
Application of Feature-Based Molecular Networking for Comparative Metabolomics and Targeted Isolation of Stereoisomers from Algicolous Fungi

 ,
,  and
and 
Abstract
:
1. Introduction
2. Results
2.1. Strain Identification and Cultivation
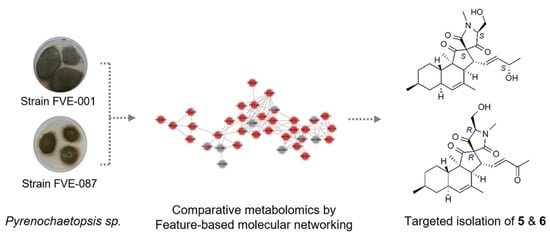
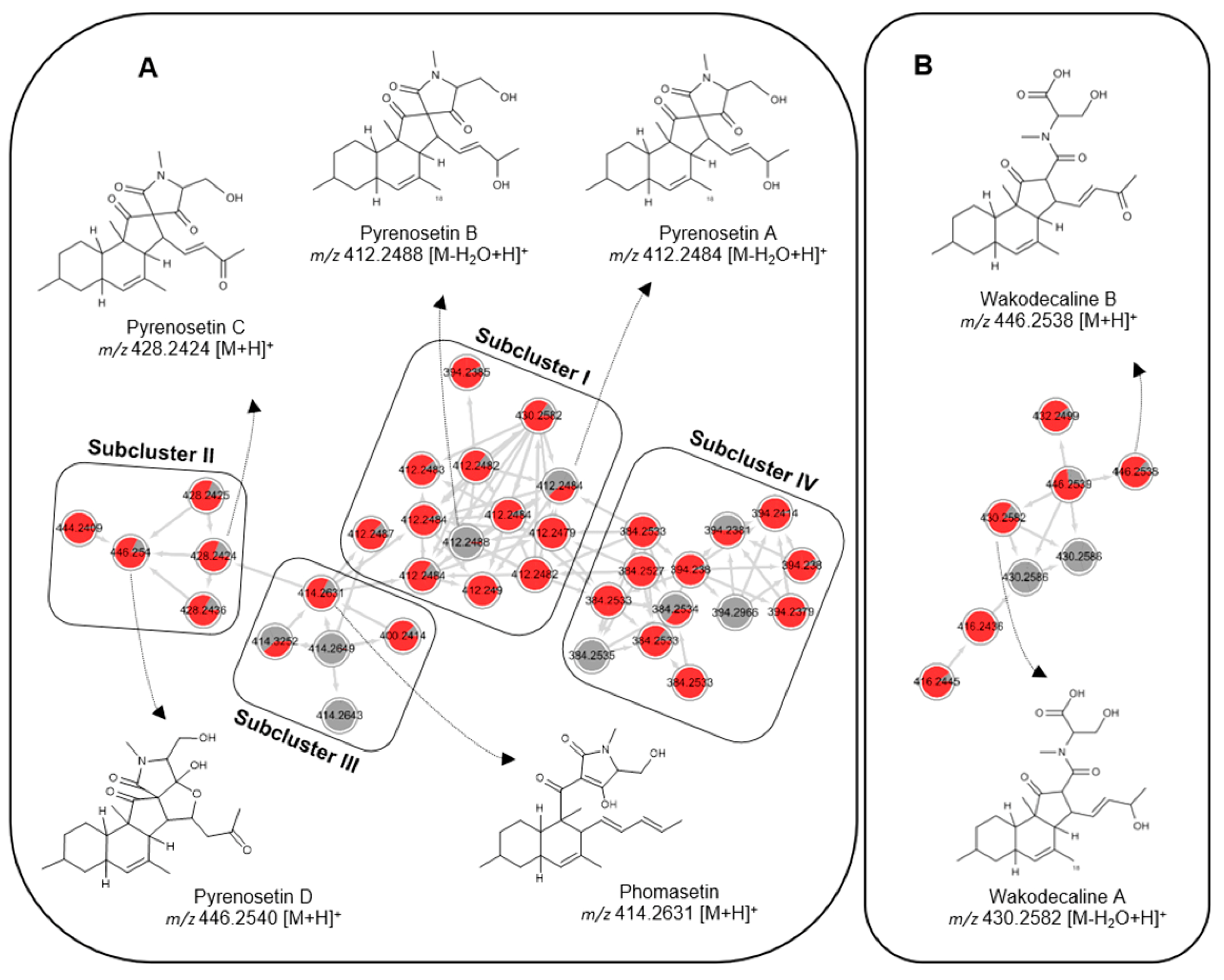
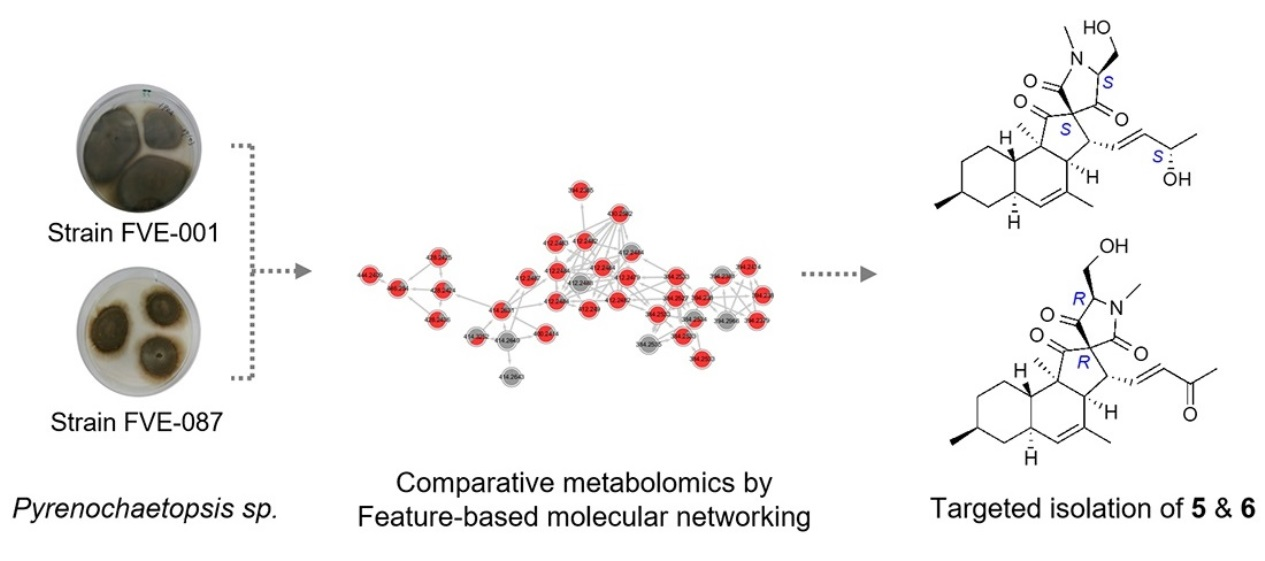
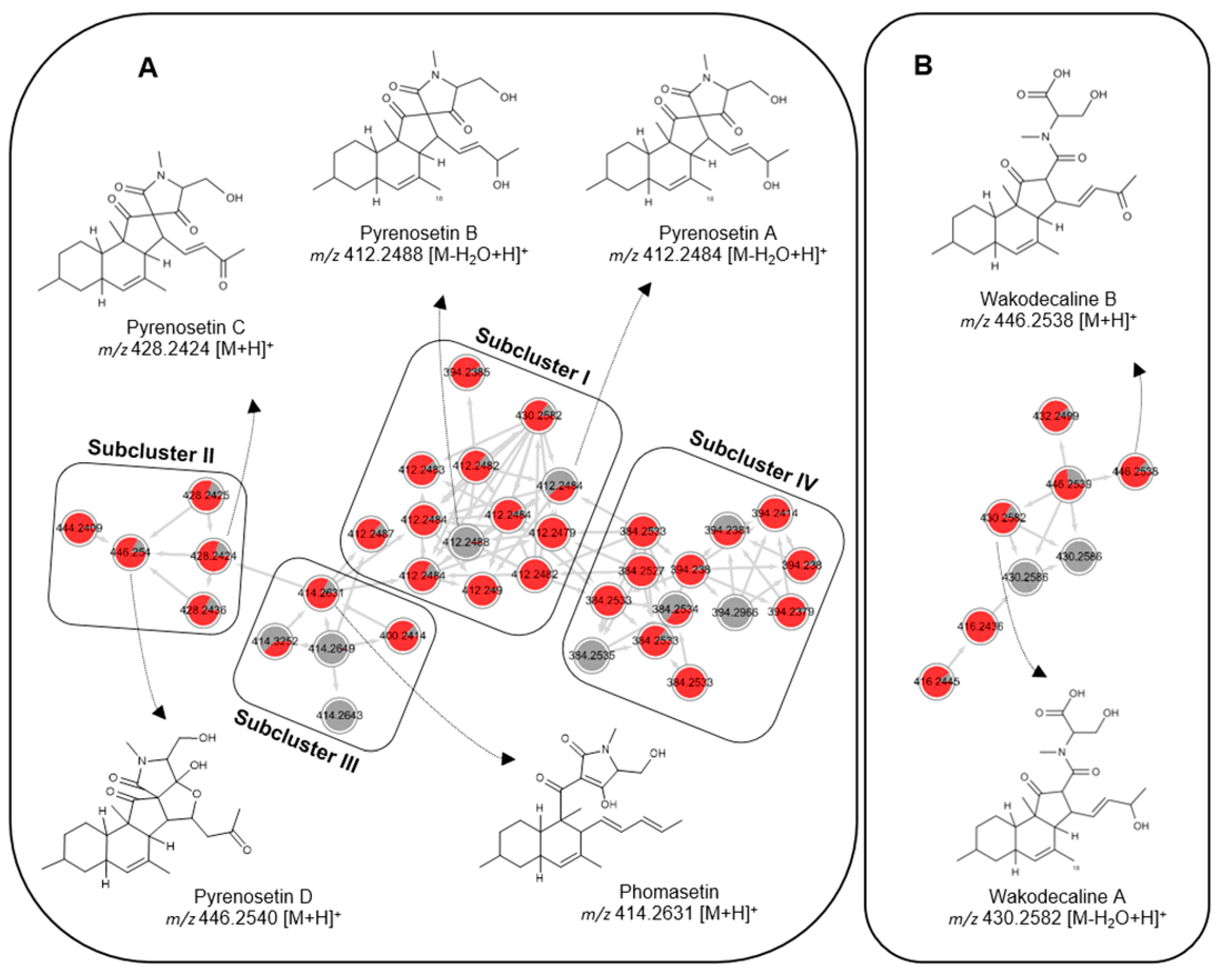
2.2. Comparative Metabolomics
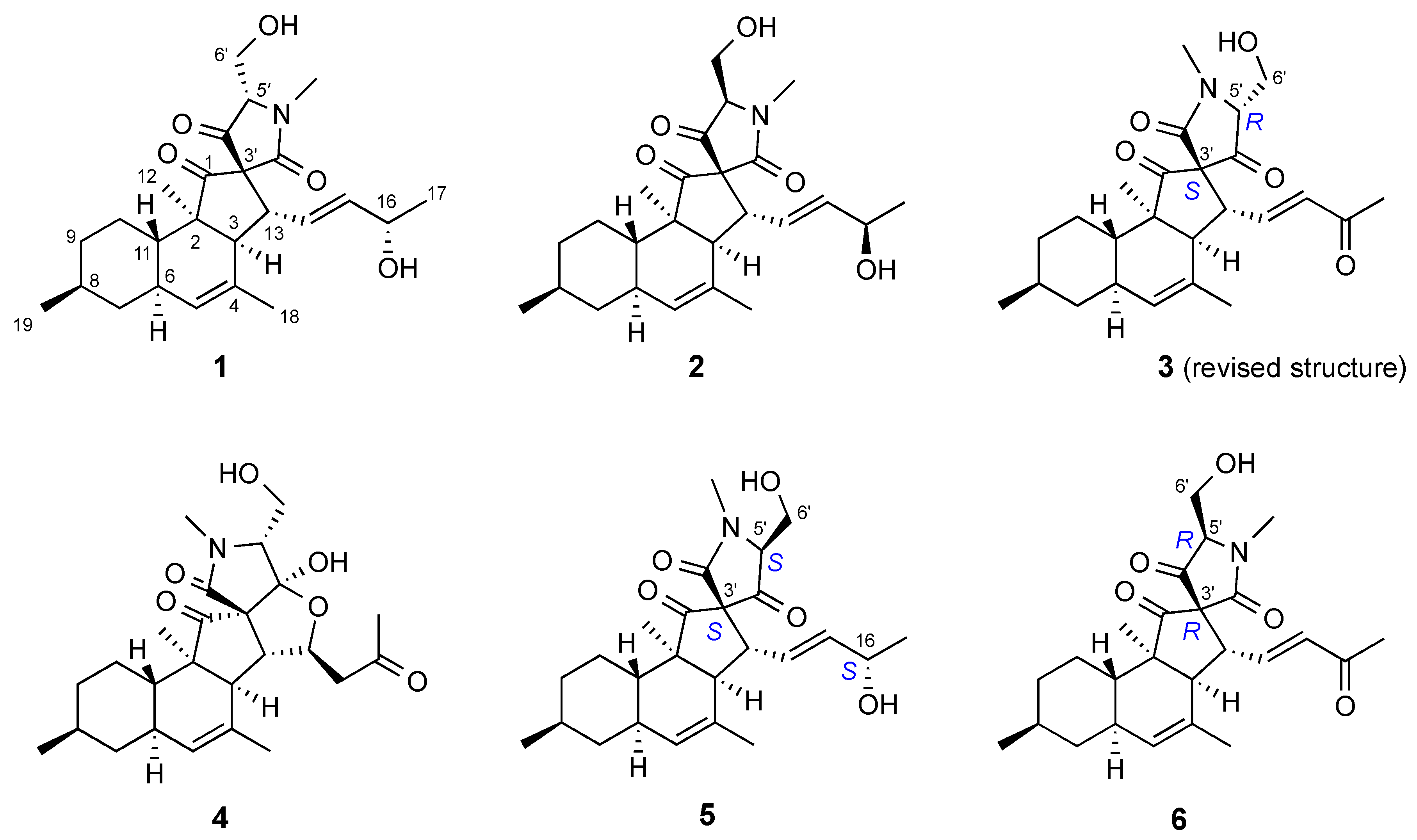
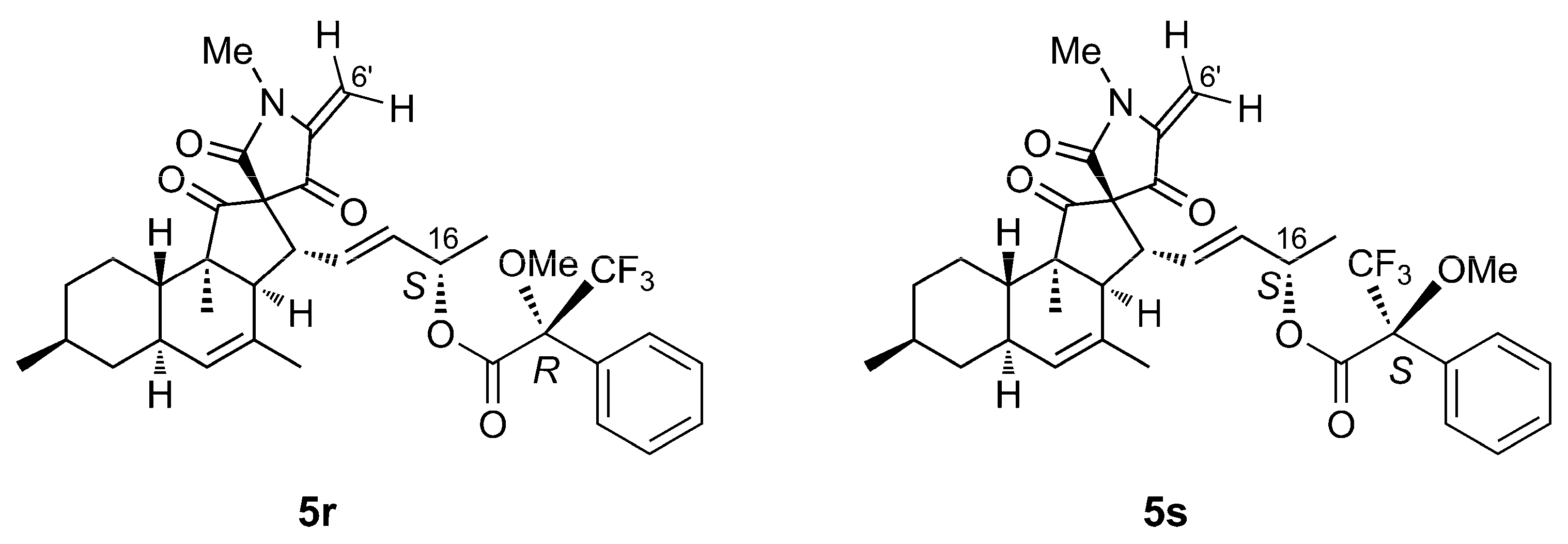
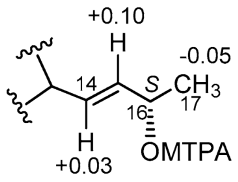
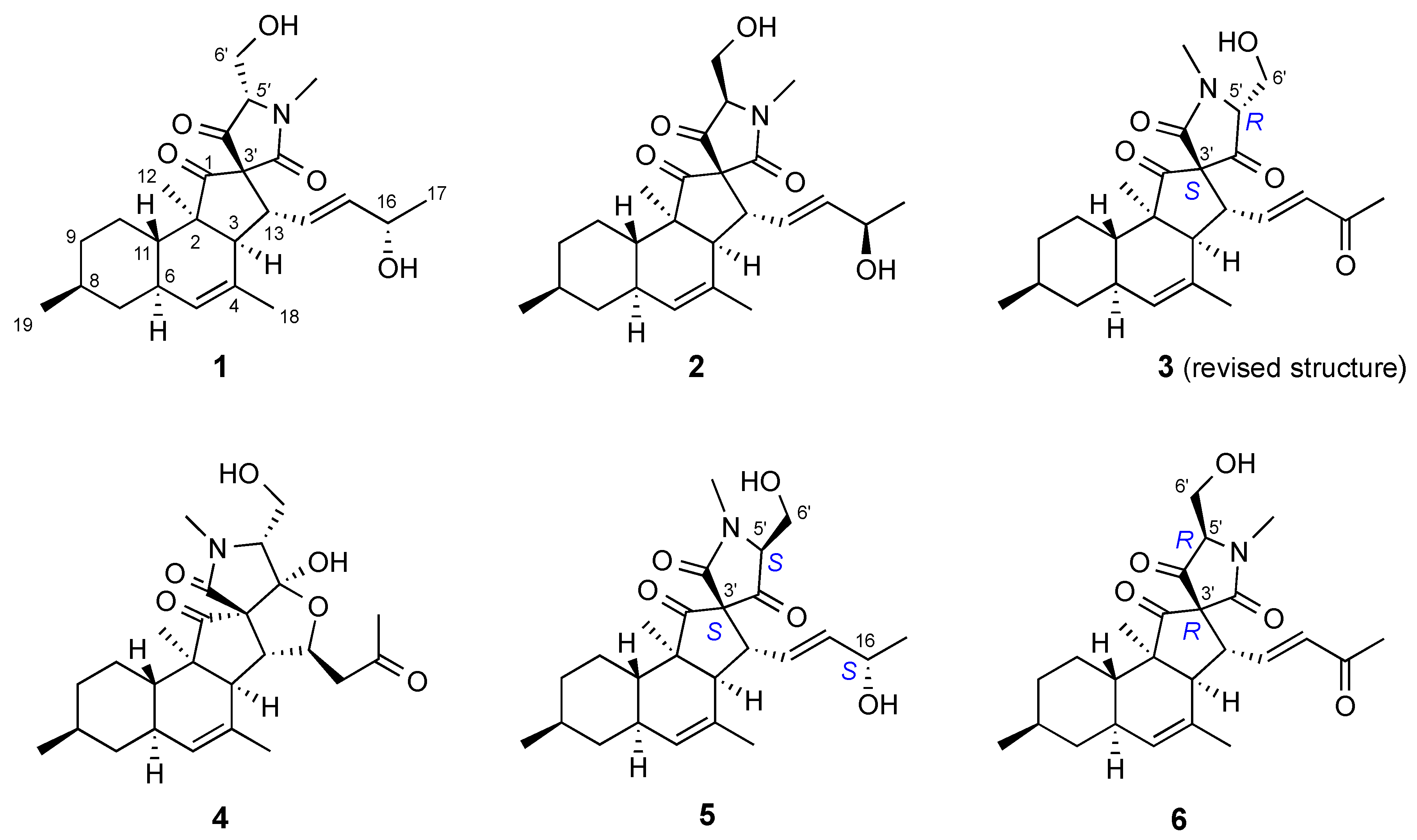
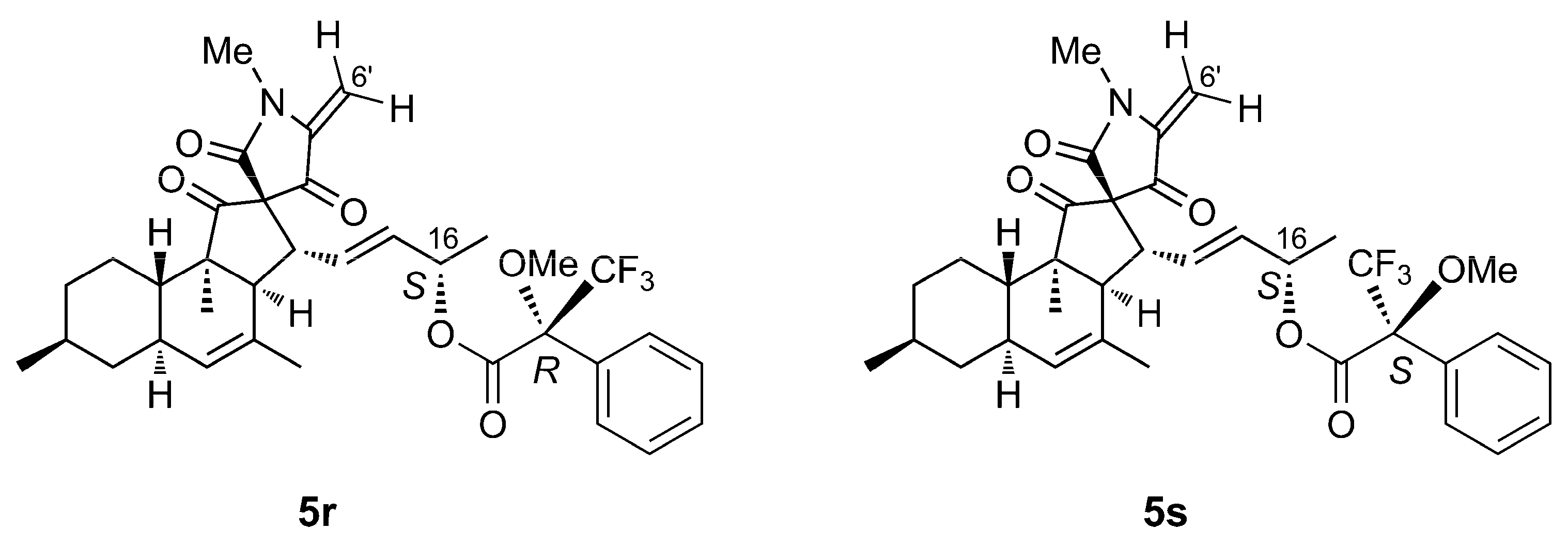
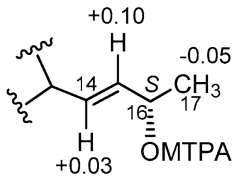
2.3. Isolation and Structure Elucidation
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Strain Identification, Cultivation and Kupchan Partition and Initial Bioassays
4.3. UPLC-QToF-MS/MS-Based Metabolome Analyses
4.4. Fractionation and Purification
4.5. Mosher’s Reaction
4.6. Computational Studies
4.7. Bioactivity Assessments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sobolevskaya, M.P.; Leshchenko, E.V.; Hoai, T.P.T.; Denisenko, V.A.; Dyshlovoy, S.A.; Kirichuk, N.N.; Khudyakova, Y.V.; Kim, N.Y.; Berdyshev, D.V.; Pislyagin, E.A.; et al. Pallidopenillines: Polyketides from the alga-derived fungus Penicillium thomii Maire KMM 4675. J. Nat. Prod. 2016, 79, 3031–3038. [Google Scholar] [CrossRef]
- Iwamoto, C.; Minoura, K.; Hagishita, S.; Nomoto, K.; Numata, A. Penostatins F–I, novel cytotoxic metabolites from a Penicillium species separated from an Enteromorpha marine alga. Perkin Trans. 1 1998, 3, 449–456. [Google Scholar] [CrossRef]
