
Transcriptomic Insights into the Diversity and Evolution of Myxozoa (Cnidaria, Endocnidozoa) Toxin-like Proteins


Abstract
:1. Introduction
2. Results
2.1. Sequencing and De Novo Assembly of Transcriptome
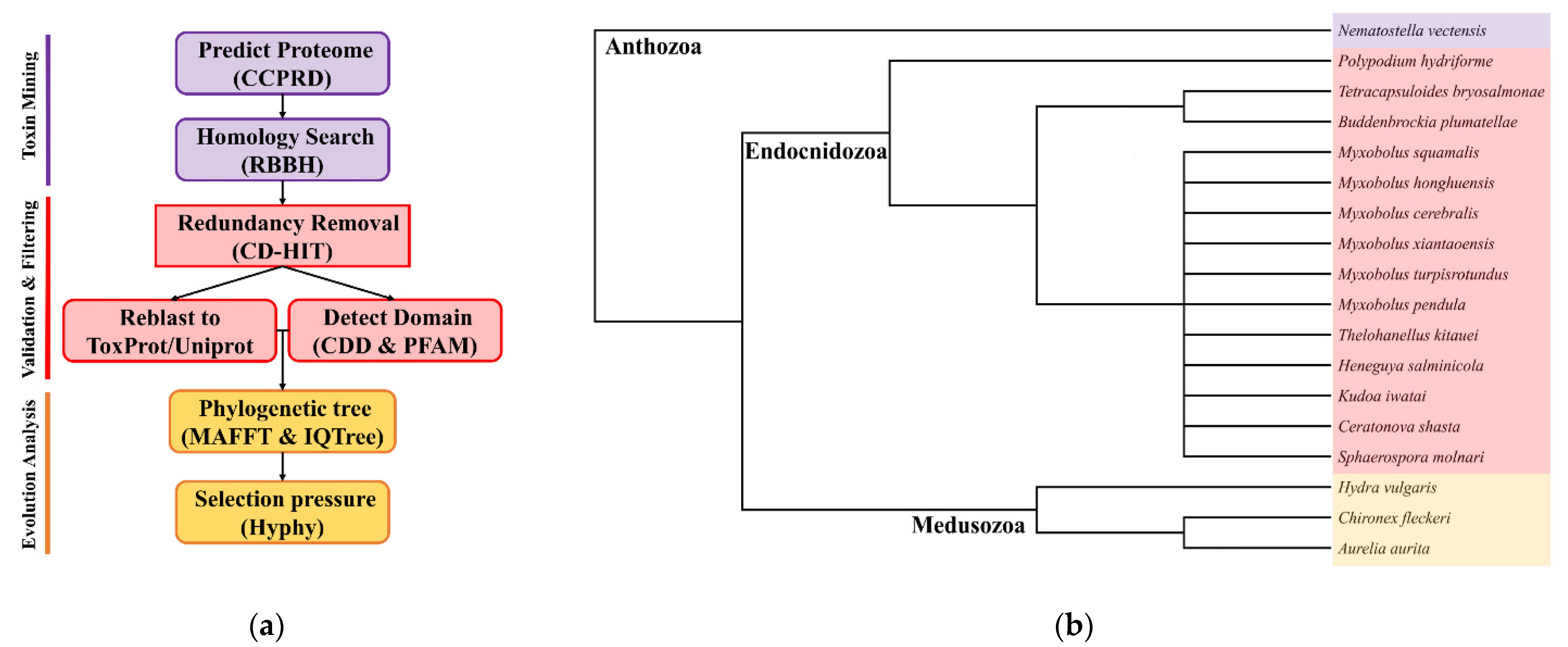
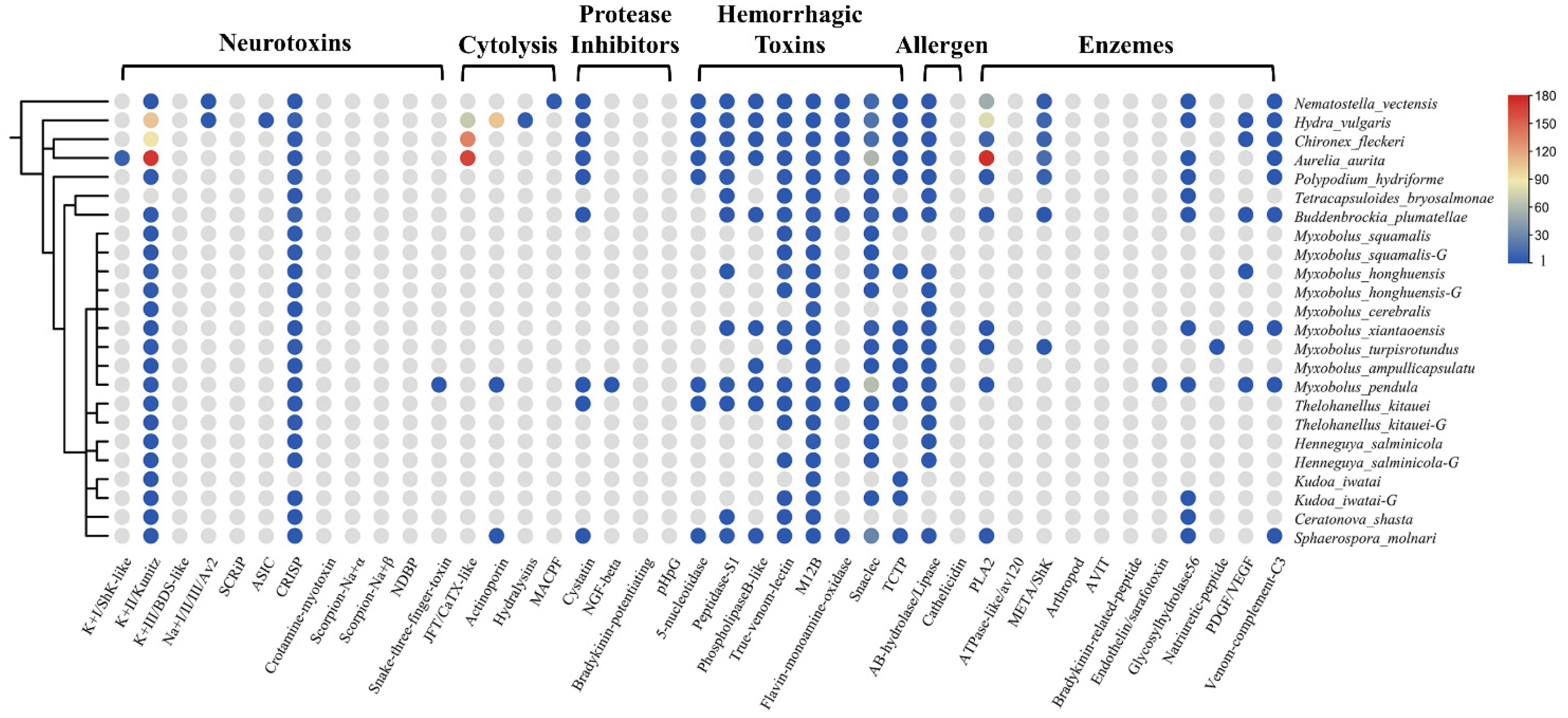
2.2. Identification of Toxin-like Proteins (TLPs) in Transcriptomes and Genomes
2.3. Phylogenetic Analysis of Toxin Families
2.3.1. Neurotoxins
2.3.2. Enzymes
2.3.3. Cytolysins
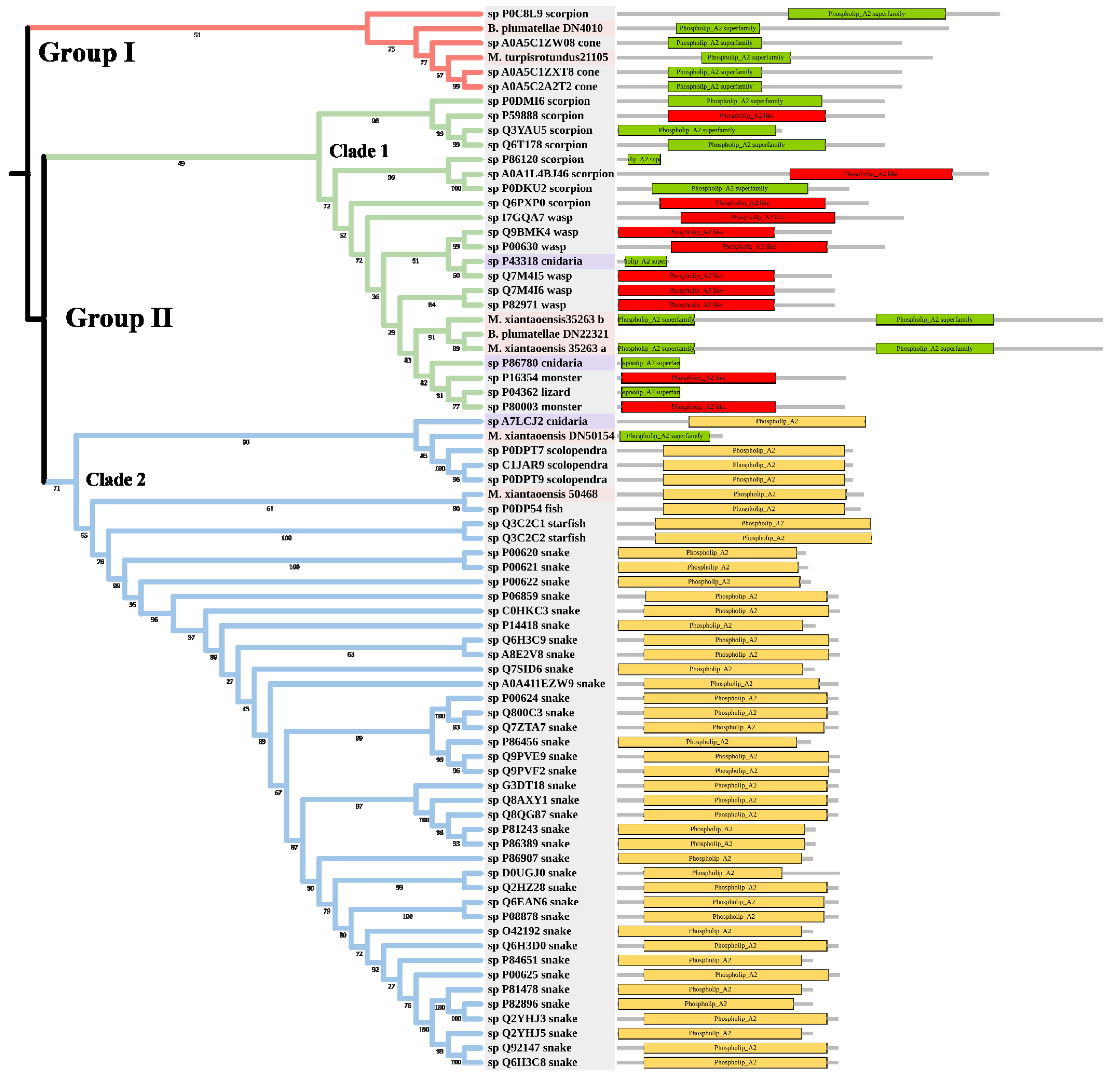
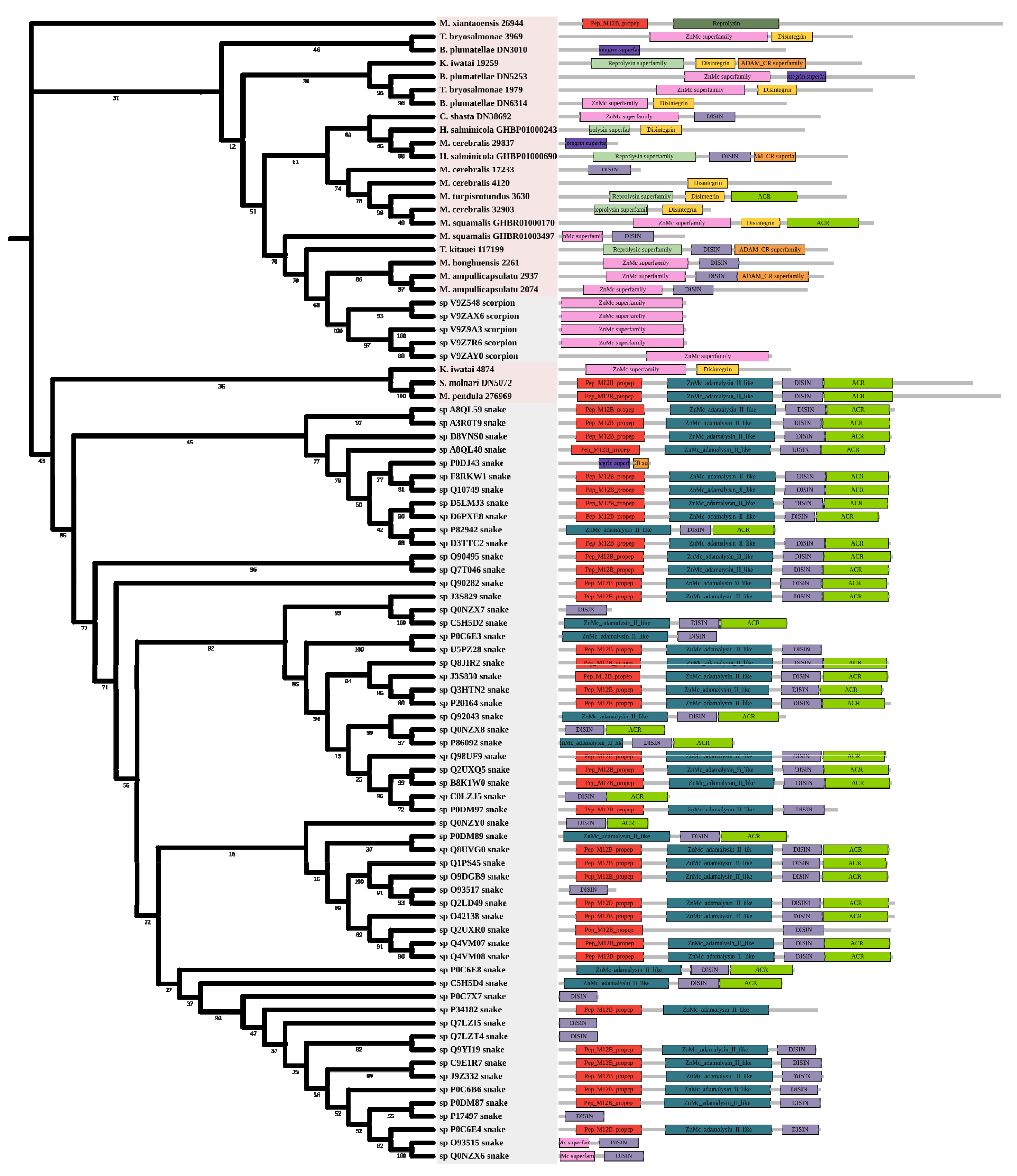
2.3.4. Hemorrhagic Toxins
2.4. Selection Pressure Analysis
3. Discussion
4. Materials and Methods
4.1. Collecting Sample
4.2. Next-Generation Sequencing and Assembly
4.3. Data Decontamination and Construction Predict Proteomes
4.4. Identification Toxin in Predict Proteomes
4.5. Alignment and Phylogenetic Analysis
4.6. Selection Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rachamim, T.; Morgenstern, D.; Aharonovich, D.; Brekhman, V.; Lotan, T.; Sher, D. The dynamically evolving nematocyst content of an anthozoan, a scyphozoan, and a hydrozoan. Mol. Biol. Evol. 2015, 32, 740–753. [Google Scholar] [CrossRef]
- Jouiaei, M.; Yanagihara, A.A.; Madio, B.; Nevalainen, T.J.; Alewood, P.F.; Fry, B.G. Ancient venom systems: A review on cnidaria toxins. Toxins 2015, 7, 2251–2271. [Google Scholar] [CrossRef] [PubMed]
- Fautin, D.G. Structural diversity, systematics, and evolution of cnidae. Toxicon 2009, 54, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Klompen, A.M.; Macrander, J.; Reitzel, A.M.; Stampar, S.N. Transcriptomic Analysis of Four Cerianthid (Cnidaria, Ceriantharia) Venoms. Mar. Drugs 2020, 18, 413. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, E.; Wilson, D.; Seymour, J. The influence of ecological factors on cnidarian venoms. Toxicon 2021, 9, 100067. [Google Scholar] [CrossRef]
- Hartigan, A.; Jaimes-Becerra, A.; Okamura, B.; Doonan, L.B.; Ward, M.; Marques, A.C.; Long, P.F. Recruitment of toxin-like proteins with ancestral venom function supports endoparasitic lifestyles of Myxozoa. PeerJ 2021, 9, e11208. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.S.; Neuhof, M.; Rubinstein, N.D.; Diamant, A.; Philippe, H.; Huchon, D.; Cartwright, P. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc. Natl. Acad. Sci. USA 2015, 112, 14912–14917. [Google Scholar] [CrossRef] [PubMed]
- Kayal, E.; Bentlage, B.; Sabrina Pankey, M.; Ohdera, A.H.; Medina, M.; Plachetzki, D.C.; Collins, A.G.; Ryan, J.F. Phylogenomics provides a robust topology of the major cnidarian lineages and insights on the origins of key organismal traits. BMC Evol. Biol. 2018, 18, 68. [Google Scholar] [CrossRef]
- Kent, M.L.; Andree, K.B.; Bartholomew, J.L.; El-Matbouli, M.; Desser, S.S.; Devlin, R.H.; Feist, S.W.; Hedrick, R.P.; Hoffmann, R.W.; Khattra, J.; et al. Recent advances in our knowledge of the Myxozoa. J. Eukaryot. Microbiol. 2001, 48, 395–413. [Google Scholar] [CrossRef]
