
Scalarane Sesterterpenoids Isolated from the Marine Sponge Hyrtios erectus and their Cytotoxicity


Abstract

1. Introduction
2. Results
2.1. Isolation of Compounds from Hyrtios Erectus
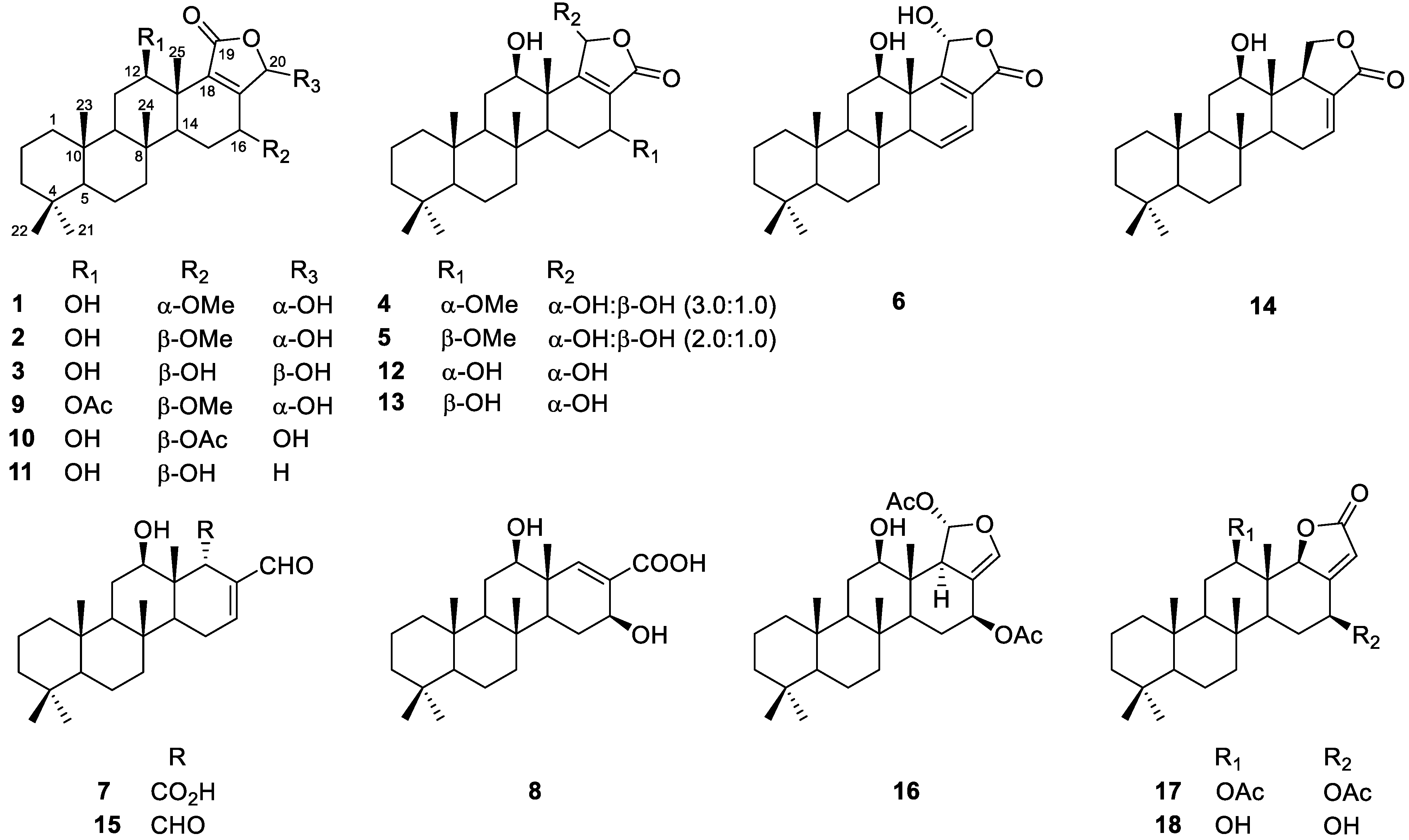
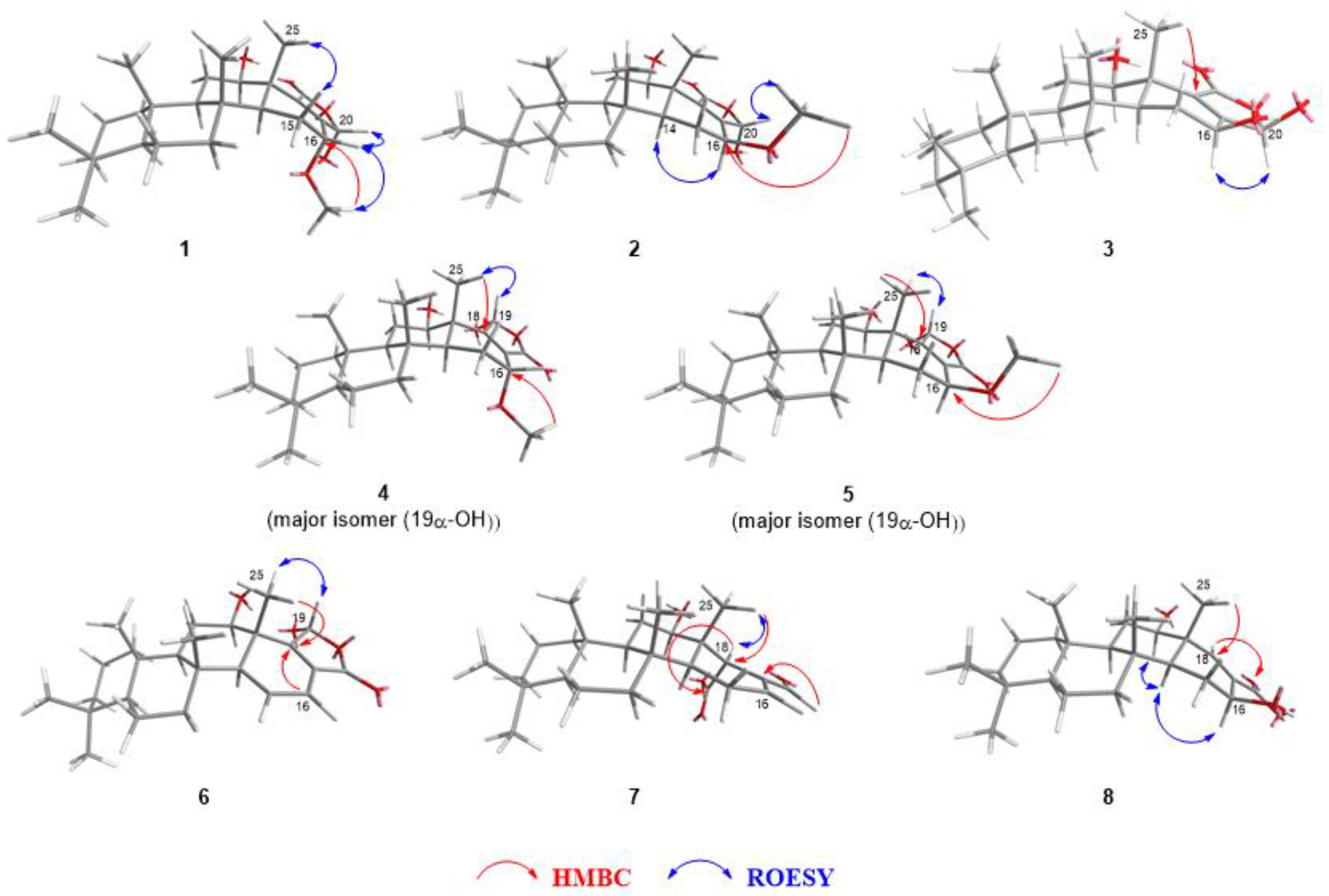
2.2. Identification of Isolated Compounds
2.3. Cytotoxicity of Isolated Scalarane Derivatives
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Sponge Material
4.3. Extraction and Isolation
4.4. Biological Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, K.; Gustafson, K.R. Sesterterpenoids: Chemistry, biology, and biosynthesis. Nat. Prod. Rep. 2021, 38, 1251–1281. [Google Scholar] [CrossRef]
- Yu, H.-B.; Chen, H.-Y.; Duan, S.; Zhu, Y.-P.; Hu, B.; He, Y.; Cheng, S.-T.; Jiao, B.-H.; Liu, X.-Y. Bioactive scalarane-type sesterterpenoids from marine sources. Chem. Biodivers. 2022, 19, e202200049. [Google Scholar] [CrossRef]
- Máximo, P.; Lourenço, A. Marine sesterterpenes: An overview. Curr. Org. Chem. 2018, 22, 2381–2393. [Google Scholar] [CrossRef]
- Fattorusso, E.; Magno, S.; Santacroce, C.; Sica, D. Scalarin, a new pentacyclic C-25 terpenoid from the sponge Cacospongia scalaris. Tetrahedron 1972, 28, 5993–5997. [Google Scholar] [CrossRef]
- Gonzalez, M.A. Scalarane sesterterpenoids. Curr. Bioact. Compd. 2010, 6, 178–206. [Google Scholar] [CrossRef]
- Roy, M.C.; Tanaka, J.; de Voogd, N.; Higa, T. New scalarane class sesterterpenes from an Indonesian sponge, Phyllospongia sp. J. Nat. Prod. 2002, 65, 1838–1842. [Google Scholar] [CrossRef]
- De Rosa, S.; Puliti, R.; Crispino, A.; de Giulio, A.; Mattia, C.A.; Mazzarella, L. A new scalarane sesterterpenoid from the marine sponge Cacospongia mollior. J. Nat. Prod. 1994, 57, 256–262. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, L.; Huang, X.; Guo, Y.; Lou, L. Scalaradial inhibition of epidermal growth factor receptor-mediated Akt phosphorylation is independent of secretory phospholipase A2. J. Pharm. Exp. Ther. 2005, 314, 1210–1217. [Google Scholar] [CrossRef]
- Zhou, M.; Peng, B.-R.; Tian, W.; Su, J.-H.; Wang, G.; Lin, T.; Zeng, D.; Sheu, J.-H.; Chen, H. 12-Deacetyl-12-epi-scalaradial, a scalarane sesterterpenoid from a marine sponge Hippospongia sp., induces HeLa cells apoptosis via MAPK/ERK pathway and modulates nuclear receptor Nur77. Mar. Drugs 2020, 18, 375. [Google Scholar] [CrossRef]