- Osterhage, C.; König, G.M.; Jones, P.G.; Wright, A.D. 5-Hydroxyramulosin, a new natural product produced by Phoma tropica, a marine-derived fungus isolated from the alga Fucus spiralis. Planta Med. 2002, 68, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.-Y.; Wang, B.-G. Mycochemistry of marine algicolous fungi. Fungal Divers. 2016, 80, 301–342. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R.; Cheng, X.C. Halimide, a Cytotoxic Marine Natural Product, and Derivatives Thereof. U.S. Patent US-6069146-A, 30 May 2000. [Google Scholar]
- Cimino, P.J.; Huang, L.; Du, L.; Wu, Y.; Bishop, J.; Dalsing-Hernandez, J.; Kotlarczyk, K.; Gonzales, P.; Carew, J.; Nawrocki, S.; et al. Plinabulin, an inhibitor of tubulin polymerization, targets KRAS signaling through disruption of endosomal recycling. Biomed. Rep. 2019, 10, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Ardehed, A.; Johansson, D.; Sundqvist, L.; Schagerstrom, E.; Zagrodzka, Z.; Kovaltchouk, N.A.; Bergstrom, L.; Kautsky, L.; Rafajlovic, M.; Pereyra, R.T.; et al. Divergence within and among seaweed siblings (Fucus vesiculosus and F. radicans) in the Baltic Sea. PLoS ONE 2016, 11, e0161266–e0161281. [Google Scholar] [CrossRef] [Green Version]
- Egan, S.; Harder, T.; Burke, C.; Steinberg, P.; Kjelleberg, S.; Thomas, T. The seaweed holobiont: Understanding seaweed–bacteria interactions. FEMS Microbiol. Rev. 2013, 37, 462–476. [Google Scholar] [CrossRef] [Green Version]
- Vallet, M.; Strittmatter, M.; Murúa, P.; Lacoste, S.; Dupont, J.; Hubas, C.; Genta-Jouve, G.; Gachon, C.M.M.; Kim, G.H.; Prado, S. Chemically-mediated interactions between macroalgae, their fungal endophytes, and protistan pathogens. Front. Microbiol. 2018, 9, 3161. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Parrot, D.; Blümel, M.; Labes, A.; Tasdemir, D. Influence of OSMAC-based cultivation in metabolome and anticancer activity of fungi associated with the brown alga Fucus vesiculosus. Mar. Drugs 2019, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Lateff, A.; Fisch, K.M.; Wright, A.D.; Konig, G.M. A new antioxidant isobenzofuranone derivative from the algicolous marine fungus Epicoccum sp. Planta Med. 2003, 69, 831–834. [Google Scholar] [CrossRef]
