
Fluorescent Labeling of Polymannuronic Acid and Its Distribution in Mice by Tail Vein Injection

 , ,
, ,
Abstract
:1. Introduction
2. Results
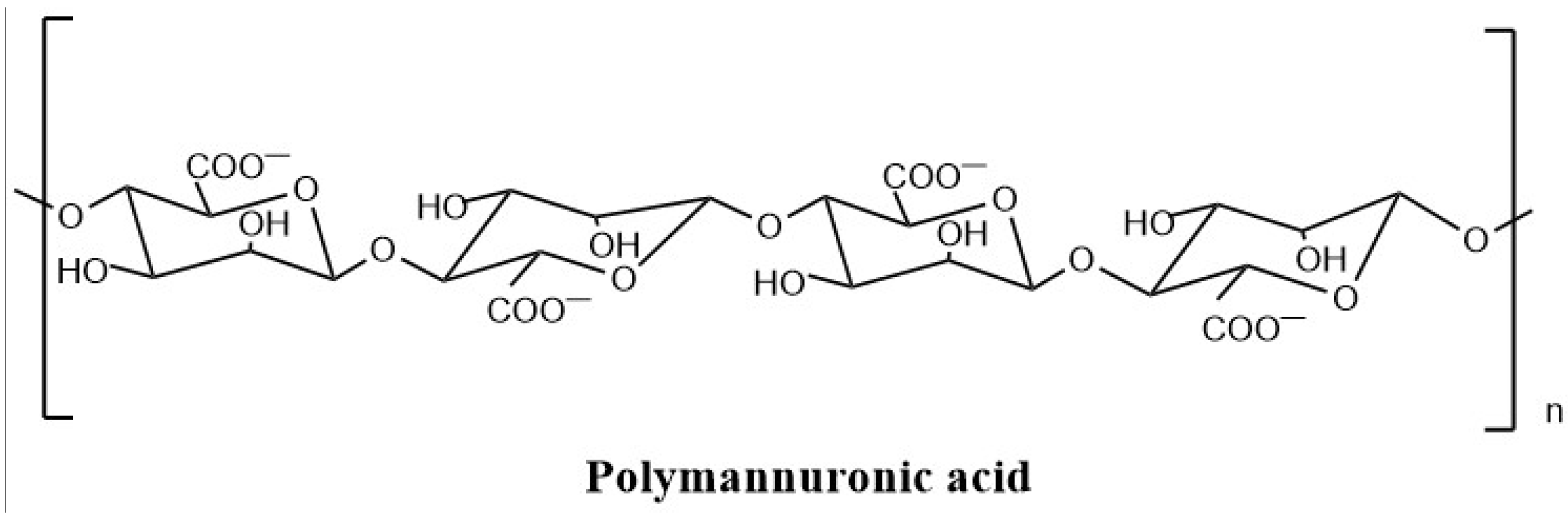
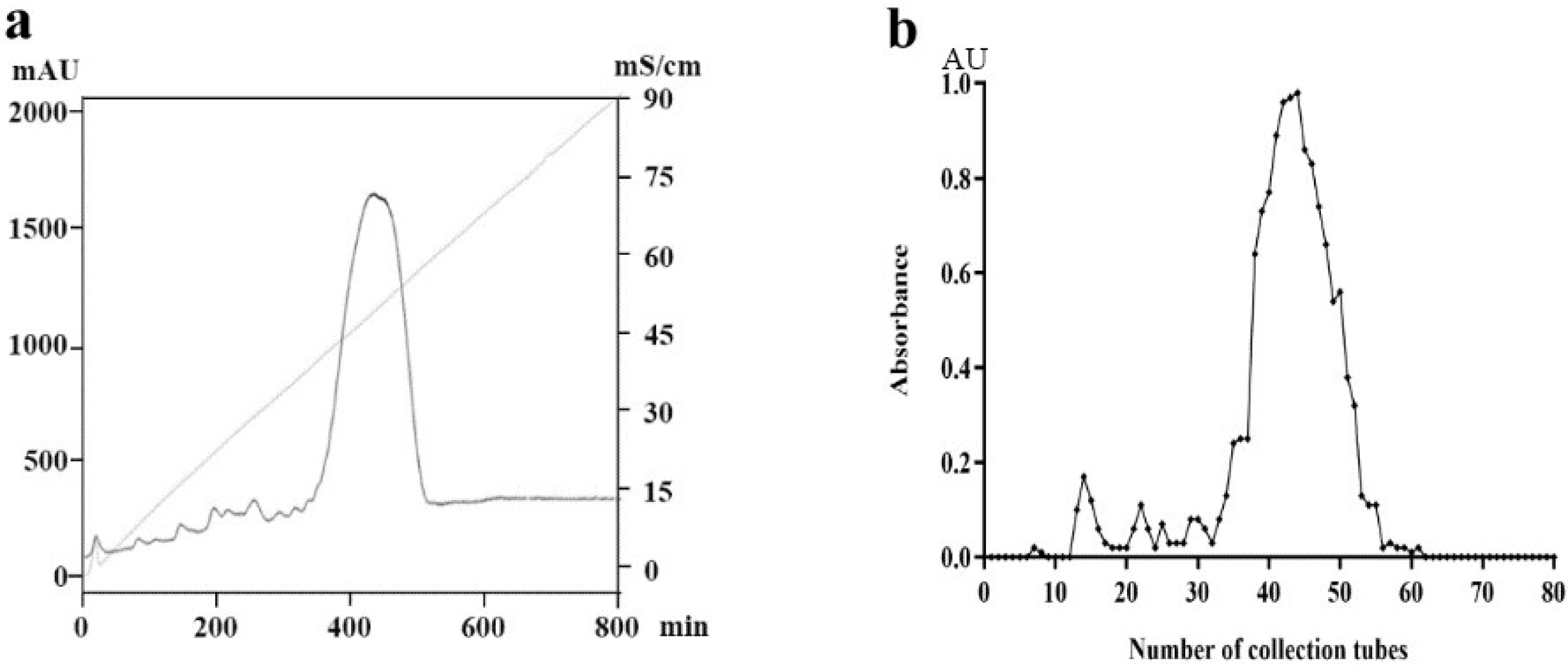
2.1. Preparation and Purification of PM
2.2. Purity and Molecular Weight Determination of PM
2.3. Validation of FITC-PM Labeling
2.4. Stability Study of FITC-PM
2.5. In Vivo Tissue Distribution and Pharmacokinetic Analysis of PM
2.5.1. Free-FITC Interference Detection
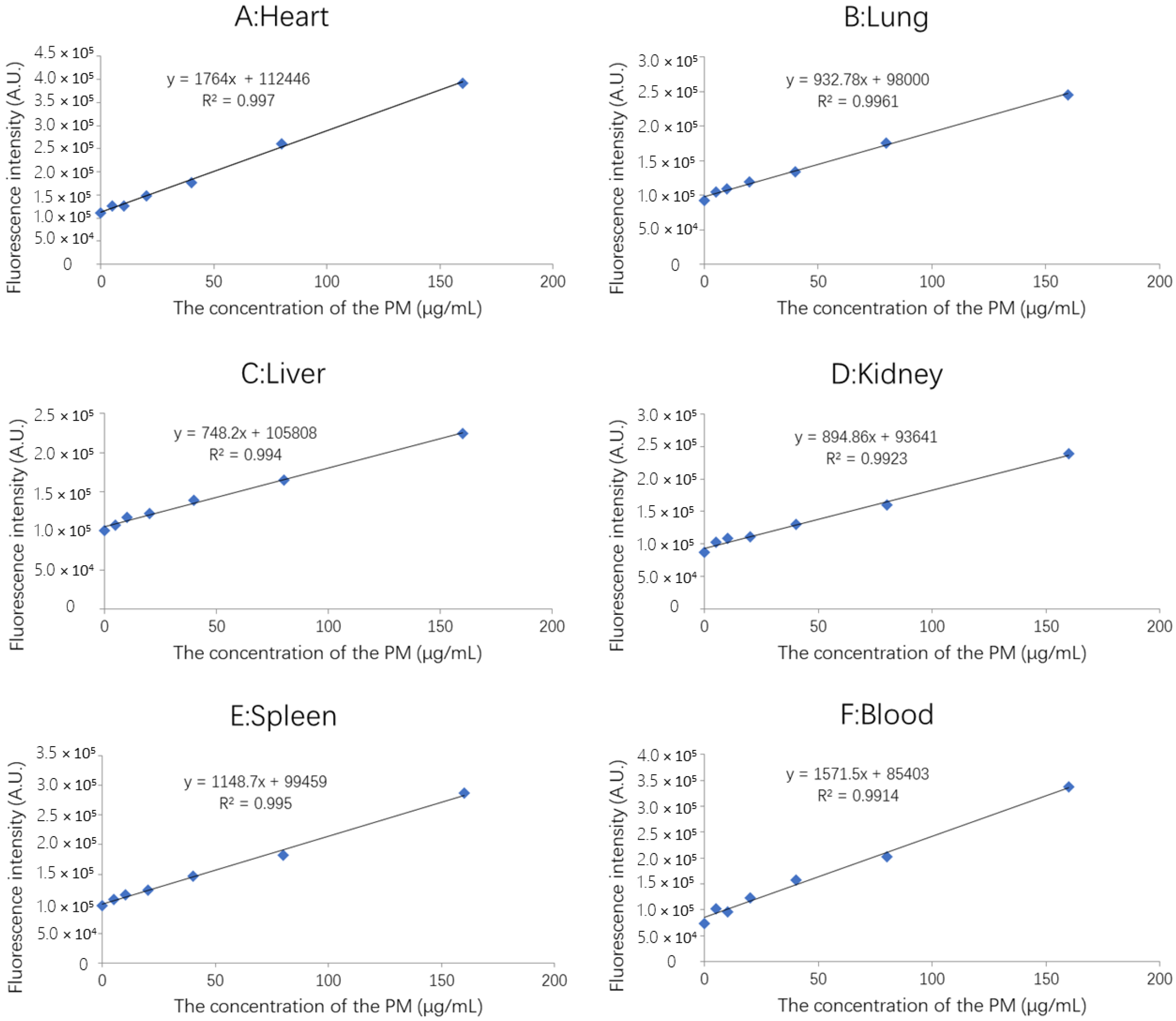
2.5.2. Standard Curve of FITC-PM in Blood and Tissues
2.5.3. Concentration and Pharmacokinetics of FITC-PM in Plasma
2.5.4. Distribution of FITC-PM in Tissues
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Preparation of PM
4.2.2. Purity Analysis and Molecular Weight Determination of PM
4.2.3. Preparation of FITC-PM
4.2.4. Validation of FITC-PM Labeling
4.2.5. Study of FITC-PM Stability
4.2.6. Tissue Distribution of PM in Mice
Grouping and Dosing of Mice
Collection and Pretreatment of Blood and Organs
Handling of Organ Homogenates
Preparation of FITC-PM Standard Curve for the Blood and Organs
Free-FITC Interference Detection
4.2.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Gao, S.K.; Yin, R.; Wang, X.C.; Jiang, H.N.; Liu, X.X.; Lv, W.; Ma, Y.; Zhou, Y.X. Structure Characteristics, Biochemical Properties, and Pharmaceutical Applications of Alginate Lyases. Mar. Drugs 2021, 19, 628. [Google Scholar] [CrossRef] [PubMed]
- Arfin, T.; Sonawane, K. Alginate: Recent Progress and Technological Prospects; Wiley: Hoboken, NJ, USA, 2019; pp. 45–57. [Google Scholar]
- Ueno, M.; Oda, T. Biological activities of alginate. Adv. Food Nutr. Res. 2014, 72. [Google Scholar] [CrossRef]