- Lom, J.; Dyková, I. Myxozoan genera: Definition and notes on taxonomy, life-cycle terminology and pathogenic species. Folia Parasitol. 2006, 53, 1–36. [Google Scholar] [CrossRef]
- Americus, B.; Lotan, T.; Bartholomew, J.L.; Atkinson, S.D. A comparison of the structure and function of nematocysts in free-living and parasitic cnidarians (Myxozoa). Int. J. Parasitol. 2020, 50, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Cannon, Q.; Wagner, E. Comparison of discharge mechanisms of cnidarian cnidae and myxozoan polar capsules. Rev. Fish. Sci. 2010, 11, 185–219. [Google Scholar] [CrossRef]
- Shpirer, E.; Diamant, A.; Cartwright, P.; Huchon, D. A genome wide survey reveals multiple nematocyst-specific genes in Myxozoa. BMC Evol. Biol. 2018, 18, 138. [Google Scholar] [CrossRef] [PubMed]
- Piriatinskiy, G.; Atkinson, S.D.; Park, S.; Morgenstern, D.; Brekhman, V.; Yossifon, G.; Bartholomew, J.L.; Lotan, T. Functional and proteomic analysis of Ceratonova shasta (Cnidaria: Myxozoa) polar capsules reveals adaptations to parasitism. Sci. Rep. 2017, 7, 9010. [Google Scholar] [CrossRef]
- Foox, J.; Ringuette, M.; Desser, S.S.; Siddall, M.E. In silico hybridization enables transcriptomic illumination of the nature and evolution of Myxozoa. BMC Genom. 2015, 16, 840. [Google Scholar] [CrossRef] [PubMed]
- Americus, B.; Hams, N.; Klompen, A.M.L.; Alama-Bermejo, G.; Lotan, T.; Bartholomew, J.L.; Atkinson, S.D. The cnidarian parasite Ceratonova shasta utilizes inherited and recruited venom-like compounds during infection. PeerJ 2021, 9, e12606. [Google Scholar] [CrossRef] [PubMed]
- Szekely, C.; Ghosh, S.; Borzak, R.; Goswami, U.; Molnar, K.; Cech, G. The occurrence of known Myxobolus and Thelohanellus species (Myxozoa, Myxosporea) from Indian major carps with the description of Myxobolus bandyopadhyayi n. sp. in West Bengal. Int. J. Parasitol. Parasites Wildl. 2021, 16, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Eiras, J.C.; Cruz, C.F.; Saraiva, A.; Adriano, E.A. Synopsis of the species of Myxobolus (Cnidaria, Myxozoa, Myxosporea) described between 2014 and 2020. Folia Parasitol. 2021, 68. [Google Scholar] [CrossRef] [PubMed]
- Eiras, J.C.; Adriano, E.A. A checklist of new species of Henneguya Thelohan, 1892 (Myxozoa: Myxosporea, Myxobolidae) described between 2002 and 2012. Syst. Parasitol. 2012, 83, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G. Myxobolus cerebralis, a worldwide cause of salmonid whirling disease. J. Aquat. Anim. Health 1990, 2, 30–37. [Google Scholar] [CrossRef]