- Flewelling, A.J.; Johnson, J.A.; Gray, C.A. Isolation and bioassay screening of fungal endophytes from North Atlantic marine macroalgae. Bot. Mar. 2013, 56, 287–297. [Google Scholar] [CrossRef]
- Flewelling, A.J.; Ellsworth, K.T.; Sanford, J.; Forward, E.; Johnson, J.A.; Gray, C.A. Macroalgal endophytes from the Atlantic coast of Canada: A potential source of antibiotic natural products? Microorganisms 2013, 1, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Zuccaro, A.; Schoch, C.L.; Spatafora, J.W.; Kohlmeyer, J.; Draeger, S.; Mitchell, J.I. Detection and identification of fungi intimately associated with the brown seaweed Fucus serratus. Appl. Environ. Microbiol. 2008, 74, 931–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccaro, A.; Summerbell, R.C.; Gams, W.; Schroers, H.-J.; Mitchell, J.I. A new Acremonium species associated with Fucus spp., and its affinity with a phylogenetically distinct marine Emericellopsis clade. Stud. Mycol. 2004, 50, 283–297. [Google Scholar]
- Fan, B.; Dewapriya, P.; Li, F.; Blümel, M.; Tasdemir, D. Pyrenosetins A-C, new decalinoylspirotetramic acid derivatives isolated by bioactivity-based molecular networking from the seaweed-derived fungus Pyrenochaetopsis sp. FVE-001. Mar. Drugs 2020, 18, 47. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.; Dewapriya, P.; Li, F.; Grauso, L.; Blümel, M.; Mangoni, A.; Tasdemir, D. Pyrenosetin D, a new pentacyclic decalinoyltetramic acid derivative from the algicolous fungus Pyrenochaetopsis sp. FVE-087. Mar. Drugs 2020, 18, 281. [Google Scholar] [CrossRef]
- Flewelling, A.J.; Johnson, J.A.; Gray, C.A. Antimicrobials from the marine algal endophyte Penicillium sp. Nat. Prod. Commun. 2013, 8, 373–374. [Google Scholar] [CrossRef] [Green Version]
