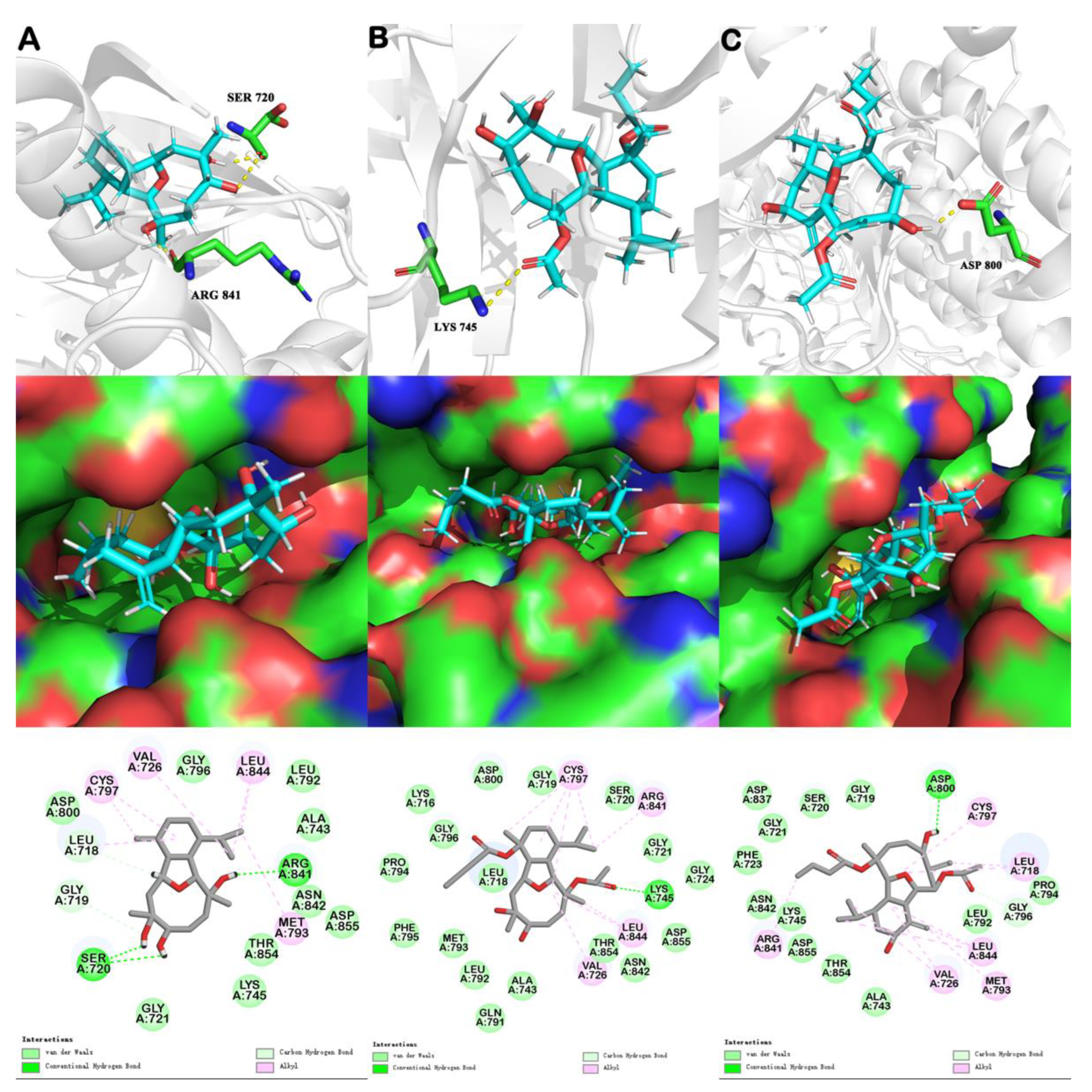
6. Molecular Docking
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chill, L.; Berrer, N.; Benayahu, Y.; Kashman, Y. Eunicellin Diterpenes from Two Kenyan Soft Corals. J. Nat. Prod. 2005, 68, 19–25. [Google Scholar] [CrossRef]
- Bernardelli, P.; Paquette, L.A. Survey of oxygenated 2,11-cyclized cembranoids of marine origin. Heterocycles 1998, 49, 531–556. [Google Scholar]
- Welford, A.J.; Collins, I. The 2,11-Cyclized Cembranoids: Cladiellins, Asbestinins, and Briarellins (Period 1998–2010). J. Nat. Prod. 2011, 74, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.S.; Yao, L.G.; Pascale, A.D.; Irace, C.; Mollo, E.; Taglialatela-Scafati, O.; Guo, Y.W. Polyoxygenated diterpenoids of the eunicellin-type from the Chinese soft coral Cladiella krempfi. Tetrahedron 2013, 69, 2214–2219. [Google Scholar] [CrossRef]
- Ru, T.; Cai, Y.S.; Li, H.; Tang, W.; Wang, H.; Guo, Y.W. Further new eunicellin-based diterpenoids from the Guangxi Weizhou soft coral Cladiella krempfi. Fitoterapia 2018, 131, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Sarma, N.S.; Chavakula, R.; Rao, I.N.; Kadirvelraj, R.; Row, T.N.G.; Saito, I. Crystal and molecular structure of sclerophytin F methyl ether from the soft coral Cladiella krempfi. J. Nat. Prod. 1993, 56, 1977–1980. [Google Scholar] [CrossRef]
- Ciavatta, M.L.; Manzo, E.; Mollo, E.; Mattia, C.A.; Tedesco, C.; Irace, C.; Guo, Y.W.; Li, X.B.; Cimino, G.; Gavagnin, M. Tritoniopsins A-D, Cladiellane-Based Diterpenes from the South China Sea Nudibranch Tritoniopsis elegans and Its Prey Cladiella krempfi. J. Nat. Prod. 2011, 74, 1902–1907. [Google Scholar] [CrossRef]
- Tai, C.J.; Chokkalingam, U.; Cheng, Y.; Shih, S.P.; Lu, M.C.; Su, J.H.; Hwang, T.L.; Sheu, J.H. Krempfielins Q and R, Two New Eunicellin-Based Diterpenoids from the Soft Coral Cladiella krempfi. Int. J. Mol. Sci. 2014, 15, 21865–21874. [Google Scholar] [CrossRef] [Green Version]
- Tai, C.J.; Su, J.H.; Huang, M.S.; Wen, Z.H.; Dai, C.F.; Sheu, J.H. Bioactive Eunicellin-Based Diterpenoids from the Soft Coral Cladiella krempfi. Mar. Drugs 2011, 9, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Raveendran, T.V.; Mol, V.L.; Parameswaran, P.S. Natural Product Antifoulants from the octocotals of Indian waters. Int. Biodeter. Biodegr. 2011, 65, 265–268. [Google Scholar] [CrossRef]
- Huang, T.Z.; Chen, B.W.; Huang, C.Y.; Hwang, T.L.; Dai, C.F.; Sheu, J.H. Eunicellin-Based Diterpenoids, Hirsutalins N-R, from the Formosan Soft Coral Cladiella hirsuta. Mar. Drugs 2014, 12, 2446–2457. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.; Sharma, P.; Zektzer, A.S.; Martin, G.E.; Ji, X.; Van der Helm, D.J. Sclerophytin C-F: Isolation and Structures of Four New Diterpenes from the Soft Coral Sclerophytum capitalis. J. Org. Chem. 1989, 54, 1896–1900. [Google Scholar] [CrossRef]