- Liu, Y.; Whipps, C.M.; Gu, Z.M.; Zeng, C.; Huang, M.J. Myxobolus honghuensis n. sp. (Myxosporea: Bivalvulida) parasitizing the pharynx of allogynogenetic gibel carp Carassius auratus gibelio (Bloch) from Honghu Lake, China. Parasitol. Res. 2012, 110, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Li, A.; Gong, X. Infection of Myxobolus turpisrotundus sp. n. in allogynogenetic gibel carp, Carassius auratus gibelio (Bloch), with revision of Myxobolus rotundus (sl) Nemeczek reported from C. auratus auratus (L.). J. Fish. Dis. 2010, 33, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiong, J.; Zhou, Z.; Huo, F.; Miao, W.; Ran, C.; Liu, Y.; Zhang, J.; Feng, J.; Wang, M. The genome of the myxosporean Thelohanellus kitauei shows adaptations to nutrient acquisition within its fish host. Genome Biol. Evol. 2014, 6, 3182–3198. [Google Scholar] [CrossRef] [PubMed]
- Okamura, B.; Gruhl, A.; Reft, A.J. Cnidarian origins of the Myxozoa. In Myxozoan Evolution, Ecology and Development; Springer: Berlin/Heidelberg, Germany, 2015; pp. 45–68. [Google Scholar]
- Klompen, A.M.L.; Kayal, E.; Collins, A.G.; Cartwright, P. Phylogenetic and Selection Analysis of an Expanded Family of Putatively Pore-Forming Jellyfish Toxins (Cnidaria: Medusozoa). Genome Biol. Evol. 2021, 13, evab081. [Google Scholar] [CrossRef]
- D’Ambra, I.; Lauritano, C. A review of toxins from cnidaria. Mar. Drugs 2020, 18, 507. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Li, S.; Siu, S.W.I.; Yang, B.; Huang, C.; Chan, J.Y.; Morlighem, J.R.L.; Wong, C.T.T.; Radis-Baptista, G.; Lee, S.M. Novel Kunitz-like peptides discovered in the Zoanthid Palythoa caribaeorum through transcriptome sequencing. J. Proteome Res. 2018, 17, 891–902. [Google Scholar] [CrossRef]
- Nicosia, A.; Bennici, C.; Biondo, G.; Costa, S.; Di Natale, M.; Masullo, T.; Monastero, C.; Ragusa, M.A.; Tagliavia, M.; Cuttitta, A. Characterization of translationally controlled tumour protein from the sea anemone Anemonia viridis and transcriptome wide identification of cnidarian homologues. Genes 2018, 9, 30. [Google Scholar] [CrossRef]
- Kini, R.M.; Doley, R. Structure, function and evolution of three-finger toxins: Mini proteins with multiple targets. Toxicon 2010, 56, 855–867. [Google Scholar] [CrossRef]
- Yahalomi, D.; Atkinson, S.D.; Neuhof, M.; Chang, E.S.; Philippe, H.; Cartwright, P.; Bartholomew, J.L.; Huchon, D. A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome. Proc. Natl. Acad. Sci. USA 2020, 117, 5358–5363. [Google Scholar] [CrossRef]
- Guo, Q.; Atkinson, S.D.; Xiao, B.; Zhai, Y.; Bartholomew, J.L.; Gu, Z. A myxozoan genome reveals mosaic evolution in a parasitic cnidarian. BMC Biol. 2022, 20, 51. [Google Scholar] [CrossRef]
- Fry, B.G.; Scheib, H.; van der Weerd, L.; Young, B.; McNaughtan, J.; Ramjan, S.F.; Vidal, N.; Poelmann, R.E.; Norman, J.A. Evolution of an arsenal: Structural and functional diversification of the venom system in the advanced snakes (Caenophidia). Mol. Cell Proteom. 2008, 7, 215–246. [Google Scholar] [CrossRef]
