
Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities



Abstract
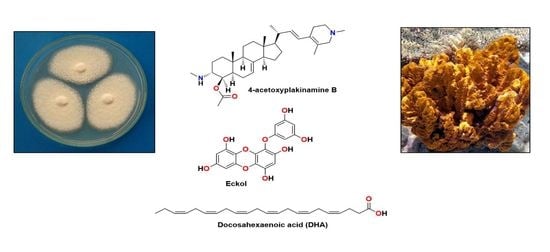
:
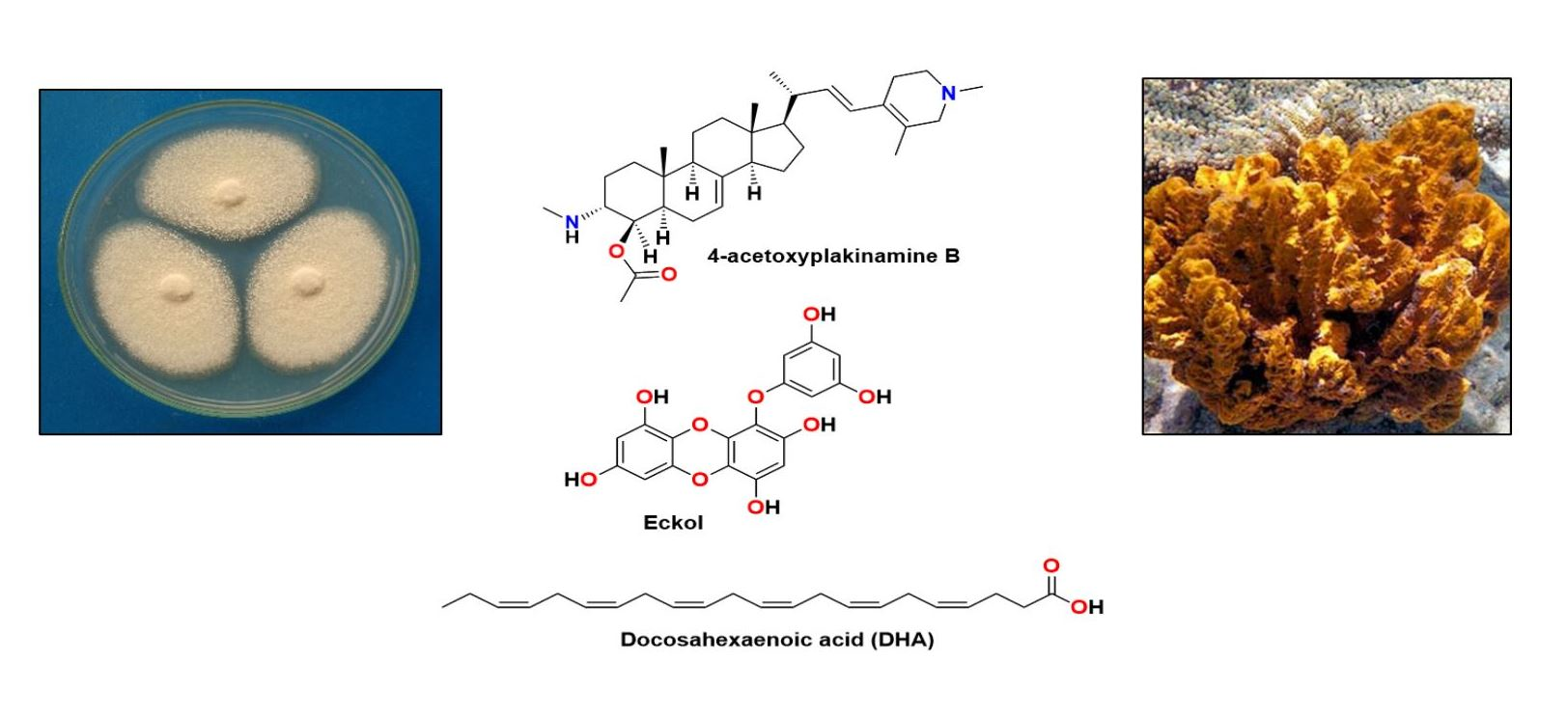{kind=link}
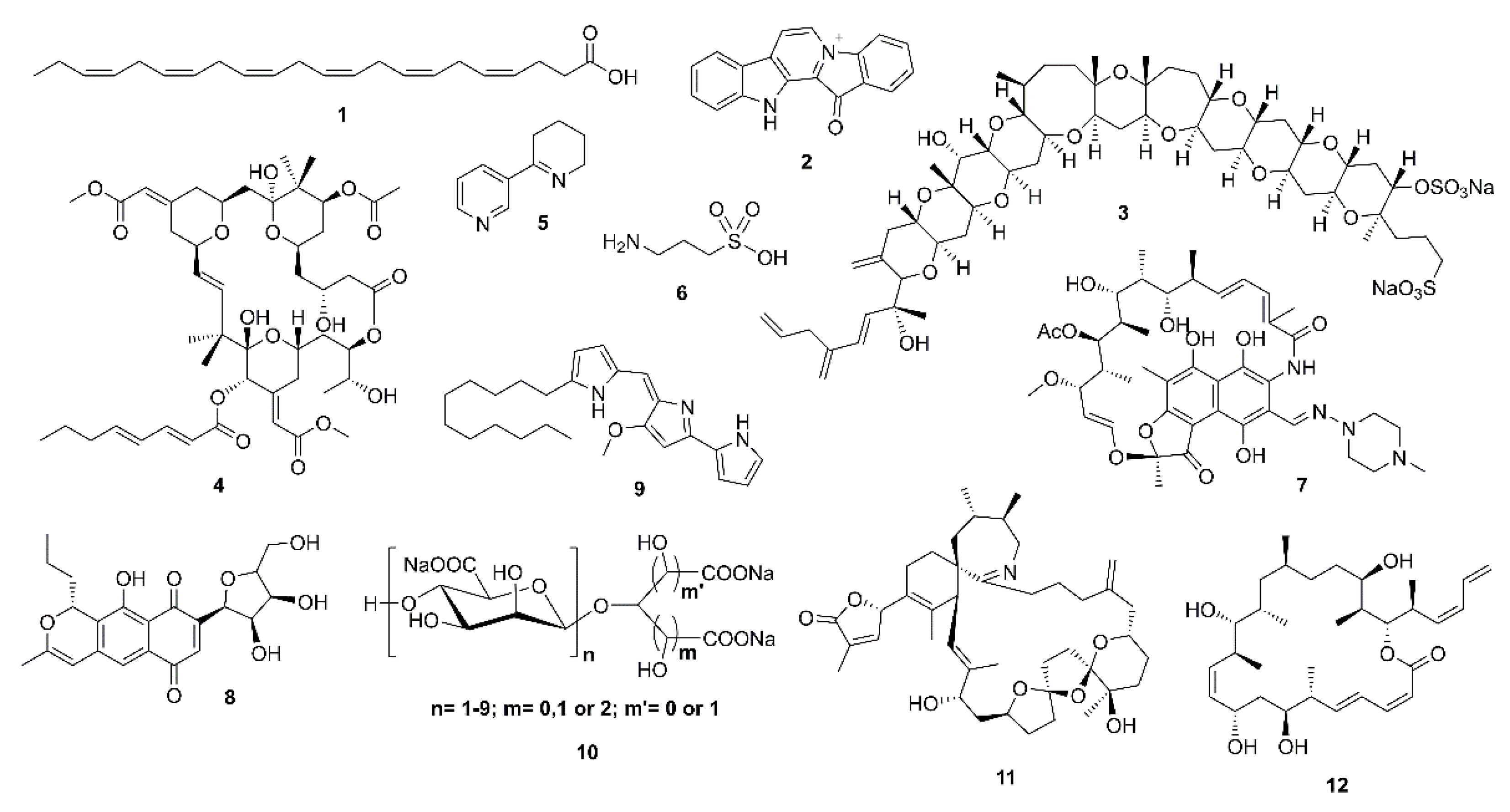{kind=link}
