
The Endo-α(1,3)-Fucoidanase Mef2 Releases Uniquely Branched Oligosaccharides from Saccharina latissima Fucoidans

 , , ,
, , ,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Sequence Analysis of the Mef2 Fucoidanase from M. eckloniae
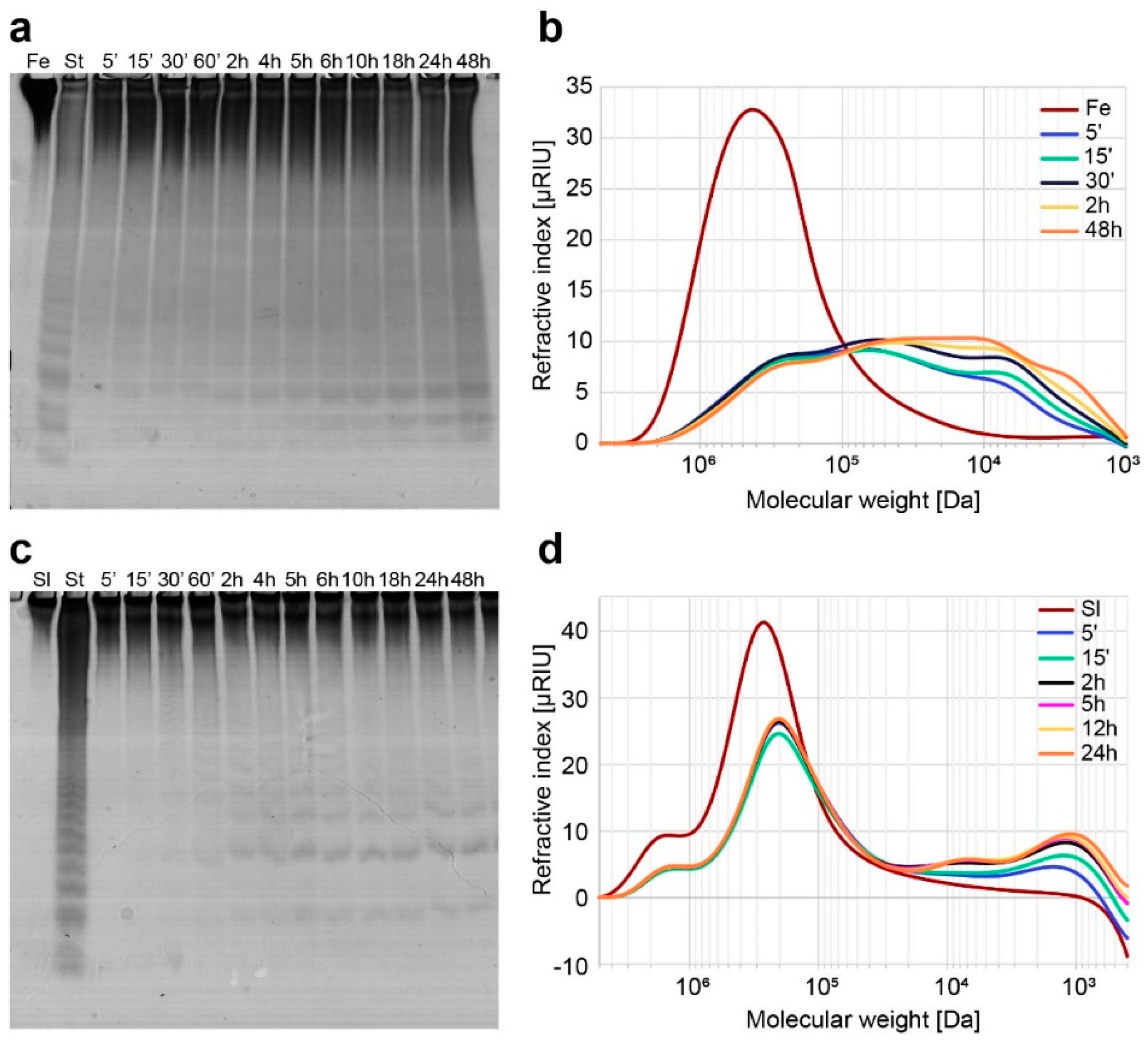
2.2. Functional Characterization of the Recombinant Mef2 Fucoidanase
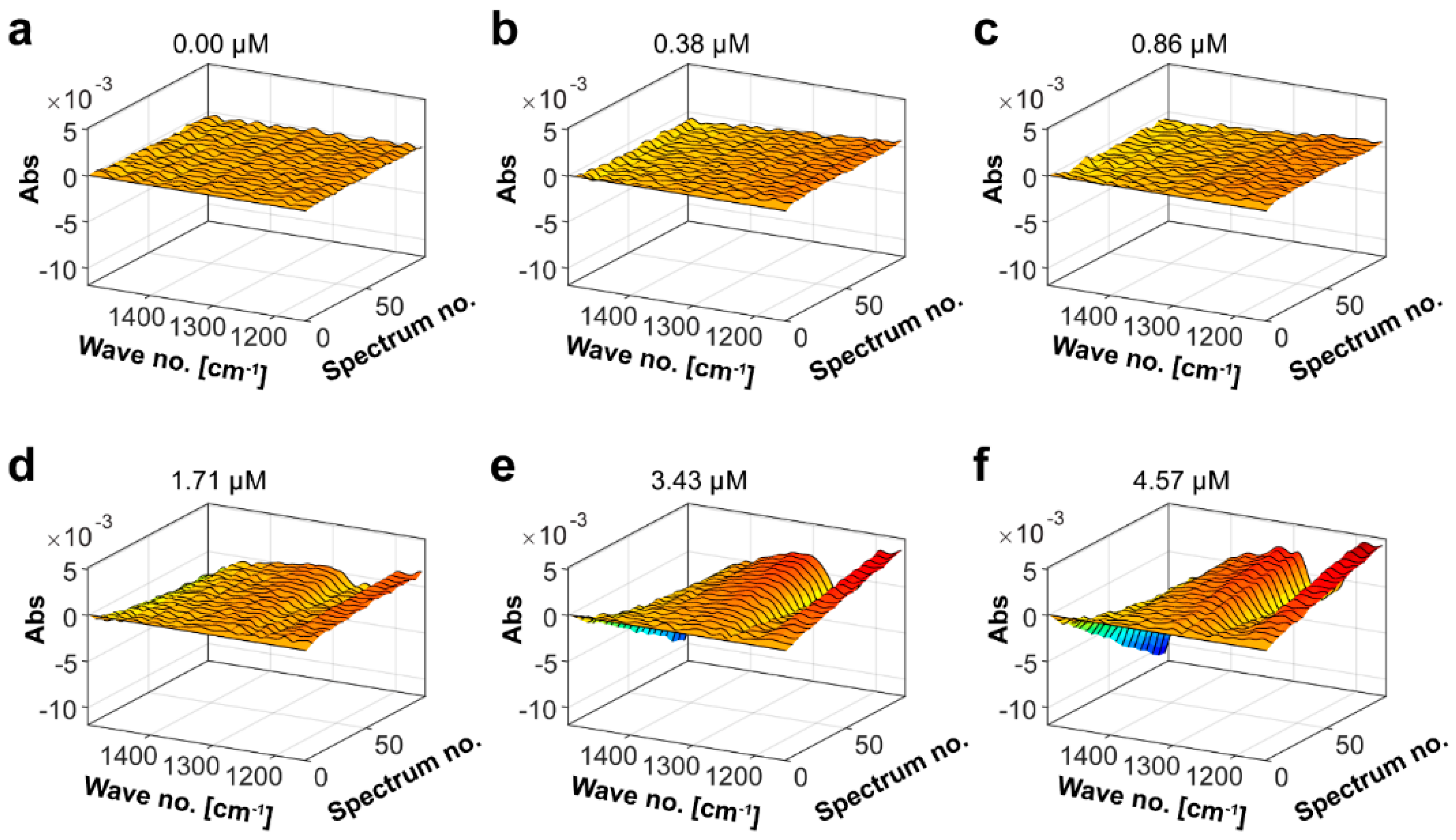
2.3. Determination of the Mef2 Fucoidanase Unit by Fourier Transform Infrared Spectroscopy (FTIR)
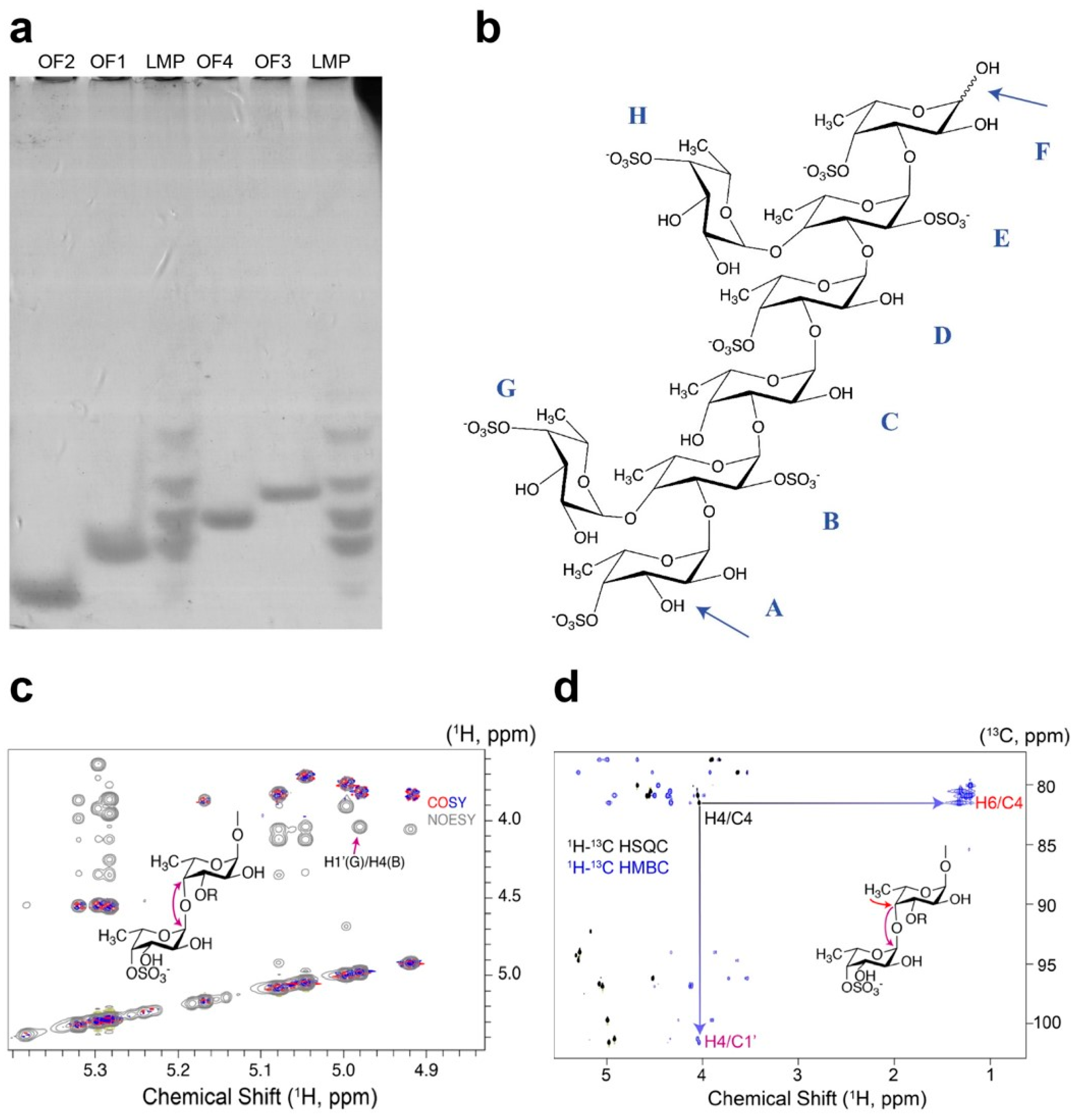
2.4. Determination of the Mef2 Linkage Specificity and Structural Elucidation of the S. latissima Fucoidan Products by NMR
3. Discussion
4. Materials and Methods
4.1. Fucoidan Substrates
4.2. Chemical Analysis of Fucoidans
4.3. Sequence Analysis of the Mef2 Gene
4.4. Construction and Cloning of the Expression Vectors
4.5. Recombinant Enzyme Expression and Purification
4.6. Endo-Fucoidanase Activity Assays
4.7. Thermal Stability of Recombinant Fucoidanase Mef2
4.8. Mef2 Kinetics by Fourier Transform InfraRed (FTIR) Spectroscopy Measurement and Parallel Factor (PARAFAC) Analysis
4.9. Enzymatic Hydrolysis of Fucoidans and Product Separation
4.10. NMR Spectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ale, M.T.; Mikkelsen, J.D.; Meyer, A.S. Important Determinants for Fucoidan Bioactivity: A Critical Review of Structure-Function Relations and Extraction Methods for Fucose-Containing Sulfated Polysaccharides from Brown Seaweeds. Mar. Drugs 2011, 9, 2106–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zvyagintseva, T.N.; Usoltseva, R.V.; Shevchenko, N.M.; Surits, V.V.; Imbs, T.I.; Malyarenko, O.S.; Besednova, N.N.; Ivanushko, L.A.; Ermakova, S.P. Structural Diversity of Fucoidans and Their Radioprotective Effect. Carbohydr. Polym. 