- Lai, K.-H.; Liu, Y.-C.; Su, J.-H.; El-Shazly, M.; Wu, C.-F.; Du, Y.-C.; Hsu, Y.-M.; Yang, J.-C.; Weng, M.-K.; Chou, C.-H.; et al. Antileukemic scalarane sesterterpenoids and meroditerpenoid from Carteriospongia (Phyllospongia) sp., induce apoptosis via dual inhibitory effects on topoisomerase II and Hsp90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef]
- Morarescu, O.; Grinco, M.; Kulciţki, V.; Shova, S.; Ungur, N. An alternative approach towards C-12 functionalized scalaranic sesterterpenoids synthesis of 17-oxo-20-norscalaran-12α,19-O-lactone. Mar. Drugs 2021, 19, 636. [Google Scholar] [CrossRef]
- Fan, W.-Y.; Wang, Z.-L.; Li, H.-C.; Fossey, J.S.; Deng, W.-P. A straightforward and efficient synthetic access to biologically active marine sesterterpenoids, sesterstatins 4 and 5. Chem. Comm. 2011, 47, 2961–2963. [Google Scholar] [CrossRef]
- Chen, X.-B.; Yuan, Q.-J.; Wang, J.; Hua, S.-K.; Ren, J.; Zeng, B.-B. Synthesis of the scalarane sesterterpenoid 16-deacetoxy-12-epi-scalarafuranacetate. J. Org. Chem. 2011, 76, 7216–7221. [Google Scholar] [CrossRef]
- Kamel, H.N.; Kim, Y.B.; Rimoldi, J.M.; Fronczek, F.R.; Ferreira, D.; Slattery, M. Scalarane sesterterpenoids: Semisynthesis and biological activity. J. Nat. Prod. 2009, 72, 1492–1496. [Google Scholar] [CrossRef]
- Ungur, N.; Kulciţki, V. Synthetic paths towards scalaranes: Assembling the scalaranic skeleton and further transformations. Phytochem. Rev. 2004, 3, 401–415. [Google Scholar] [CrossRef]
- Shady, N.H.; El-Hossary, E.M.; Fouad, M.A.; Gulder, T.A.M.; Kamel, M.S.; Abdelmohsen, U.R. Bioactive natural products of marine sponges from the genus Hyrtios. Molecules 2017, 22, 781–801. [Google Scholar] [CrossRef]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Takuma, S. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Bourguet-Kondracki, M.L.; Martin, M.T.; Guyot, M. A new β-carboline alkaloid isolated from the marine sponge Hyrtios erecta. Tetrahedron Lett. 1996, 37, 3457–3460. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Melody, N.; Cichacz, Z.A.; Herald, D.L.; Hoard, M.S.; Pettit, R.K.; Chapuis, J.C. Antineoplastic agents. 397: Isolation and structure of sesterstatins 4 and 5 from Hyrtios erecta (the Republic of Maldives). Bioorg. Med. Chem. Lett. 1998, 8, 2093–2098. [Google Scholar] [CrossRef]