- Mohsenzadegan, M.; Moghbeli, F.; Mirshafiey, A.; Farajollahi, M.M. Anti-tumor effect of M2000 (beta-d-mannuronic acid) on the expression of inflammatory molecules in the prostate cancer cell. Immunopharmacol. Immunotoxicol. 2021, 43, 419–430. [Google Scholar] [CrossRef]
- Zhao, X.; Li, B.F.; Xue, C.H.; Sun, L.P. Effect of molecular weight on the antioxidant property of low molecular weight alginate from Laminaria japonica. J. Appl. Phycol. 2012, 24, 295–300. [Google Scholar] [CrossRef]
- Zeng, X.; Lan, Y.; Zeng, P.; Guo, Z.; Hao, C.; Zhang, L. Polyguluronate sulfate, polymannuronate sulfate, and their oligosaccharides have antithrombin III- and heparin cofactor II-independent anticoagulant activity. J. Ocean Univ. China 2017, 16, 346–350. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Kurachi, M.; Yamaguchi, K.; Oda, T. Induction of multiple cytokine secretion from RAW264.7 cells by alginate oligosaccharides. Biosci. Biotechnol. Biochem. 2007, 71, 238–241. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, N.A.G.; Razavi, A.; Mirshafiey, A. Beta-D-Mannuronic Acid (M2000) as a Landmark in Pharmacology. Curr. Drug Discov. Technol. 2021, 18, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Sun, G.Q.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.Q.; Chu, X.K.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef]
- Yang, J.H.; Bang, M.A.; Jang, C.H.; Jo, G.H.; Jung, S.K.; Ki, S.H. Alginate oligosaccharide enhances LDL uptake via regulation of LDLR and PCSK9 expression. J. Nutr. Biochem. 