- Mohyeldin, M.M.; Akl, M.R.; Siddique, A.B.; Hassan, H.M.; Sayed, K.A.E. The marine-derived pachycladin diterpenoids as novel inhibitors of wild-type and mutant EGFR. Biochem. Pharmacol. 2017, 126, 51–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, G.Z.; Zhang, Y.; Fang, L.C.; Li, C.C.; Luo, T.P.; Yang, Z. Collective Synthesis of Cladiellins Based on the Gold-Catalyzed Cascade Reaction of 1,7-Diynes. Angew. Chem. Int. Ed. 2014, 53, 1837–1840. [Google Scholar] [CrossRef]
- Sun, L.L.; Li, X.W.; Guo, Y.W. Ximaoornatins A-C, Polyoxygenated Diterpenoids from the Hainan Soft Coral sinularia ornate. Mar. Drugs 2022, 22, 218. [Google Scholar] [CrossRef]
- Miyamoto, T.; Yamada, K.; Ikeda, N.; Komori, T.; Higuchi, R. Bioactive terpenoids from Octocorallia, I. Bioactive diterpenoids: Iitophynols A and B from the mucus of the soft coral Litophyton sp. J. Nat. Prod. 1994, 57, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.S.; Delion, L.; Farrugia, L. Synthesis of Four Diastereomers of Sclerophytin F and Structural Reassignment of Several Sclerophytin Natural Products. Chem. Eur. J. 2015, 21, 4772–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.J.; Zhao, P.; Yang, J.; Ma, R.; Yan, X.E.; Yang, S.Y.; Yang, J.W.; Yun, C.H. Structural insights into drug development strategy targeting EGFR T790M/C797S. Oncotarget 2018, 9, 13652–13665. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and Futuer Development of a Precise and Rapid Dye Method for Measuring Cell Growth/cell Kill. J. Immunol. Methods 1989, 199, 203–210. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Park, J.; Hassan, A.H.E.; Ra, H.; Pae, A.N.; Lee, J.; Park, B.G.; Moon, B.; Park, H.M.; Roh, E.J. Discovery of 1-(3-(benzyloxy) pyridine-2-yl)-3-(2-(piperazin-1-yl) ethyl) urea: A new modulator for amyloid beta-induced mitochondrial dysfunction. Eur. J. Med. Chem. 2017, 128, 56–69. [Google Scholar] [CrossRef]
- Wang, S.B.; Liu, H.; Wang, X.K.; Lei, K.; Li, G.Y.; Li, J.; Liu, R.M.; Quan, Z.S. Synthesis of 1,3,4-oxadiazole derivatives with anticonvulsant activity and their binding to the GABAA receptor. Eur. J. Med. Chem. 2020, 206, 112672. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Elnagar, A.Y.; Khanfar, M.A.; Sallam, A.A.; Mohammed, R.; Shaala, L.A.; Youssef, D.T.A.; Hifnawy, M.S.; Sayed, K.A.E. Design of semisynthetic analogues and 3D-QSAR study of eunicellin-based diterpenoids as prostate cancer migration and invasion inhibitors. Eur. J. Med. Chem. 2011, 46, 1122–1130. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (mult., J, Hz) a | δC (mult) b | δH (mult., J, Hz) a | δC (mult) b | |
| 1 | 2.23, m | 43.8, d | 2.24, m | 43.8, d |
| 2 | 3.84, s | 91.1, d | 3.86, s | 91.0, d |