- Miyaoka, H.; Nishijima, S.; Mitome, H.; Yamada, Y. Three new scalarane sesterterpenoids from the Okinawan sponge Hyrtios erectus. J. Nat. Prod. 2000, 63, 1369–1372. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Cichacz, Z.A. Antineoplastic agents. 542. Isolation and structure of sesterstatin 6 from the Indian Ocean sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1253–1255. [Google Scholar] [CrossRef]
- Elhady, S.S.; Al-Abd, A.M.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. Antiproliferative scalarane-based metabolites from the Red Sea sponge Hyrtios erectus. Mar. Drugs 2016, 14, 130. [Google Scholar] [CrossRef]
- Pettit, G.R.; Cichacz, Z.A.; Tan, R.; Herald, D.L.; Melody, N.; Hoard, M.S.; Doubek, D.L.; Hooper, J.N.A. Antineoplastic agents-385. The isolation and structure of a scalarane-type sesterterpene from the Indian Ocean porifera Hyrtios erecta. Collect. Czech. Chem. C 1998, 63, 1671–1677. [Google Scholar] [CrossRef]
- Mahidol, C.; Prawat, H.; Sangpetsiripan, S.; Ruchirawat, S. Bioactive scalaranes from the Thai sponge Hyrtios gumminae. J. Nat. Prod. 2009, 72, 1870–1874. [Google Scholar] [CrossRef]
- Walker, R.P.; Thompson, J.E.; Faulkner, D.J. Sesterterpenes from Spongia idia. J. Org. Chem. 1980, 45, 4976–4979. [Google Scholar] [CrossRef]
- Crews, P.; Bescansa, P. Sesterterpenes from a common marine sponge, Hyrtios erecta. J. Nat. Prod. 1986, 49, 1041–1052. [Google Scholar] [CrossRef]
- Lu, Q.; Faulkner, D.J. Two new sesterterpenoids and a new 9,11-secosterol from Spongia matamata. J. Nat. Prod. 1997, 60, 195–198l. [Google Scholar] [CrossRef]
- Ryu, G.; Matsunaga, S.; Fusetani, N. Three new cytotoxic sesterterpenes from the marine sponge Hyrtios cf. erectus. J. Nat. Prod. 1996, 59, 515–517. [Google Scholar] [CrossRef]
- Sun, J.-B.; Hong, L.-L.; Shang, R.-Y.; Liu, H.-Y.; Zhang, L.; Liu, L.-Y.; Zhao, L.; Zhang, W.; Sun, F.; Jiao, W.-H.; et al. Dysiscalarones A-E, scalarane sesterterpenoids with nitric oxide production inhibitory activity from marine sponge Dysidea granulosa. Bioorg. Chem. 2021, 111, 104791–104799. [Google Scholar] [CrossRef]