2021, 273, 118551. [Google Scholar] [CrossRef] [PubMed]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Kelly, M.; Sanderson, C.J.; Nifantiev, N.E.; Usov, A.I. Further Studies on the Composition and Structure of a Fucoidan Preparation from the Brown Alga Saccharina latissima. Carbohydr. Res. 2010, 345, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Imbs, T.I.; Shevchenko, N.M.; Semenova, T.L.; Sukhoverkhov, S.V.; Zvyagintseva, T.N. Compositional Heterogeneity of Sulfated Polysaccharides Synthesized by the Brown Alga Costaria costata. Chem. Nat. Compd. 2011, 47, 96–97. [Google Scholar] [CrossRef]
- Skriptsova, A.V.; Shevchenko, N.M.; Tarbeeva, D.V.; Zvyagintseva, T.N. Comparative Study of Polysaccharides from Reproductive and Sterile Tissues of Five Brown Seaweeds. Mar. Biotechnol. 2012, 14, 304–311. [Google Scholar] [CrossRef]
- Bak, U.G.; Gregersen, Ó.; Infante, J. Technical Challenges for Offshore Cultivation of Kelp Species: Lessons Learned and Future Directions. Bot. Mar. 2020, 63, 341–353. [Google Scholar] [CrossRef]
- Kwak, J.-Y. Fucoidan as a Marine Anticancer Agent in Preclinical Development. Mar. Drugs 2014, 12, 851–870. [Google Scholar] [CrossRef]
- Fitton, J.H. Therapies from Fucoidan; Multifunctional Marine Polymers. Mar. Drugs 2011, 9, 1731–1760. [Google Scholar] [CrossRef]
- Jin, W.; Zhang, Q.; Wang, J.; Zhang, W. A Comparative Study of the Anticoagulant Activities of Eleven Fucoidans. Carbohydr. Polym. 2013, 91, 1–6. [Google Scholar] [CrossRef]
- Ustyuzhanina, N.; Ushakova, N.; Zyuzina, K.; Bilan, M.; Elizarova, A.; Somonova, O.; Madzhuga, A.; Krylov, V.; Preobrazhenskaya, M.; Usov, A.; et al. Influence of Fucoidans on Hemostatic System. Mar. Drugs 2013, 11, 2444–2458. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Oda, T.; Yu, Q.; Jin, J.-O. Fucoidan from Macrocystis pyrifera Has Powerful Immune-Modulatory Effects Compared to Three Other Fucoidans. Mar. Drugs 2015, 13, 1084–1104. [Google Scholar] [CrossRef] [Green Version]