2015, 26, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.C.; Cao, Q.; Wang, Y.; Xiao, H.; Zhao, J.R.; Zhang, Q.; Ji, A.G.; Song, S.L. Advances in Research on the Bioactivity of Alginate Oligosaccharides. Mar. Drugs 2020, 18, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bai, X.; Hu, B.; Xing, M.C.; Cao, Q.; Ji, A.G.; Song, S.L. Transport Mechanisms of Polymannuronic Acid and Polyguluronic Acid Across Caco-2 Cell Monolayers. Pharmaceutics 2020, 12, 167. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wang, R.; Gao, J.; Peng, J.; Li, H.; Wang, L. Progress in pharmacokinetics of polysaccharides. Chin. J. Clin. Pharmacol. Ther. 2021, 26, 230–235. [Google Scholar]
- Yu-Hao, D.; Chun, C.; Xiong, F.; Rui-Hai, L. Study on the pharmacokinetics of mulberry fruit polysaccharides through fluorescence labeling. Int. J. Biol. Macromol. 2021, 186, 462–471. [Google Scholar] [CrossRef]
- Li, F.; Wei, Y.; Zhao, J.; Zhang, L.; Li, Q. In vivo pharmacokinetic study of a Cucurbita moschata polysaccharide after oral administration. Int. J. Biol. Macromol. 2022, 203, 19–28. [Google Scholar] [CrossRef]
- Pozharitskaya, O.N.; Shikov, A.N.; Faustova, N.M.; Obluchinskaya, E.D.; Kosman, V.M.; Vuorela, H.; Makarov, V.G. Pharmacokinetic and Tissue Distribution of Fucoidan from Fucus vesiculosus after Oral Administration to Rats. Mar. Drugs 2018, 16, 132. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yu, G.L.; Guan, H.S.; Yue, N.; Zhang, Z.Q.; Li, H.H. Preparation of low-molecular-weight polyguluronate sulfate and its anticoagulant and anti-inflammatory activities. Carbohydr. Polym. 2007, 69, 272–279. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Z.M.; Yang, X.W.; Pan, X.T.; Yin, L.L.; Huang, X.; Li, Q.; Shu, Y.M.; Zhang, Q.L.; Wang, K.P. A sensitive and rapid radiolabelling method for the in vivo pharmacokinetic study of lentinan. Food Funct. 2018, 9, 3114–3125. [Google Scholar] [CrossRef]