- Peng, B.-R.; Lai, K.-H.; Lee, G.-H.; Yu, S.S.-F.; Duh, C.-Y.; Su, J.-H.; Zheng, L.-G.; Hwang, T.-L.; Sung, P.-J. Scalarane-type sesterterpenoids from the marine sponge Lendenfeldia sp. alleviate inflammation in human neutrophils. Mar. Drugs 2021, 19, 561–577. [Google Scholar] [CrossRef]
- Jaspars, M.; Jackson, E.; Lobkovsky, E.; Clardy, J.; Diaz, M.C.; Crews, P. Using scalarane sesterterpenes to examine a sponge taxonomic anomaly. J. Nat. Prod. 1997, 60, 556–561. [Google Scholar] [CrossRef]
- Tsuchiya, N.; Sato, A.; Hata, T.; Sato, N.; Sasagawa, K.; Kobayashi, T. Cytotoxic scalarane sesterterpenes from a sponge, Hyrtios erecta. J. Nat. Prod. 1998, 61, 468–473. [Google Scholar] [CrossRef]
- Jeon, J.-E.; Bae, J.; Lee, K.J.; Oh, K.-B.; Shin, J. Scalarane sesterterpenes from the sponge Hyatella sp. J. Nat. Prod 2011, 74, 847–851. [Google Scholar] [CrossRef]
- Hernández-Guerrero, C.J.; Zubía, E.; Ortega, M.J.; Carballo, J.L. Sesterterpene metabolites from the sponge Hyatella intestinalis. Tetrahedron 2006, 62, 5392–5400. [Google Scholar] [CrossRef]
- Valter, R.E. Ring–chain isomeric conversions of aldehydo-carboxylic and keto-carboxylic acids and their derivatives. Russ. Chem. Rev. 1973, 42, 464–476. [Google Scholar] [CrossRef]
- Thuring, J.W.J.F.; Klunder, A.J.H.; Nefkens, G.H.L.; Wegman, M.A.; Zwanenburg, B. Lipase catalyzed dynamic kinetic resolution of some 5-hydroxy-2(5H)-furanones. Tetrahedron Lett. 1996, 37, 4759–4760. [Google Scholar] [CrossRef][Green Version]
- Van der Deen, H.; Cuiper, A.D.; Hof, R.P.; van Oeveren, A.; Feringa, B.L.; Kellogg, R.M. Lipase-catalyzed second-order asymmetric transformations as resolution and synthesis strategies for chiral 5-(acyloxy)-2(5H)-furanone and pyrrolinone synthons. J. Am. Chem. Soc. 1996, 118, 3801–3803. [Google Scholar] [CrossRef][Green Version]
- Poskonin, V.V.; Badovskaya, L.A. Unusual conversion of 5-hydroxy-2(5H)furanone in aqueous solution. Chem. Heterocycl. Compd. 2003, 39, 594–597. [Google Scholar] [CrossRef]
- Tilvi, S.; Khan, S.; Majik, S.M. γ-Hydroxybutenolide containing marine natural products and their synthesis: A review. Curr. Org. Chem. 2019, 23, 2436–2468. [Google Scholar] [CrossRef]
- Wonganuchitmeta, S.-n.; Yuenyongsawad, S.; Keawpradub, N.; Plubrukarn, A. Antitubercular Sesterterpenes from the Thai Sponge Brachiaster sp. J. Nat. Prod. 2004, 67, 1767–1770. [Google Scholar] [CrossRef]