- de Gruyter, J.; Woudenberg, J.H.C.; Aveskamp, M.M.; Verkley, G.J.M.; Groenewald, J.Z.; Crous, P.W. Systematic reappraisal of species in Phoma section Paraphoma, Pyrenochaeta and Pleurophoma. Mycologia 2010, 102, 1066–1081. [Google Scholar] [CrossRef]
- da Silva, R.R.; da Rosa, N.G.; de Oliveira, L.C.G.; Juliano, M.A.; Juliano, L.; Rosa, J.C.; Cabral, H. Biochemical properties and catalytic specificity of a novel neutral serine peptidase secreted by fungus Pyrenochaetopsis sp. Appl. Biochem. Biotechnol. 2019, 187, 1158–1172. [Google Scholar] [CrossRef]
- Li, J.; Jiang, H.; Li, L.; Zhang, X.; Chen, J. The effect of disease and season to hepatopancreas and intestinal mycobiota of Litopenaeus vannamei. Front. Microbiol. 2019, 10, 889. [Google Scholar] [CrossRef]
- Gnavi, G.; Garzoli, L.; Poli, A.; Prigione, V.; Burgaud, G.; Varese, G.C. The culturable mycobiota of Flabellia petiolata: First survey of marine fungi associated to a Mediterranean green alga. PLoS ONE 2017, 12, e0175941–e0175960. [Google Scholar] [CrossRef] [PubMed]
- Nogawa, T.; Kato, N.; Shimizu, T.; Okano, A.; Futamura, Y.; Takahashi, S.; Osada, H. Wakodecalines A and B, new decaline metabolites isolated from a fungus Pyrenochaetopsis sp. RK10-F058. J. Antibiot. 2017, 71, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.B.; Park, E.J.; da Silva, R.R.; Kim, H.W.; Dorrestein, P.C.; Sung, S.H. Targeted Isolation of Neuroprotective dicoumaroyl neolignans and lignans from Sageretia theezans using in silico molecular network annotation propagation-based dereplication. J. Nat. Prod. 2018, 81, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Hoye, T.R.; Jeffrey, C.S.; Shao, F. Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat. Protoc. 2007, 2, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Lopez, N.; Cano-Lira, J.F.; Guarro, J.; Sutton, D.A.; Wiederhold, N.; Crous, P.W.; Stchigel, A.M. Coelomycetous Dothideomycetes with emphasis on the families Cucurbitariaceae and Didymellaceae. Stud. Mycol. 2018, 90, 1–69. [Google Scholar] [CrossRef]