- Chung, E.-H.; Lee, K.-S.; Han, J.-H.; Je, Y.-H.; Chang, J.-H.; Roh, J.-Y. Molecular cloning of two cDNAs encoding an insecticidal toxin from the spider, Araneus ventricosus, and construction of a recombinant baculovirus expressing a spider toxin. Int. J. Ind. Entomol. 2002, 4, 43–49. [Google Scholar]
- Hu, H.; Bandyopadhyay, P.K.; Olivera, B.M.; Yandell, M. Characterization of the Conus bullatus genome and its venom-duct transcriptome. BMC Genom. 2011, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Graf, R.; Ferber, M.; Zweckstetter, M.; Imperial, J.; Garrett, J.E.; Olivera, B.M.; Terlau, H.; Becker, S. Production of recombinant Conkunitzin-S1 in Escherichia coli. Protein Expr. Purif. 2006, 47, 640–644. [Google Scholar] [CrossRef]
- Pollastri, M.P.; Adade, C.M.; Carvalho, A.L.O.; Tomaz, M.A.; Costa, T.F.R.; Godinho, J.L.; Melo, P.A.; Lima, A.P.C.A.; Rodrigues, J.C.F.; Zingali, R.B.; et al. Crovirin, a snake venom cysteine-rich secretory protein (CRISP) with promising activity against trypanosomes and leishmania. PLoS Negl. Trop. Dis. 2014, 8, e3252. [Google Scholar]
- Yamazaki, Y.; Brown, R.L.; Morita, T. Purification and cloning of toxins from elapid venoms that target cyclic nucleotide-gated ion channels. Biochemistry 2002, 41, 11331–11337. [Google Scholar] [CrossRef]
- Brown, R.L.; Haley, T.L.; West, K.A.; Crabb, J.W. Pseudechetoxin: A peptide blocker of cyclic nucleotide-gated ion channels. Proc. Natl. Acad. Sci. USA 1999, 96, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, T.J.; Peuravuori, H.J.; Quinn, R.J.; Llewellyn, L.E.; Benzie, J.A.; Fenner, P.J.; Winkel, K.D. Phospholipase A2 in cnidaria. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2004, 139, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Mariottini, G.L.; Pane, L. Cytotoxic and cytolytic cnidarian venoms. A review on health implications and possible therapeutic applications. Toxins 2013, 6, 108–151. [Google Scholar] [CrossRef] [PubMed]
- Talvinen, K.A.; Nevalainen, T.J. Cloning of a novel phospholipase A2 from the cnidarian Adamsia carciniopados. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2002, 132, 571–578. [Google Scholar] [CrossRef]
- Hessinger, D.A.; Lenhoff, H.M. Mechanism of hemolysis induced by nematocyst venom: Roles of phospholipase A and direct lytic factor. Arch. Biochem. Biophys. 1976, 173, 603–613. [Google Scholar] [CrossRef]
- Fedorov, S.; Dyshlovoy, S.; Monastyrnaya, M.; Shubina, L.; Leychenko, E.; Kozlovskaya, E.; Jin, J.O.; Kwak, J.Y.; Bode, A.M.; Dong, Z.; et al. The anticancer effects of actinoporin RTX-A from the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 2010, 55, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Sajevic, T.; Leonardi, A.; Krizaj, I. Haemostatically active proteins in snake venoms. Toxicon 2011, 57, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.M.; Maroun, R.C. Snake venom serine proteinases: Sequence homology vs. substrate specificity, a paradox to be solved. Toxicon 2005, 45, 1115–1132. [Google Scholar] [CrossRef] [PubMed]