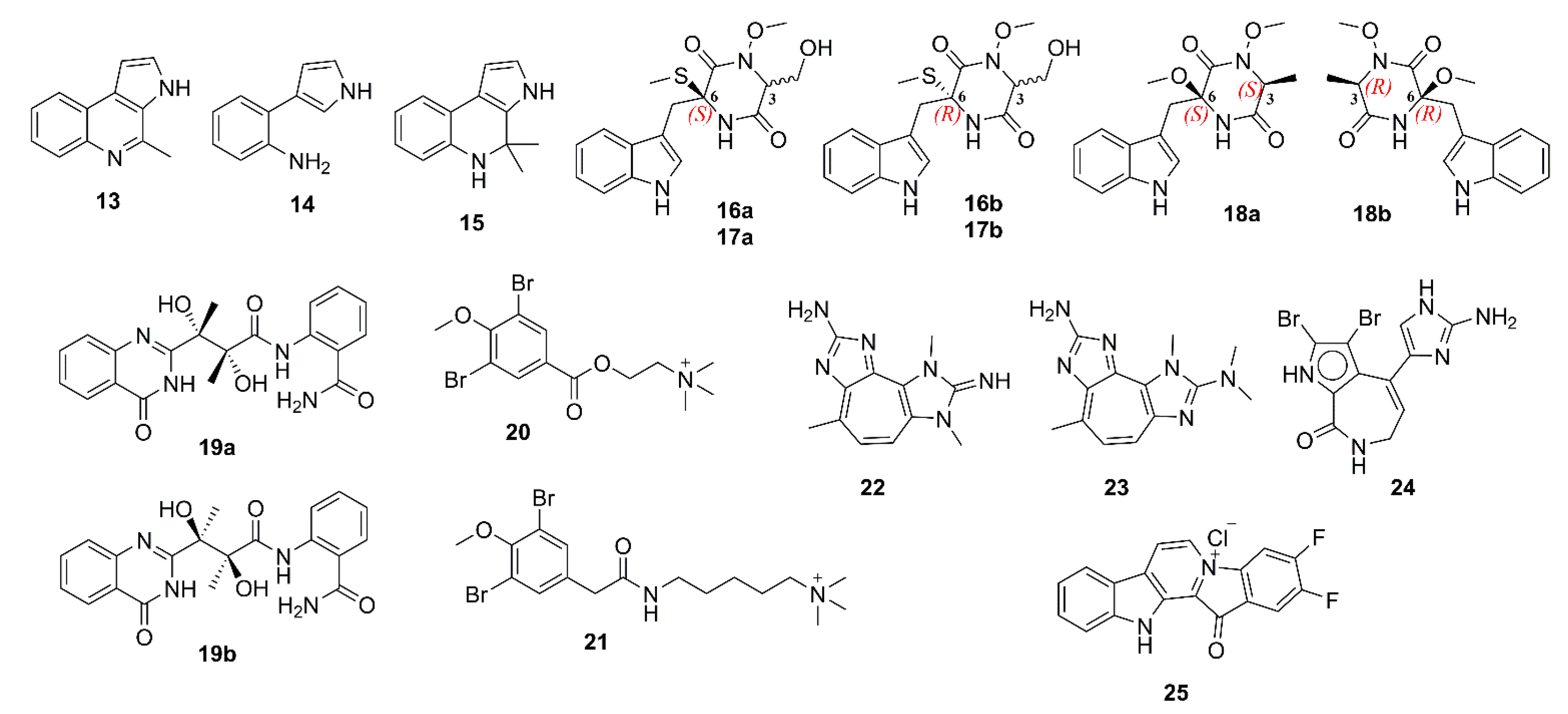{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