- Dörschmann, P.; Mikkelsen, M.D.; Thi, T.N.; Roider, J.; Meyer, A.S.; Klettner, A. Effects of a Newly Developed Enzyme-Assisted Extraction Method on the Biological Activities of Fucoidans in Ocular Cells. Mar. Drugs 2020, 18, 282. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Mikkelsen, M.D.; Ptak, S.H.; Hejbøl, E.K.; Ohmes, J.; Thi, T.N.; Nguyen Ha, V.T.; Fretté, X.; Fuchs, S.; Meyer, A.; et al. Efficacy of Marine Bioactive Compound Fucoidan for Bone Regeneration and Implant Fixation in Sheep. J. Biomed. Mater. Res. Part A 2022, 110, 861–872. [Google Scholar] [CrossRef]
- Ohmes, J.; Mikkelsen, M.D.; Nguyen, T.T.; Tran, V.H.N.; Meier, S.; Nielsen, M.S.; Ding, M.; Seekamp, A.; Meyer, A.S.; Fuchs, S. Depolymerization of Fucoidan with Endo-Fucoidanase Changes Bioactivity in Processes Relevant for Bone Regeneration. Carbohydr. Polym. 2022, 286, 119286. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, Y.; Shen, J.; Xue, C. Expression and Characterization of a Novel β-Porphyranase from Marine Bacterium Wenyingzhuangia fucanilytica: A Biotechnological Tool for Degrading Porphyran. J. Agric. Food Chem. 2019, 67, 9307–9313. [Google Scholar] [CrossRef]
- Bakunina, I.Y.; Shevchenko, L.S.; Nedashkovskaya, O.I.; Shevchenko, N.M.; Alekseeva, S.A.; Mikhailov, V.V.; Zvyagintseva, T.N. Screening of Marine Bacteria for Fucoidanases. Microbiology 2000, 69, 370–376. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The Carbohydrate-Active Enzymes Database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Colin, S.; Deniaud, E.; Jam, M.; Descamps, V.; Chevolot, Y.; Kervarec, N.; Yvin, J.C.; Barbeyron, T.; Michel, G.; Kloareg, B. Cloning and Biochemical Characterization of the Fucanase FcnA: Definition of a Novel Glycoside Hydrolase Family Specific for Sulfated Fucans. Glycobiology 2006, 16, 1021–1032. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Rasin, A.B.; Kusaykin, M.I.; Kalinovsky, A.I.; Miansong, Z.; Changheng, L.; Malyarenko, O.; Zueva, A.O.; Zvyagintseva, T.N.; Ermakova, S.P. Structure, Enzymatic Transformation, Anticancer Activity of Fucoidan and Sulphated Fucooligosaccharides from Sargassum horneri. Carbohydr. Polym. 2017, 175, 654–660. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Ustyuzhanina, N.E.; Kusaykin, M.I.; Krylov, V.B.; Shashkov, A.S.; Dmitrenok, A.S.; Usoltseva, R.V.; Zueva, A.O.; Nifantiev, N.E.; Zvyagintseva, T.N. Expression and Biochemical Characterization and Substrate Specificity of the Fucoidanase from Formosa algae. Glycobiology 2017, 27, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Silchenko, A.S.; Kusaykin, M.I.; Kurilenko, V.V.; Zakharenko, A.M.; Isakov, V.V.; Zaporozhets, T.S.; Gazha, A.K.; Zvyagintseva, T.N. Hydrolysis of Fucoidan by Fucoidanase Isolated from the Marine Bacterium Formosa algae. Mar. Drugs 2013, 11, 2413–2430. [Google Scholar] [CrossRef] [Green Version]
- Vuillemin, M.; Silchenko, A.S.; Cao, H.T.T.; Kokoulin, M.S.; Trang, V.T.D.; Holck, J.; Ermakova, S.P.; Meyer, A.S.; Mikkelsen, M.D. Functional Characterization of a New GH107 Endo-α-(1,4)-Fucoidanase from the Marine Bacterium Formosa haliotis. Mar. Drugs 2020, 18, 562. [Google Scholar] [CrossRef]
- Trang, V.T.D.; Mikkelsen, M.D.; Vuillemin, M.; Meier, S.; Cao, H.T.T.; Muschiol, J.; Perna, V.; Nguyen, T.T.; Tran, V.H.N.; Holck, J.; et al. The Endo-α(1,4) Specific Fucoidanase Fhf2 From Formosa haliotis Releases Highly Sulfated Fucoidan Oligosaccharides. Front. Plant Sci. 2022, 13, 823668. [Google Scholar] [CrossRef]
- Zueva, A.O.; Silchenko, A.S.; Rasin, A.B.; Kusaykin, M.I.; Usoltseva, R.V.; Kalinovsky, A.I.; Kurilenko, V.V.; Zvyagintseva, T.N.; Thinh, P.D.; Ermakova, S.P. Expression and Biochemical Characterization of Two Recombinant Fucoidanases from the Marine Bacterium Wenyingzhuangia fucanilytica CZ1127T. Int. J. Biol. Macromol. 2020, 164, 3025–3037. [Google Scholar] [CrossRef]
- Takayama, M.; Koyama, N.; Sakai, T.; Kato, I. Enzymes Capable of Degrading a Sulfated-Fucose-Containing Polysaccharide and Their Encoding Genes. U.S. Patent US6489155B1, 3 December 2002. [Google Scholar]