- Sartim, M.A.; Sampaio, S.V. Snake venom galactoside-binding lectins: A structural and functional overview. J. Venom. Anim. Toxins Incl. Trop. Dis. 2015, 21, 35. [Google Scholar] [CrossRef] [PubMed]
- Earl, S.T.; Robson, J.; Trabi, M.; de Jersey, J.; Masci, P.P.; Lavin, M.F. Characterisation of a mannose-binding C-type lectin from Oxyuranus scutellatus snake venom. Biochimie 2011, 93, 519–527. [Google Scholar] [CrossRef]
- Lopes-Ferreira, M.; Magalhaes, G.S.; Fernandez, J.H.; Junqueira-de-Azevedo Ide, L.; Le Ho, P.; Lima, C.; Valente, R.H.; Moura-da-Silva, A.M. Structural and biological characterization of Nattectin, a new C-type lectin from the venomous fish Thalassophryne nattereri. Biochimie 2011, 93, 971–980. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A customizable platform for evolutionary hypothesis testing using phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less Is More: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-wide identification of episodic selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Murrell, B.; Smith, M.D.; Kosakovsky Pond, S.L.; Scheffler, K. RELAX: Detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 2015, 32, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, D.; Zhai, Y.; Gu, Z. CCPRD: A novel analytical framework for the comprehensive proteomic reference database construction of nonmodel organisms. ACS Omega 2020, 5, 15370–15384. [Google Scholar] [CrossRef]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2016, 135, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Zancolli, G.; Casewell, N.R. Venom systems as models for studying the origin and regulation of evolutionary novelties. Mol. Biol. Evol. 2020, 37, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Flo, M.; Margenat, M.; Duran, R.; Gonzalez-Sapienza, G.; Grana, M.; Parkinson, J.; Maizels, R.M.; Salinas, G.; Alvarez, B.; et al. A family of diverse Kunitz inhibitors from Echinococcus granulosus potentially involved in host-parasite cross-talk. PLoS ONE 2009, 4, e7009. [Google Scholar] [CrossRef] [PubMed]
- Britos, L.; Lalanne, A.I.; Castillo, E.; Cota, G.; Senorale, M.; Marin, M. Mesocestoides corti (syn. vogae, cestoda): Characterization of genes encoding cysteine-rich secreted proteins (CRISP). Exp. Parasitol. 2007, 116, 95–102. [Google Scholar] [CrossRef]
- Price, D.R.; Bell, H.A.; Hinchliffe, G.; Fitches, E.; Weaver, R.; Gatehouse, J.A. A venom metalloproteinase from the parasitic wasp Eulophus pennicornis is toxic towards its host, tomato moth (Lacanobia oleracae). Insect Mol. Biol. 2009, 18, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ranasinghe, S.; McManus, D.P. Structure and function of invertebrate Kunitz serine protease inhibitors. Dev. Comp. Immunol. 