- Flores-Bocanegra, L.; Raja, H.A.; Bacon, J.W.; Maldonado, A.C.; Burdette, J.E.; Pearce, C.J.; Oberlies, N.H. Cytotoxic naphthoquinone analogues, including heterodimers, and their structure elucidation using LR-HSQMBC NMR experiments. J. Nat. Prod. 2020, 84, 771–778. [Google Scholar] [CrossRef]
- Jang, J.-H.; Asami, Y.; Jang, J.-P.; Kim, S.-O.; Moon, D.O.; Shin, K.-S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H. Fusarisetin A, an acinar morphogenesis inhibitor from a soil fungus, Fusarium sp. FN080326. J. Am. Chem. Soc. 2011, 133, 6865–6867. [Google Scholar] [CrossRef]
- Zhao, D.; Han, X.; Wang, D.; Liu, M.; Gou, J.; Peng, Y.; Liu, J.; Li, Y.; Cao, F.; Zhang, C. Bioactive 3-decalinoyltetramic acids derivatives from a marine-derived strain of the fungus Fusarium equiseti D39. Front. Microbiol. 2019, 10, 1285. [Google Scholar] [CrossRef]
- Yamada, T.; Kikuchi, T.; Tanaka, R. Altercrasin A, a novel decalin derivative with spirotetramic acid, produced by a sea urchin-derived Alternaria sp. Tetrahedron Lett. 2015, 56, 1229–1232. [Google Scholar] [CrossRef]
- Yamada, T.; Tanaka, A.; Nehira, T.; Nishii, T.; Kikuchi, T. Altercrasins A-E, decalin derivatives, from a sea-urchin-derived Alternaria sp.: Isolation and structural analysis including stereochemistry. Mar. Drugs 2019, 17, 218. [Google Scholar] [CrossRef] [Green Version]
- Pornpakakul, S.; Roengsumran, S.; Deechangvipart, S.; Petsom, A.; Muangsin, N.; Ngamrojnavanich, N.; Sriubolmas, N.; Chaichit, N.; Ohta, T. Diaporthichalasin, a novel CYP3A4 inhibitor from an endophytic Diaporthe sp. Tetrahedron Lett. 2007, 48, 651–655. [Google Scholar] [CrossRef]