- Bae, S.S.; Kwon, K.K.; Yang, S.H.; Lee, H.S.; Kim, S.J.; Lee, J.H. Flagellimonas Eckloniae Gen. Nov., Sp. Nov., a Mesophilic Marine Bacterium of the Family Flavobacteriaceae, Isolated from the Rhizosphere of Ecklonia kurome. Int. J. Syst. Evol. Microbiol. 2007, 57, 1050–1054. [Google Scholar] [CrossRef]
- Nielsen, H.; Tsirigos, K.D.; Brunak, S.; von Heijne, G. A Brief History of Protein Sorting Prediction. Protein J. 2019, 38, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Veith, P.D.; Glew, M.D.; Gorasia, D.G.; Reynolds, E.C. Type IX Secretion: The Generation of Bacterial Cell Surface Coatings Involved in Virulence, Gliding Motility and the Degradation of Complex Biopolymers. Mol. Microbiol. 2017, 106, 35–53. [Google Scholar] [CrossRef]
- Cao, H.T.T.; Mikkelsen, M.D.; Lezyk, M.J.; Bui, L.M.; Tran, V.T.T.; Silchenko, A.S.; Kusaykin, M.I.; Pham, T.D.; Truong, B.H.; Holck, J.; et al. Novel Enzyme Actions for Sulphated Galactofucan Depolymerisation and a New Engineering Strategy for Molecular Stabilisation of Fucoidan Degrading Enzymes. Mar. Drugs 2018, 16, 422. [Google Scholar] [CrossRef] [Green Version]
- Vickers, C.; Liu, F.; Abe, K.; Salama-Alber, O.; Jenkins, M.; Springate, C.M.K.; Burke, J.E.; Withers, S.G.; Boraston, A.B. Endo-Fucoidan Hydrolases from Glycoside Hydrolase Family 107 (GH107) Display Structural and Mechanistic Similarities to -L-Fucosidases from GH29. J. Biol. Chem. 2018, 285, 4281–4295. [Google Scholar] [CrossRef] [Green Version]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Thuy, T.T.T.; Thanh Van, T.T.; Ly, B.M.; Nifantiev, N.E.; Usov, A.I. Preliminary Investigation of a Highly Sulfated Galactofucan Fraction Isolated from the Brown Alga Sargassum polycystum. Carbohydr. Res. 2013, 377, 48–57. [Google Scholar] [CrossRef]
- Tran, V.H.N.; Perna, V.; Mikkelsen, M.D.; Thi Nguyen, T.; Thi Dieu Trang, V.; Baum, A.; Thi Thuy Cao, H.; Thi Thanh Van, T.; Meyer, A.S. A New FTIR Assay for Quantitative Measurement of Endo-Fucoidanase Activity. Enzyme Microb. Technol. 2022, 110035. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Mikkelsen, M.D.; Nguyen Tran, V.H.; Dieu Trang, V.T.; Rhein-Knudsen, N.; Holck, J.; Rasin, A.B.; Thuy Cao, H.T.; Thanh Van, T.T.; Meyer, A.S. Enzyme-Assisted Fucoidan Extraction from Brown Macroalgae Fucus distichus Subsp. evanescens and Saccharina latissima. Mar. Drugs 2020, 18, 296. [Google Scholar] [CrossRef] [PubMed]