2013, 39, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Morlighem, J.R.; Zhou, H.; Lima, E.P.; Gomes, P.B.; Cai, J.; Lou, I.; Perez, C.D.; Lee, S.M.; Radis-Baptista, G. The transcriptome of the zoanthid Protopalythoa variabilis (Cnidaria, Anthozoa) predicts a basal repertoire of toxin-like and venom-auxiliary polypeptides. Genome Biol. Evol. 2016, 8, 3045–3064. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, A.; Mikov, A.; Cammarata, M.; Colombo, P.; Andreev, Y.; Kozlov, S.; Cuttitta, A. The Anemonia viridis venom: Coupling biochemical purification and RNA-Seq for translational research. Mar. Drugs 2018, 16, 407. [Google Scholar] [CrossRef]
- Vogel, C.; Teichmann, S.A.; Pereira-Leal, J. The relationship between domain duplication and recombination. J. Mol. Biol. 2005, 346, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Undheim, E.A.; Jones, A.; Clauser, K.R.; Holland, J.W.; Pineda, S.S.; King, G.F.; Fry, B.G. Clawing through evolution: Toxin diversification and convergence in the ancient lineage Chilopoda (centipedes). Mol. Biol. Evol. 2014, 31, 2124–2148. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, H.; Xue, W.; Yue, Y.; Liu, S.; Xing, R.; Li, P. Jellyfish venomics and venom gland transcriptomics analysis of Stomolophus meleagris to reveal the toxins associated with sting. J. Proteom. 2014, 106, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Jouiaei, M.; Casewell, N.R.; Yanagihara, A.A.; Nouwens, A.; Cribb, B.W.; Whitehead, D.; Jackson, T.N.; Ali, S.A.; Wagstaff, S.C.; Koludarov, I.; et al. Firing the sting: Chemically induced discharge of cnidae reveals novel proteins and peptides from box jellyfish (Chironex fleckeri) venom. Toxins 2015, 7, 936–950. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.F.; Reuille, R.L.; Ming, L.-J.; Livingston, B.T. Overexpression and mechanistic characterization of blastula protease 10, a metalloprotease involved in sea urchin embryogenesis and development. J. Biol. Chem. 2006, 281, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genom. Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Kordiš, D.; Gubenšek, F. Adaptive evolution of animal toxin multigene families. Gene 2000, 261, 43–52. [Google Scholar] [CrossRef]
- Chang, D.; Duda, T.F., Jr. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 2012, 29, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Binford, G.J.; Bodner, M.R.; Cordes, M.H.; Baldwin, K.L.; Rynerson, M.R.; Burns, S.N.; Zobel-Thropp, P.A. Molecular evolution, functional variation, and proposed nomenclature of the gene family that includes sphingomyelinase D in sicariid spider venoms. Mol. Biol. Evol. 2009, 26, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, H.; Moran, Y.; Gordon, D.; Turkov, M.; Kahn, R.; Gurevitz, M. Positions under positive selection-key for selectivity and potency of scorpion alpha-toxins. Mol. Biol. Evol. 2010, 27, 1025–1034. [Google Scholar] [CrossRef]