- Kittiwisut, S.; Rohena, C.C.; Yuenyongsawad, S.; Mooberry, S.L.; Plubrukarn, A. Antiproliferative effects of 12-oxoheteronemin vs heteronemin. Nat. Prod. Commun. 2014, 9, 359–360. [Google Scholar] [CrossRef]
- Cheng, J.; Nakano, C.; Shi, G.L.; Hoshino, T. Further insight into polycyclization cascades of acyclic geranylfarnesol and its acetate by squalene-hopene cyclase from Alicyclobacillus acidocaldarius. Nat. Prod. Commun. 2016, 11, 163–167. [Google Scholar] [CrossRef]
- Shinozaki, J.; Shibuya, M.; Ebizuka, Y.; Masuda, K. Cyclization of all-E- and 2Z-geranylfarnesols by a bacterial triterpene synthase: Insight into sesterterpene biosynthesis in Aleuritopteris Ferns. Biosci. Biotechnol. Biochem. 2013, 77, 2278–2282. [Google Scholar] [CrossRef][Green Version]
- Christianson, D.W. Structural and chemical biology of terpenoid cyclases. Chem. Rev. 2017, 117, 11570–11648. [Google Scholar] [CrossRef]
- Santana-Molina, C.; Rivas-Marin, E.; Rojas, A.M.; Devos, D.P. Origin and evolution of polycyclic triterpene synthesis. Mol. Biol. Evol. 2020, 37, 1925–1941. [Google Scholar] [CrossRef]
- Syrén, P.-O.; Henche, S.; Eichler, A.; Nestl, B.M.; Hauer, B. Squalene-hopene cyclases—Evolution, dynamics and catalytic scope. Curr. Opin. Struct. 2016, 41, 73–82. [Google Scholar] [CrossRef]
- Siedenburg, G.; Jendrossek, D. Squalene-hopene cyclases. Appl. Environ. Microbiol. 2011, 77, 3905–3915. [Google Scholar] [CrossRef]
- Kwon, O.S.; Kim, D.; Kim, C.K.; Sun, J.; Sim, C.J.; Oh, D.C.; Lee, S.K.; Oh, K.B.; Shin, J. Cytotoxic scalarane sesterterpenes from the sponge Hyrtios erectus. Mar. Drugs 2020, 18, 253–270. [Google Scholar] [CrossRef]
- Keller, C. Die Spongienfauna des rothen Meeres (I. Hälfte). Z. Wiss. Zool. 1889, 48, 311–405. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | 3 b | |||
|---|---|---|---|---|---|---|
| δCc, Type | δH (J in Hz) | δCc, Type | δH (J in Hz) | δCc, Type | δH (J in Hz) | |
| 1 | 39.7, CH2 | 1.65, m | 40.0, CH2 | 1.72, m | 40.3, CH2 | 1.61, d (13.2) |
| 0.70, m d | 0.77, m d | 0.73, m | ||||
| 2 | 18.2, CH2 | 1.53, m d | 18.7, CH2 | 1.59, m d | 19.3, CH2 | 1.54, m |
| 1.36, m d | 1.42, m d | 1.35, m d | ||||
| 3 | 42.1, CH2 | 1.30, m | 42.3, CH2 | 1.38, d (13.8) | 42.8, CH2 | 1.33, m d |
| 1.06, m | 1.09, m d | 1.13, dt (13.8, 4.2) | ||||
| 4 | 33.3, C | 33.4, C | 33.8, C | |||