- Zhang, E.; Chu, F.L.; Zhao, T.D.; Chai, Y.C.; Liang, H.; Song, S.L.; Ji, A.G. Determination of fucoidan in rat plasma by HPLC and its application in pharmacokinetics. Pak. J. Pharm. Sci. 2020, 33, 1–9. [Google Scholar]
- Bai, X.; Zhang, E.; Hu, B.; Liang, H.; Song, S.L.; Ji, A.G. Study on Absorption Mechanism and Tissue Distribution of Fucoidan. Molecules 2020, 25, 1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Value |
|---|---|---|
| Ke | 1/h | 0.24 |
| t1/2 | h | 2.85 |
| Tmax | h | 0.50 |
| Cmax | μg/mL | 235.17 |
| C0 | μg/mL | 389.98 |
| AUC0–t | μg/mL·h | 567.37 |
| AUC0–∞ | μg/mL·h | 631.48 |
| AUC0–t/0–∞ | 0.90 | |
| AUMC0–∞ | μg/mL·h2 | 1843.15 |
| MRT0–∞ | h | 2.92 |
| Vz | mL | 325.46 |
| CL | mL/h | 79.18 |
| Vss | mL | 231.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Wei, Q.; Wang, K.; Yang, Q.; Wang, Y.; Ji, A.; Chen, G. Fluorescent Labeling of Polymannuronic Acid and Its Distribution in Mice by Tail Vein Injection. Mar. Drugs 2022, 20, 289. https://doi.org/10.3390/md20050289
Song S, Wei Q, Wang K, Yang Q, Wang Y, Ji A, Chen G. Fluorescent Labeling of Polymannuronic Acid and Its Distribution in Mice by Tail Vein Injection. Marine Drugs. 2022; 20(5):289. https://doi.org/10.3390/md20050289
Chicago/Turabian StyleSong, Shuliang, Qiang Wei, Ke Wang, Qiong Yang, Yu Wang, Aiguo Ji, and Guanjun Chen. 2022. "Fluorescent Labeling of Polymannuronic Acid and Its Distribution in Mice by Tail Vein Injection" Marine Drugs 20, no. 5: 289. https://doi.org/10.3390/md20050289
APA StyleSong, S., Wei, Q., Wang, K., Yang, Q., Wang, Y., Ji, A., & Chen, G. (2022). Fluorescent Labeling of Polymannuronic Acid and Its Distribution in Mice by Tail Vein Injection. Marine Drugs, 20(5), 289. https://doi.org/10.3390/md20050289