- Fry, B.G.; Wuster, W.; Kini, R.M.; Brusic, V.; Khan, A.; Venkataraman, D.; Rooney, A.P. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003, 57, 110–129. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Moran, Y. The rise and fall of an evolutionary innovation: Contrasting strategies of venom evolution in ancient and young animals. PLoS Genet. 2015, 11, e1005596. [Google Scholar] [CrossRef] [PubMed]
- Surm, J.M.; Smith, H.L.; Madio, B.; Undheim, E.A.B.; King, G.F.; Hamilton, B.R.; van der Burg, C.A.; Pavasovic, A.; Prentis, P.J. A process of convergent amplification and tissue-specific expression dominates the evolution of toxin and toxin-like genes in sea anemones. Mol. Ecol. 2019, 28, 2272–2289. [Google Scholar] [CrossRef] [PubMed]
- Naldoni, J.; Zatti, S.A.; da Silva, M.R.M.; Maia, A.A.M.; Adriano, E.A. Morphological, ultrastructural, and phylogenetic analysis of two novel Myxobolus species (Cnidaria: Myxosporea) parasitizing bryconid fish from Sao Francisco River, Brazil. Parasitol. Int. 2019, 71, 27–36. [Google Scholar] [CrossRef]
- Lom, J.; Arthur, J. A guideline for the preparation of species descriptions in Myxosporea. J. Fish. Dis. 1989, 12, 151–156. [Google Scholar] [CrossRef]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2010.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; A Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinform. 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinform. 2003, 19, 651–652. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simao, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Jungo, F.; Bougueleret, L.; Xenarios, I.; Poux, S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon 2012, 60, 551–557. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39 (Suppl. 2), W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Minh, B.Q.; A Schmidt, H.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corre-sponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Shank, S.D.; Spielman, S.; Li, M.; Muse, S.V.; Pond, S.L.K. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Contigs | Unigene | Mean Length | N50 | CEGMA | BUSCO |
|---|---|---|---|---|---|---|
| T. kitauei | 90,775 | 88,081 | 590 | 1118 | 67.3% | 57.3% |
| M. xiantaoensis | 98,837 | 66,662 | 844 | 1368 | 77.8% | 78.4% |
| M. ampullicapsulatus | 276,231 | 174,559 | 380 | 381 | 71.8% | 64.7% |
| M. turpisrotundus | 171,878 | 153,649 | 453 | 759 | 62.1% | 57.3% |
| M. honghuensis | 27,502 | 23,995 | 6013 | 857 | 59.6% | 59.6% |
| Class/Toxins | Neurotoxins | Cytolysins | Protease Inhibitors | Hemorrhagic Toxins | Allergen | Enzymes | ALL |
|---|---|---|---|---|---|---|---|
| N. vctensis | 4/4.5% | 3/3.4% | 1/1.1% | 21/23.9% | 2/2.3% | 57/64.8% | 88 |
| H. vulgaris | 115/27.4% | 179/42.7% | 2/0.5% | 31/7.4% | 2/0.5% | 90/21.5% | 419 |
| C. fleckeri | 93/34.1% | 136/49.8% | 1/0.4% | 25/9.2% | 2/0.7% | 16/5.9% | 273 |