- Kato, N.; Nogawa, T.; Takita, R.; Kinugasa, K.; Kanai, M.; Uchiyama, M.; Osada, H.; Takahashi, S. Control of the stereochemical course of [4+2] cycloaddition during trans-decalin formation by Fsa2-family enzymes. Angew. Chem. Int. Ed. 2018, 57, 9754–9758. [Google Scholar] [CrossRef] [PubMed]
- Katajamaa, M.; Miettinen, J.; Orešič, M. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. bioRxiv 2019, 17, 905–908. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Mohimani, H.; Gurevich, A.; Shlemov, A.; Mikheenko, A.; Korobeynikov, A.; Cao, L.; Shcherbin, E.; Nothias, L.-F.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 4035. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Li, Y.; Grauso, L.; Scarpato, S.; Cacciola, N.A.; Borrelli, F.; Zidorn, C.; Mangoni, A. Stable catechol keto tautomers in cytotoxic heterodimeric cyclic diarylheptanoids from the seagrass Zostera marina. Org. Lett. 2021, 23, 7134–7138. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | Pyrenosetin E (5) | Pyrenosetin B (2) | Pyrenosetin F (6) | Pyrenosetin C (3) |
|---|---|---|---|---|
| δH, J in Hz | δH, J in Hz | δH, J in Hz | δH, J in Hz | |
| 1 | - | - | - | - |
| 2 | - | - | - | - |
| 3 | 2.57, d (11.5) | 2.73, d (11.4) | 2.82, d (11.3) | 2.66, d (11.3) |
| 4 | - | - | - | - |
| 5 | 5.22, br s | 5.22, br s | 5.26, br s | 5.28, br s |
| 6 | 1.82, m | 1.82, m | 1.85, m | 1.83, m |
| 7 eq | 1.79, m | 1.80, m | 1.82, m | 1.82, m |
| 7ax | 0.85, q (11.9) | 0.84, m | 0.86, q (12.0) | 0.88, m |
| 8 | 1.42, m | 1.44, m | 1.45, m | 1.44, m |
| 9 eq | 1.72, m | 1.73, m | 1.74, m | 1.73, m |
| 9 ax | 0.97, dq (3.3, 12.2) | 0.93, m | 0.92, q (3.5, 12.4) | 0.99, m |
| 10 eq | 1.43, m | 1.40, m | 1.37, m | 1.41, m |
| 10 ax | 1.05, dq (3.0, 12.5) | 1.07, dq (12.8, 3.4) | 1.08, dq (3.3, 12.6) | 1.04, m |