| 5 | 56.6, CH | 0.70, m d | 56.9, CH | 0.77, m d | 57.1, CH | 0.71, m |
| 6 | 18.6, CH2 | 1.53, m d | 18.3, CH2 | 1.59, m d | 18.9, CH2 | 1.44, m |
| 1.36, m d | 1.42, m d | 1.28, m | ||||
| 7 | 42.7, CH2 | 1.76, m d | 41.8, CH2 | 1.73, m | 41.9, CH2 | 1.68, d (12.6) |
| 0.86, m | 0.91, m | 0.63, m d | ||||
| 8 | 36.8, C | 37.6, C | 37.6, C | |||
| 9 | 57.8, CH | 0.84, m d | 58.1, CH | 0.85, m | 58.2, CH | 0.65, d (12.6) |
| 10 | 37.4, C | 37.3, C | 37.9, C | |||
| 11 | 25.5, CH2 | 1.76, m d | 25.6, CH2 | 1.83, m | 26.6, CH2 | 1.96, m d |
| 1.42, m | 1.49, m | 1.62, d (10.8) | ||||
| 12 | 75.0, CH | 3.61, dd (10.8, 4.2) | 75.2, CH | 3.60, dd (10.8, 4.2) | 76.2, CH | 3.65, dd (10.8, 4.2) |
| 13 | 41.4, C | 42.8, C | 43.7, C | |||
| 14 | 49.7, CH | 1.22, m | 54.0, CH | 1.09, md | 54.9, CH | 1.00, d (12.0) |
| 15 | 21.7, CH2 | 2.06, d (14.4) | 23.6, CH2 | 2.30, dd (12.0, 7.2) | 28.8, CH2 | 2.39, dd (12.6, 7.2) |
| 1.54, m | 1.53, m | 1.99, m d | ||||
| 16 | 69.7, CH | 3.98, d (4.2) | 74.5, CH | 4.10, dd (10.2, 7.2) | 66.4, CH | 5.12, dd (10.2, 7.2) |
| 17 | 158.3, C | 160.0, C | 165.3, C | |||
| 18 | 140.8, C | 140.0, C | 139.2, C | |||
| 19 | 173.7, C | 172.7, C | 174.8, C | |||
| 20 | 97.7, CH | 6.06, s | 96.9, CH | 6.15, s | 99.4, CH | 6.96, s |
| 21 | 33.3, CH3 | 0.78, s | 33.5, CH3 | 0.84, s d | 33.9, CH3 | 0.87, s |
| 22 | 21.3, CH3 | 0.75, s | 21.4, CH3 | 0.81, s | 21.8, CH3 | 0.80, s |
| 23 | 15.9, CH3 | 0.78, s | 16.1, CH3 | 0.84, sd | 16.5, CH3 | 0.77, s |
| 24 | 17.2, CH3 | 0.83, s | 17.5, CH3 | 0.90, s | 17.8, CH3 | 0.85, s |
| 25 | 14.7, CH3 | 1.02, s | 16.5, CH3 | 1.15, s | 17.2, CH3 | 1.34, s |
| 16-OMe | 57.4, CH3 | 3.35, s | 57.7, CH3 | 3.47, s | ||
| Position | 4 | 5 | ||
|---|---|---|---|---|
| δCb, Type | δH (J in Hz) | δC b, Type | δH (J in Hz) | |
| 1 | 39.9, CH2 | 1.60, m c | 40.0, CH2 | 1.68, m |
| 1.43, m c | 0.78, m c | |||
| 2 | 18.3/18.7 d, CH2 | 1.60, m c | 18.7, CH2 | 1.57, m c |
| 1.43, m c | 1.42, m c | |||
| 3 | 42.2, CH2 | 1.37, m | 42.1, CH2 | 1.37, m |
| 1.15, m | 1.13, m | |||
| 4 | 33.4, C | 33.3, C | ||
| 5 | 56.6, CH | 0.80, m c | 56.7, CH | 0.78, m c |
| 6 | 18.7/18.3 d, CH2 | 1.68, d (12.6) | 18.3, CH2 | 1.57, m c |
| 0.80, m c | 1.42, m c | |||
| 7 | 41.5, CH2 | 1.79, d (12.6) | 41.5, CH2 | 1.82, m |
| 0.94, d (12.6) c | 0.90, m | |||