| A. aurita | 180/29.5% | 165/27.0% | 2/0.3% | 72/11.8% | 2/0.3% | 190/31.1% | 611 |
| P. hydriforme | 8/22.2% | 0/0.0% | 1/2.8% | 16/44.4% | 1/2.8% | 10/27.8% | 36 |
| T. bryosalmonae | 2/16.7% | 0/0.0% | 0/0.0% | 6/50.0% | 3/25.0% | 1/8.3% | 12 |
| B. plumatellae | 10/27.0% | 0/0.0% | 1/2.7% | 20/54.1% | 2/5.4% | 4/10.8% | 37 |
| M. squamalis | 5/45.5% | 0/0.0% | 0/0.0% | 6/54.5% | 0/0.0% | 0/0.0% | 11 |
| M. squamalis-G 1 | 5/55.6% | 0/0.0% | 0/0.0% | 4/44.4% | 0/0.0% | 0/0.0% | 9 |
| M. honghuensis | 5/26.3% | 0/0.0% | 0/0.0% | 13/68.4% | 1/5.3% | 0/0.0% | 19 |
| M. honghuensis-G 1 | 6/50.0% | 0/0.0% | 0/0.0% | 5/41.7% | 1/8.3% | 0/0.0% | 12 |
| M. cerebralis | 2/25.0% | 0/0.0% | 0/0.0% | 5/62.5% | 1/12.5% | 0/0.0% | 8 |
| M. xiantaoensis | 3/14.3% | 0/0.0% | 0/0.0% | 14/66.7% | 2/9.5% | 2/9.5% | 21 |
| M. turpisrotundus | 7/38.9% | 0/0.0% | 0/0.0% | 7/38.9% | 1/5.6% | 3/16.7% | 18 |
| M. ampullicapsulatu | 2/22.2% | 0/0.0% | 0/0.0% | 6/66.7% | 1/11.1% | 0/0.0% | 9 |
| M. pendula | 6/6.8% | 1/1.1% | 2/2.3% | 74/84.1% | 2/2.3% | 3/3.4% | 88 |
| T. kitauei | 6/30.0% | 0/0.0% | 1/5.0% | 12/60.0% | 1/5.0% | 0/0.0% | 20 |
| T. kitauei-G 1 | 5/50.0% | 0/0.0% | 0/0.0% | 4/40.0% | 1/10.0% | 0/0.0% | 10 |
| H. salminicola | 3/33.3% | 0/0.0% | 0/0.0% | 5/55.6% | 1/11.1% | 0/0.0% | 9 |
| H. salminicola-G 1 | 4/50.0% | 0/0.0% | 0/0.0% | 3/37.5% | 1/12.5% | 0/0.0% | 8 |
| K. iwatai | 1/25.0% | 0/0.0% | 0/0.0% | 3/75.0% | 0/0.0% | 0/0.0% | 4 |
| K. iwatai-G 1 | 2/22.2% | 0/0.0% | 0/0.0% | 6/66.7% | 0/0.0% | 1/11.1% | 9 |
| C. shasta | 2/28.6% | 0/0.0% | 0/0.0% | 4/57.1% | 0/0.0% | 1/14.3% | 7 |
| S. molnari | 3/6.0% | 1/2.0% | 1/2.0% | 40/80.0% | 2/4.0% | 3/6.0% | 50 |
| Method/Family | Kunitz | CRISP | M12B |
|---|---|---|---|
| Codeml | 0.05767 | 0.09041 | 0.09904 |
| FEL | −32 | −98 | −29 |
| MEME | 1 | 3 | 4 |
| aBSREL | 0 | 2 | 1 |
| BUSTED | found no evidence | found evidence | found no evidence |
| RELAX | K 1 | K 1 | K 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, B.; Guo, Q.; Zhai, Y.; Gu, Z. Transcriptomic Insights into the Diversity and Evolution of Myxozoa (Cnidaria, Endocnidozoa) Toxin-like Proteins. Mar. Drugs 2022, 20, 291. https://doi.org/10.3390/md20050291
Xiao B, Guo Q, Zhai Y, Gu Z. Transcriptomic Insights into the Diversity and Evolution of Myxozoa (Cnidaria, Endocnidozoa) Toxin-like Proteins. Marine Drugs. 2022; 20(5):291. https://doi.org/10.3390/md20050291
Chicago/Turabian StyleXiao, Bin, Qingxiang Guo, Yanhua Zhai, and Zemao Gu. 2022. "Transcriptomic Insights into the Diversity and Evolution of Myxozoa (Cnidaria, Endocnidozoa) Toxin-like Proteins" Marine Drugs 20, no. 5: 291. https://doi.org/10.3390/md20050291
APA StyleXiao, B., Guo, Q., Zhai, Y., & Gu, Z. (2022). Transcriptomic Insights into the Diversity and Evolution of Myxozoa (Cnidaria, Endocnidozoa) Toxin-like Proteins. Marine Drugs, 20(5), 291. https://doi.org/10.3390/md20050291