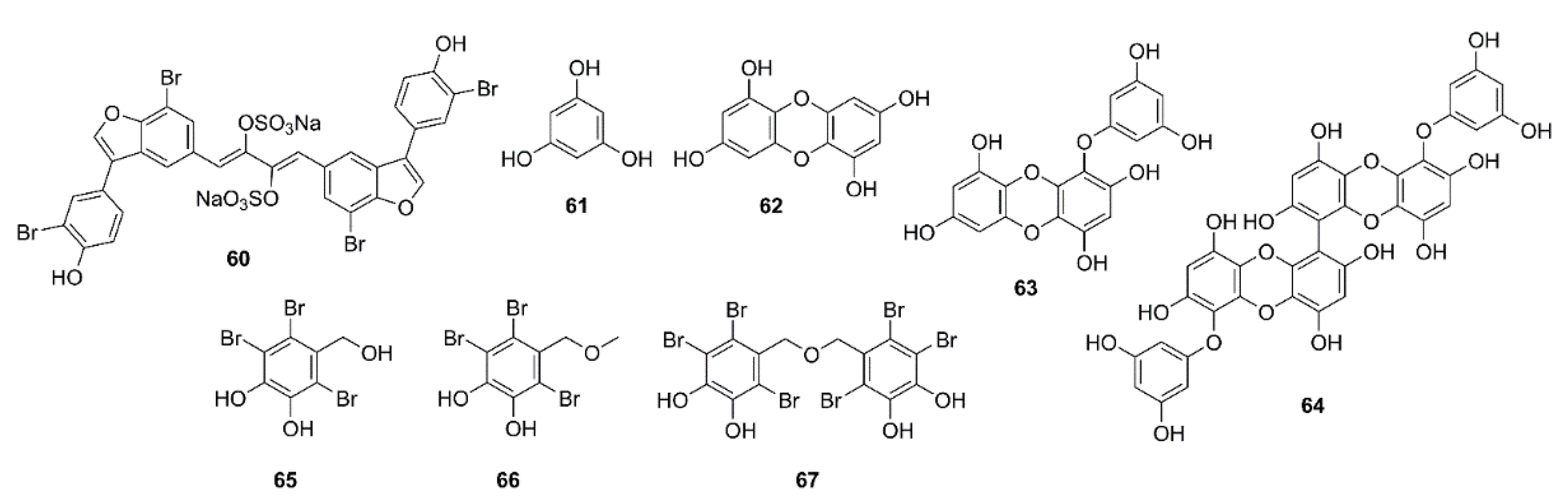
2. Marine-Derived Compounds That Inhibit Acetylcholinesterase (AChE) and Butyrylcholinesterase (BuChE) Activities
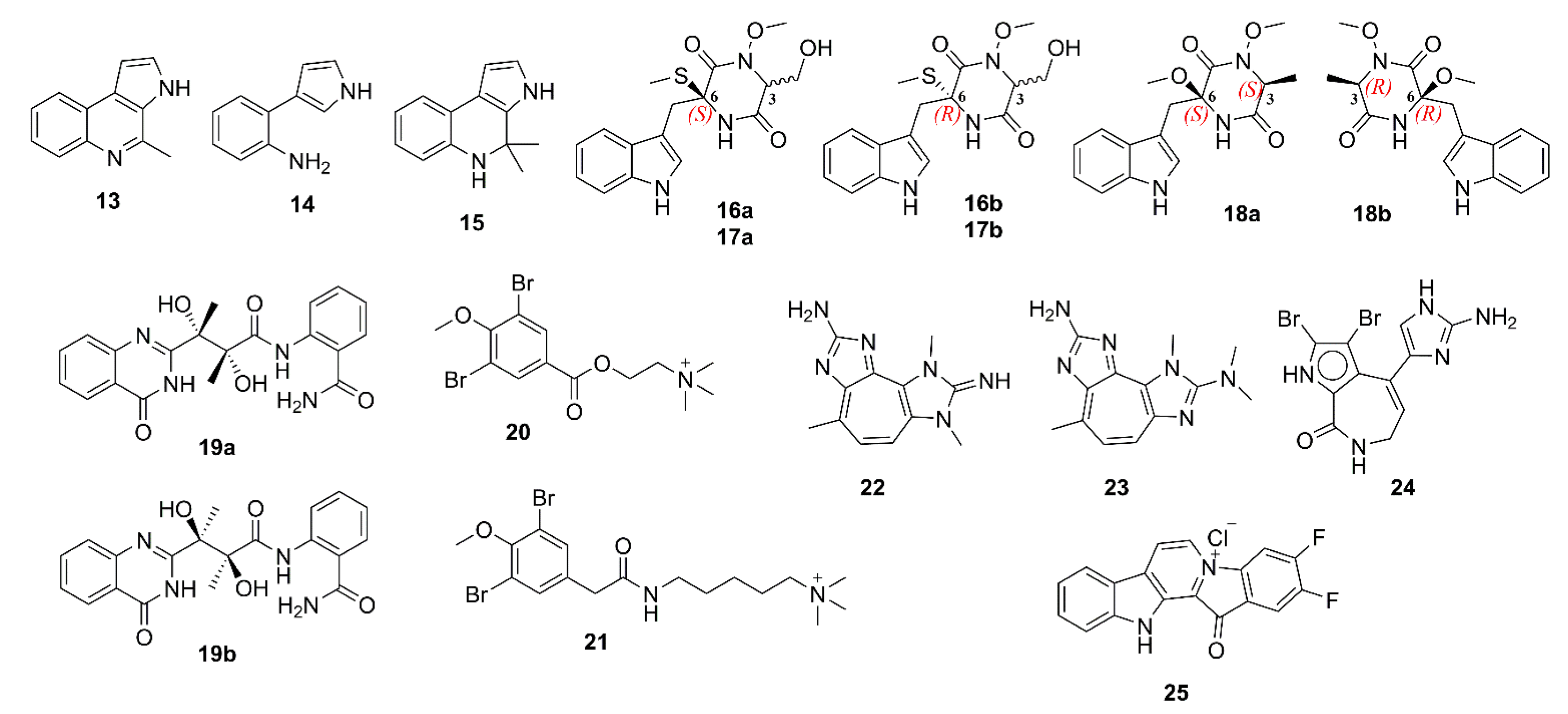