| 8 | 37.6, C | 37.5, C | ||
| 9 | 58.7, CH | 0.94, d (12.6) c | 58.7, CH | 0.87, m c |
| 10 | 37.0, C | 37.6, C | ||
| 11 | 25.8, CH2 | 1.82, m | 26.0, CH2 | 1.79, m |
| 1.55, d (12.6) | 1.52, m | |||
| 12 | 74.1, CH | 3.80, dd (10.8, 4.5) | 74.4, CH | 3.80, dd (11.4, 4.2) |
| 13 | 45.3, C | 44.7, C | ||
| 14 | 49.0, CH | 1.44, m | 53.4, CH | 1.42, mc |
| 15 | 22.5, CH2 | 2.06, d (15.0) | 24.6, CH2 | 2.25, m |
| 1.50, dd (13.5, 3.9) | 1.63, m | |||
| 16 | 69.2, CH | 4.00, d (3.6) | 73.8, CH | 4.09, dd (9.3, 7.5) |
| 17 | 126.9, C | 128.6, C | ||
| 18 | 170.7, C | 169.1, C | ||
| 19 | 95.9, CH | 6.12, d (1.2) | 95.4, CH | 6.18, s |
| 20 | 170.4, C | 169.2, C | ||
| 21 | 33.4, CH3 | 0.81, s | 33.4, CH3 | 0.84, s c |
| 22 | 21.4, CH3 | 0.85, s c | 21.4, CH3 | 0.81, s |
| 23 | 16.3, CH3 | 0.85, s c | 16.7, CH3 | 0.84, s c |
| 24 | 17.9, CH3 | 0.90, s | 17.6, CH3 | 0.91, s |
| 25 | 15.4, CH3 | 1.11, s | 16.6, CH3 | 1.21, s |
| 16-OMe | 57.7, CH3 | 3.46, s | 58.0, CH3 | 3.54, s |
| Position | 6 | 7 | 8 | |||
|---|---|---|---|---|---|---|
| δCb, Type | δH (J in Hz) | δCb, Type | δH (J in Hz) | δCb, Type | δH (J in Hz) | |
| 1 | 40.2, CH2 | 1.55, m c | 41.9, CH2 | 1.63, d (13.2) | 40.4, CH2 | 1.60, d (13.2) |
| 0.66, m | 0.89, m | 0.72, m | ||||
| 2 | 19.3, CH2 | 1.55, m c | 28.1, CH2 | 1.75, t (11.4) c | 19.4, CH2 | 1.54, m |
| 1.33, m c | 1.40, m | 1.33, m c | ||||
| 3 | 42.7, CH2 | 1.33, m c | 40.1, CH2 | 1.51, m c | 41.7, CH2 | 1.73, d (13.2) |
| 1.10, m | 0.48, t (9.6) | 0.76, m c | ||||
| 4 | 33.8, C | 33.7, C | 38.1, C | |||
| 5 | 57.0, CH | 0.68, m | 56.5, CH | 0.55, d (12.6) | 57.2, CH | 0.76, m c |
| 6 | 18.6, CH2 | 1.47, m | 19.3, CH2 | 1.28, m c | 19.0, CH2 | 1.47, d (13.8) |
| 1.35, m | 1.33, m c | |||||
| 7 | 41.3, CH2 | 1.84, d (12.6) | 42.8, CH2 | 1.31, m c | 42.8, CH2 | 1.36, m |
| 0.74, m | 1.16, dd (13.2, 9.6) | |||||
| 8 | 37.9, C | 38.2, C | 38.0, C | |||
| 9 | 58.1, CH | 0.81, m | 58.7, CH | 0.95, m | 59.4, CH | 0.86, m |
| 10 | 33.9, C | 37.8, C | 34.0, C | |||
| 11 | 28.1, CH2 | 1.96, d (11.4) | 28.0, CH2 | 1.96, dd (11.6, 3.6) | 27.9, CH2 | 2.01, m |
| 1.74, q (12.0) | 1.75, t (11.4) c | 1.79, m | ||||
| 12 | 72.8, CH | 4.57, dd (11.4, 4.2) | 76.6, CH | 4.24, dd (11.4, 3.6) | 77.0, CH | 3.74, dd (10.8, 3.6) |
| 13 | 44.9, C | 42.5, C | 43.9, C | |||