- Bilan, M.I.; Kusaykin, M.I.; Grachev, A.A.; Tsvetkova, E.A.; Zvyagintseva, T.N.; Nifantiev, N.E.; Usov, A.I. Effect of Enzyme Preparation from the Marine Mollusk. Biochemistry 2005, 70, 1321–1326. [Google Scholar] [CrossRef]
- Sakai, T.; Kawai, T.; Kato, I. Isolation and Characterization of a Fucoidan-Degrading Marine Bacterial Strain and Its Fucoidanase. Mar. Biotechnol. 2004, 6, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Bilan, M.I.; Grachev, A.A.; Ustuzhanina, N.E.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a Fucoidan from the Brown Seaweed Fucus evanescens C. Ag. Carbohydr. Res. 2002, 337, 719–730. [Google Scholar] [CrossRef]
- Chizhov, A.O.; Dell, A.; Morris, H.R.; Haslam, S.M.; McDowell, R.A.; Shashkov, A.S.; Nifant’ev, N.E.; Khatuntseva, E.A.; Usov, A.I. A Study of Fucoidan from the Brown Seaweed Chorda filum. Carbohydr. Res. 1999, 320, 108–119. [Google Scholar] [CrossRef]
- Holtkamp, A.D. Isolation, Characterisation, Modification and Application of Fucoidan from Fucus vesiculosus. Analysis 2009, 179. [Google Scholar] [CrossRef]
- Hemmingson, J.A.; Falshaw, R.; Furneaux, R.H.; Thompson, K. Structure and Antiviral Activity of the Galactofucan Sulfates Extracted from Undaria pinnatifida (Phaeophyta). J. Appl. Phycol. 2006, 18, 185. [Google Scholar] [CrossRef]
- Silchenko, A.S.; Kusaykin, M.I.; Zakharenko, A.M.; Menshova, R.V.; Khanh, H.H.N.; Dmitrenok, P.S.; Isakov, V.V.; Zvyagintseva, T.N. Endo-1,4-Fucoidanase from Vietnamese Marine Mollusk Lambis sp. Which Producing Sulphated Fucooligosaccharides. J. Mol. Catal. B Enzym. 2014, 102, 154–160. [Google Scholar] [CrossRef]