| 11 | 1.56, dt (2.9, 12.0) | 1.42, m | 1.41, m | 1.64, td (11.0, 2.7) |
| 12 | 0.98, s | 1.00, s | 1.02, s | 1.01, s |
| 13 | 3.42, dd (11.5, 9.3) | 3.26, dd (11.4, 9.4) | 3.40, dd (11.3, 9.5) | 3.57, dd (11.4, 9.8) |
| 14 | 5.82, dd (15.4, 9.3) | 5.97, dd (15.3, 9.4) | 7.09, dd (16.1, 9.5) | 6.85, dd (15.9, 9.8) |
| 15 | 5.55, dd (15.4, 7.5) | 5.50, dd (15.3, 8.0) | 6.02, d (16.1) | 6.18, d (15.9) |
| 16 | 4.17, quintet (6.6) | 4.18, m | - | - |
| 17 | 1.18, d (6.3) | 1.19, d (6.2) | 2.22, s | 2.22, s |
| 18 | 1.70, s | 1.69, br s | 1.69, s | 1.68, br s |
| 19 | 0.89, d (6.5) | 0.91, d (6.5) | 0.91, d (6.5) | 0.90, d (6.2) |
| 2′ | - | - | - | - |
| 3′ | - | - | - | - |
| 4′ | - | - | - | - |
| 5′ | 3.85, m | 3.94, dd (2.7, 1.9) | 4.00, t (2.6) | 3.61, dd (4.9, 2.7) |
| 6′ a | 4.10, br. d (11.7) | 4.08, m | 4.03, d (11.3) | 4.10, m |
| 6′ b | 3.87, m | 3.86, dd (12.4, 2.7) | 3.84, d (11.3) | 3.94, m |
| 7′ | 3.09, s | 3.07, s | 3.06, s | 3.11, s |
| C | Pyrenosetin E (5) δC | Pyrenosetin B (2) δC | Pyrenosetin F (6) δC | Pyrenosetin C (3) δC |
|---|---|---|---|---|
| 1 | 209.6 (C) | 209.8 (C) | 208.5 (C) | 212.1 (C) |
| 2 | 54.3 (C) | 54.1 (C) | 54.1 (C) | 54.7 (C) |
| 3 | 53.4 (CH) | 52.8 (CH) | 52.9 (CH) | 53.6 (CH) |
| 4 | 132.1 (C) | 132.3 (C) | 131.5 (C) | 130.9 (C) |
| 5 | 127.8 (CH) | 127.6 (CH) | 128.4 (CH) | 128.8 (CH) |
| 6 | 37.6 (CH) | 37.6 (CH) | 37.7 (CH) | 37.6 (CH) |
| 7 | 42.0 (CH2) | 42.0 (CH2) | 41.9 (CH2) | 41.8 (CH2) |
| 8 | 33.0 (CH) | 32.9 (CH) | 32.9 (CH) | 32.9 (CH) |
| 9 | 35.4 (CH2) | 35.3 (CH2) | 35.3 (CH2) | 35.2 (CH2) |
| 10 | 25.3 (CH2) | 25.3 (CH2) | 25.3 (CH2) | 25.2 (CH2) |
| 11 | 37.8 (CH) | 38.0 (CH) | 38.0 (CH) | 37.4 (CH) |
| 12 | 15.4 (CH3) | 15.2 (CH3) | 15.3 (CH3) | 15.2 (CH3) |
| 13 | 49.4 (CH) | 51.0 (CH) | 50.0 (CH) | 50.6 (CH) |
| 14 | 131.2 (CH) | 130.5 (CH) | 146.1 (CH) | 144.4 (CH) |