| 14 | 57.9, CH | 2.19, brs | 47.8, CH | 2.29, m c | 53.1, CH | 1.17, d (12.6) |
| 15 | 133.6, CH | 6.11, dd (9.6, 1.8) | 25.4, CH2 | 2.48, m | 28.1, CH2 | 2.37, dd (12.6, 7.2) |
| 2.29, m c | 2.01, m | |||||
| 16 | 118.4, CH | 6.57, dd (9.6, 2.4) | 153.7, CH | 7.07, m | 69.4, CH | 5.13, dd (8.7, 7.5) |
| 17 | 126.3, C | 140.1, C | 133.0, C | |||
| 18 | 167.3, C | 52.1, CH | 4.27, s | 149.7, CH | 7.94, s | |
| 19 | 99.1, CH | 7.02, s | 176.6, C | 172.0, C | ||
| 20 | 170.8, C | 194.4, CH | 9.71, s | 33.9, CH3 | 0.88, s | |
| 21 | 33.8, CH3 | 0.85, s | 33.8, CH3 | 0.81, s c | 22.0, CH3 | 0.81, s c |
| 22 | 21.9, CH3 | 0.80, s c | 22.0, CH3 | 0.77, s | 16.9, CH3 | 0.81, s c |
| 23 | 16.6, CH3 | 0.80, s c | 17.3, CH3 | 0.81, s c | 18.2, CH3 | 0.91, s |
| 24 | 19.7, CH3 | 1.01, s | 17.6, CH3 | 0.93, s | 16.9, CH3 | 1.40, s |
| 25 | 12.5, CH3 | 1.26, s | 16.8, CH3 | 1.13, s | ||
| Compound | Cell Line | Compounds | Cell Line | ||
|---|---|---|---|---|---|
| HeLa | MCF-7 | HeLa | MCF-7 | ||
| 1 | 53.4 | 27.3 | 11 | >80.0 | 49.4 |
| 2 | 46.2 | 26.2 | 12 | 41.9 | 24.3 |
| 3 | 60.1 | 29.9 | 13 | 65.2 | 40.8 |
| 4 | 61.3 | 45.9 | 14 | 58.4 | 30.9 |
| 5 | 70.7 | 76.4 | 15 | >80.0 | 51.3 |
| 6 | 59.3 | 33.8 | 16 | 46.3 | 20.0 |
| 7 | >80.0 | >80.0 | 17 | 46.9 | 36.5 |
| 8 | >80.0 | >80.0 | 18 | >80.0 | >80.0 |
| 9 | >80.0 | 43.8 | |||
| 10 | 45.7 | 27.7 | staurosporine | 0.18 | 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, H.N.K.; Kim, M.J.; Lee, Y.-J. Scalarane Sesterterpenoids Isolated from the Marine Sponge Hyrtios erectus and their Cytotoxicity. Mar. Drugs 2022, 20, 604. https://doi.org/10.3390/md20100604
Tran HNK, Kim MJ, Lee Y-J. Scalarane Sesterterpenoids Isolated from the Marine Sponge Hyrtios erectus and their Cytotoxicity. Marine Drugs. 2022; 20(10):604. https://doi.org/10.3390/md20100604
Chicago/Turabian StyleTran, Huynh Nguyen Khanh, Min Jin Kim, and Yeon-Ju Lee. 2022. "Scalarane Sesterterpenoids Isolated from the Marine Sponge Hyrtios erectus and their Cytotoxicity" Marine Drugs 20, no. 10: 604. https://doi.org/10.3390/md20100604
APA StyleTran, H. N. K., Kim, M. J., & Lee, Y.-J. (2022). Scalarane Sesterterpenoids Isolated from the Marine Sponge Hyrtios erectus and their Cytotoxicity. Marine Drugs, 20(10), 604. https://doi.org/10.3390/md20100604