- Manns, D.; Nyffenegger, C.; Saake, B.; Meyer, A.S. Impact of Different Alginate Lyases on Combined Cellulase–Lyase Saccharification of Brown Seaweed. RSC Adv. 2016, 6, 45392–45401. [Google Scholar] [CrossRef] [Green Version]
- Manns, D.; Deutschle, A.L.; Saake, B.; Meyer, A.S. Methodology for Quantitative Determination of the Carbohydrate Composition of Brown Seaweeds (Laminariaceae). RSC Adv. 2014, 4, 25736–25746. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.G.; McCandless, E.L. Simple, Rapid, Turbidometric Determination of Inorganic Sulfate and/or Protein. Anal. Biochem. 1978, 90, 802–808. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Dubois, M.; Gilles, K.A.; Hamilton, J.K.; Rebers, P.A.; Smith, F. Colorimetric Method for Determination of Sugars and Related Substances. Anal. Chem. 1956, 28, 350–356. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
 |
| Residue | Chemical Shifts (ppm) | ||||||
|---|---|---|---|---|---|---|---|
| H1/C1 | H2/C2 | H3/C3 | H4/C4 | H5/C5 | H6/C6 | ||
| I | α-L-Fucp(3Ac, 4SO3−)-(1→ | 5.282 | 3.785 | 4.099 | 4.589 | 4.510 | 1.258 |
| 100.3 | 69.0 | 68.6 | 81.1 | 66.5 | 15.8 | ||
| J | -3)-β-D-Galp(2Ac, 4SO3−)-(1→ | 4.773 | 3.731 | 3.908 | 4.692 | 3.798 | 3.802 |
| 102.8 | 71.6 | 76.5 | 77.3 | 74.4 | 61.0 | ||
| K | -4,6)-β-D-Galp(3SO3−)-(1→ | 4.580 | 3.815 | 4.426 | 4.540 | 3.930 | 4.188/3.924 |
| 103.1 | 68.9 | 79.9 | 73.8 | 73.2 | 70.3 | ||
| Residue\Atom | Chemical Shifts (ppm) | ||||||
|---|---|---|---|---|---|---|---|
| H1/C1 | H2/C2 | H3/C3 | H4/C4 | H5/C5 | H6/C6 | ||
| A | α-L-Fucp(4SO3−)-(1- | 5.036 | 3.71 | 3.982 | 4.561 | 4.458 | 1.211 |
| 96.87 | 68.5 | 69.06 | 80.87 | 66.6 | 15.84 | ||
| B | →3,4)-α-L-Fucp(2SO3−)-(1- | 5.283 | 4.556 | 4.122 | 4.037 | 4.331 | 1.325 |
| 94.05 | 73.2 | 73.2 | 81.46 | 67.96 | 15.47 | ||
| C | →3)-α-D-Fucp-(1- | 4.995 | 3.764 | 3.858 | 3.954 | 4.25 | 1.181 |
| 99.81 | 66.96 | 75.67 | 68.62 | 66.7 | 15.59 | ||
| D | →3)-α- D-Fucp(4SO3−)-(1- | 5.076 | 3.833 | 3.905 | 4.68 | 4.475 | 1.211 |
| 96.74 | 67.53 | 77.9 | 80.08 | 66.54 | 15.84 | ||