| 3 | 84.7, s | 84.5, s | ||
| 4a | 1.79, m | 29.8, t | 1.88, m | 29.4, t |
| 4b | 2.23, m | 2.26, m | ||
| 5a | 1.74, m | 35.9, t | 1.50, m | 30.4, t |
| 5b | 2.20, m | 2.16, m | ||
| 6 | 4.71, d (8.1) | 68.4, d | 4.93, dd (3.6, 11.3) | 81.6, d |
| 7 | 149.4, s | 145.0, s | ||
| 8 | 5.28, s | 79.2, d | 5.25, s | 78.6, d |
| 9 | 4.68, d (10.5) | 83.7, d | 4.65, d (10.7) | 83.8, d |
| 10 | 2.94, dd (7.7, 10.9) | 46.6, d | 2.96, dd (7.7, 11.1) | 46.4, d |
| 11 | 146.1, s | 145.8, s | ||
| 12 | 4.46, s | 71.5, d | 4.47, s | 71.5, d |
| 13a | 1.27, m | 30.9, t | 1.29, m | 31.1, t |
| 13b | 1.94, m | 1.92, m | ||
| 14 | 1.87, m | 35.6, d | 1.89, m | 35.5, d |
| 15 | 1.60, s | 22.7, q | 1.61, s | 22.7, q |
| 16a | 5.37, s | 120.2, t | 5.51, s | 121.6, t |
| 16b | 5.64, s | 5.66, s | ||
| 17a | 4.89, brs | 116.4, t | 4.91, brs | 116.7, t |
| 17b | 5.15, brs | 5.15, brs | ||
| 18 | 1.92, m | 27.2, d | 1.89, m | 27.2, d |
| 19 | 0.98, d (7.0) | 21.8, q | 0.99, d (6.9) | 21.8, q |
| 20 | 0.76, d (6.8) | 15.5, q | 0.76, d (6.8) | 15.4, q |
| 1′ | 172.7, s | 172.7, s | ||
| 2′ | 2.13, m | 37.5, t | 2.14, m | 37.5, t |
| 3′ | 1.59, m | 18.6, t | 1.58, m | 18.6, t |
| 4′ | 0.93, t (7.4) | 13.8, q | 0.92, t (7.4) | 13.8, q |
| 1′′ | 170.8, s | 171.3, s | ||
| 2′′ | 2.11, s | 21.6, q | 2.06, s | 21.6, q |
| EGFR Crystal Structure | Compound ID | Number of Hydrogen Bonds | Binding Energy (kcal mol−1) | Van der Waals Energy (kcal mol−1) | -CDOCKER ENERGY (kcal mol−1) | -CDOCKER INTERACTION ENERGY (kcal mol−1) |
|---|---|---|---|---|---|---|
| 5X2A | sclerophytin A | 3 | –1.74 | –10.52 | –31.78 | 43.13 |
| pachycladin A | 1 | –12.68 | –15.40 | –10.59 | 49.68 | |
| litophynol C (1) | 1 | 8.57 | –17.40 | –26.10 | 51.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Yao, L.-G.; Guo, Y.-W.; Li, X.-W. New Cladiellin-Type Diterpenoids from the South China Sea Soft Coral Cladiella krempfi: Structures and Molecular Docking Analysis in EGFRs. Mar. Drugs 2022, 20, 381. https://doi.org/10.3390/md20060381
Jin Y, Yao L-G, Guo Y-W, Li X-W. New Cladiellin-Type Diterpenoids from the South China Sea Soft Coral Cladiella krempfi: Structures and Molecular Docking Analysis in EGFRs. Marine Drugs. 2022; 20(6):381. https://doi.org/10.3390/md20060381
Chicago/Turabian StyleJin, Yang, Li-Gong Yao, Yue-Wei Guo, and Xu-Wen Li. 2022. "New Cladiellin-Type Diterpenoids from the South China Sea Soft Coral Cladiella krempfi: Structures and Molecular Docking Analysis in EGFRs" Marine Drugs 20, no. 6: 381. https://doi.org/10.3390/md20060381
APA StyleJin, Y., Yao, L.-G., Guo, Y.-W., & Li, X.-W. (2022). New Cladiellin-Type Diterpenoids from the South China Sea Soft Coral Cladiella krempfi: Structures and Molecular Docking Analysis in EGFRs. Marine Drugs, 20(6), 381. https://doi.org/10.3390/md20060381