| 15 | 136.8 (CH) | 137.8 (CH) | 134.1 (CH) | 133.9 (CH) |
| 16 | 69.0 (CH) | 69.0 (CH) | 198.8 (C) | 197.6 (C) |
| 17 | 22.8 (CH3) | 22.9 (CH3) | 26.8 (CH3) | 27.6 (CH3) |
| 18 | 23.9 (CH3) | 23.9 (CH3) | 23.8 (CH3) | 23.7 (CH3) |
| 19 | 22.4 (CH3) | 22.4 (CH3) | 22.4 (CH3) | 22.4 (CH3) |
| 2′ | 169.1 (C) | 168.6 (C) | 167.7 (C) | 167.8 (C) |
| 3′ | 74.4 (C) | 73.8 (C) | 73.7 (C) | 72.7 (C) |
| 4′ | 204.8 (C) | 205.0 (C) | 204.1 (C) | 206.4 (C) |
| 5′ | 68.8 (CH) | 69.4 (CH) | 69.1 (CH) | 69.8 (CH) |
| 6′ | 57.9 (CH2) | 58.3 (CH2) | 58.4 (CH2) | 60.3 (CH2) |
| 7′ | 28.0 (CH3) | 27.7 (CH3) | 27.8 (CH3) | 28.5 (CH3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, B.; Grauso, L.; Li, F.; Scarpato, S.; Mangoni, A.; Tasdemir, D. Application of Feature-Based Molecular Networking for Comparative Metabolomics and Targeted Isolation of Stereoisomers from Algicolous Fungi. Mar. Drugs 2022, 20, 210. https://doi.org/10.3390/md20030210
Fan B, Grauso L, Li F, Scarpato S, Mangoni A, Tasdemir D. Application of Feature-Based Molecular Networking for Comparative Metabolomics and Targeted Isolation of Stereoisomers from Algicolous Fungi. Marine Drugs. 2022; 20(3):210. https://doi.org/10.3390/md20030210
Chicago/Turabian StyleFan, Bicheng, Laura Grauso, Fengjie Li, Silvia Scarpato, Alfonso Mangoni, and Deniz Tasdemir. 2022. "Application of Feature-Based Molecular Networking for Comparative Metabolomics and Targeted Isolation of Stereoisomers from Algicolous Fungi" Marine Drugs 20, no. 3: 210. https://doi.org/10.3390/md20030210
APA StyleFan, B., Grauso, L., Li, F., Scarpato, S., Mangoni, A., & Tasdemir, D. (2022). Application of Feature-Based Molecular Networking for Comparative Metabolomics and Targeted Isolation of Stereoisomers from Algicolous Fungi. Marine Drugs, 20(3), 210. https://doi.org/10.3390/md20030210