| Eα | →3,4)-α- D-Fucp(2SO3−)-(1- | 5.32 | 4.552 | 4.12 | 4.05 | 4.34 | 1.29 |
| 94.28 | 73.2 | 73.2 | 80.85 | 67.95 | 15.65 | ||
| Eβ | →3,4)-β- D-Fucp(2SO3−)-(1- | 5.297 | 4.545 | 4.114 | 4.054 | 4.35 | 1.29 |
| 94.69 | 73.2 | 72.8 | 80.85 | 67.95 | 15.65 | ||
| Fα | →3)-α- D-Fucp | 5.161 | 3.872 | 3.871 | 3.986 | 4.118 | 1.165 |
| 92.25 | 66.56 | 75.36 | 68.41 | 66.07 | 15.61 | ||
| Fβ | →3)-β- D-Fucp | 4.517 | 3.539 | 3.633 | 3.92 | 3.72 | 1.203 |
| 96.25 | 70.12 | 78.93 | 68.1 | 70.67 | 15.583 | ||
| G | α- D-Fucp(4SO3−)-(1→ | 4.974 | 3.824 | 4.103 | 4.584 | 4.37 | 1.254 |
| 101.6 | 69.05 | 68.03 | 80.95 | 67.172 | 16.14 | ||
| H | α- D-Fucp(4SO3−)-(1→ | 4.918 | 3.831 | 4.074 | 4.546 | 4.338 | 1.307 |
| 101.34 | 69.38 | 68.37 | 80.55 | 67.33 | 16.38 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, V.H.N.; Nguyen, T.T.; Meier, S.; Holck, J.; Cao, H.T.T.; Van, T.T.T.; Meyer, A.S.; Mikkelsen, M.D. The Endo-α(1,3)-Fucoidanase Mef2 Releases Uniquely Branched Oligosaccharides from Saccharina latissima Fucoidans. Mar. Drugs 2022, 20, 305. https://doi.org/10.3390/md20050305
Tran VHN, Nguyen TT, Meier S, Holck J, Cao HTT, Van TTT, Meyer AS, Mikkelsen MD. The Endo-α(1,3)-Fucoidanase Mef2 Releases Uniquely Branched Oligosaccharides from Saccharina latissima Fucoidans. Marine Drugs. 2022; 20(5):305. https://doi.org/10.3390/md20050305
Chicago/Turabian StyleTran, Vy Ha Nguyen, Thuan Thi Nguyen, Sebastian Meier, Jesper Holck, Hang Thi Thuy Cao, Tran Thi Thanh Van, Anne S. Meyer, and Maria Dalgaard Mikkelsen. 2022. "The Endo-α(1,3)-Fucoidanase Mef2 Releases Uniquely Branched Oligosaccharides from Saccharina latissima Fucoidans" Marine Drugs 20, no. 5: 305. https://doi.org/10.3390/md20050305
APA StyleTran, V. H. N., Nguyen, T. T., Meier, S., Holck, J., Cao, H. T. T., Van, T. T. T., Meyer, A. S., & Mikkelsen, M. D. (2022). The Endo-α(1,3)-Fucoidanase Mef2 Releases Uniquely Branched Oligosaccharides from Saccharina latissima Fucoidans. Marine Drugs, 20(5), 305. https://doi.org/10.3390/md20050305