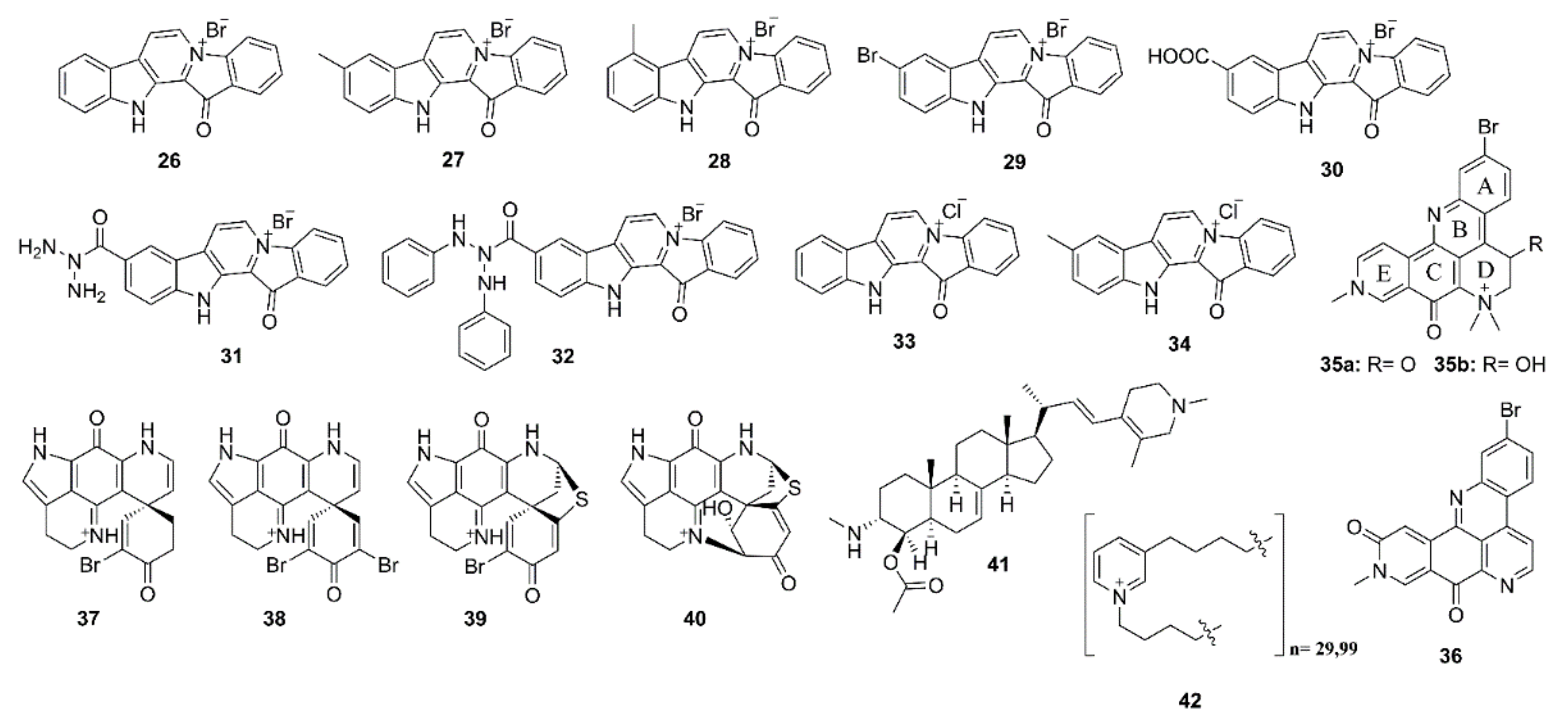
2.1. Alkaloids
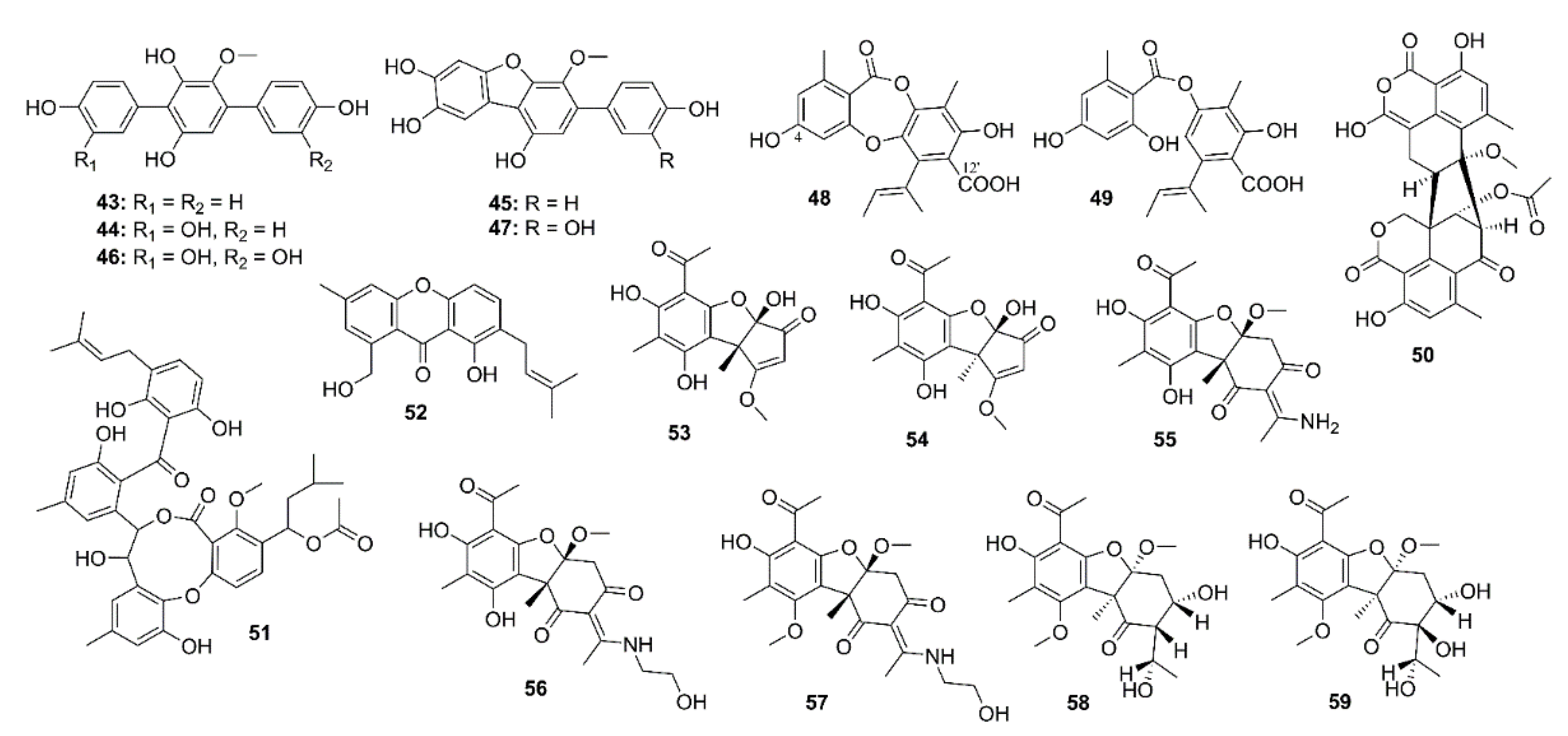
2.2. Phenolics
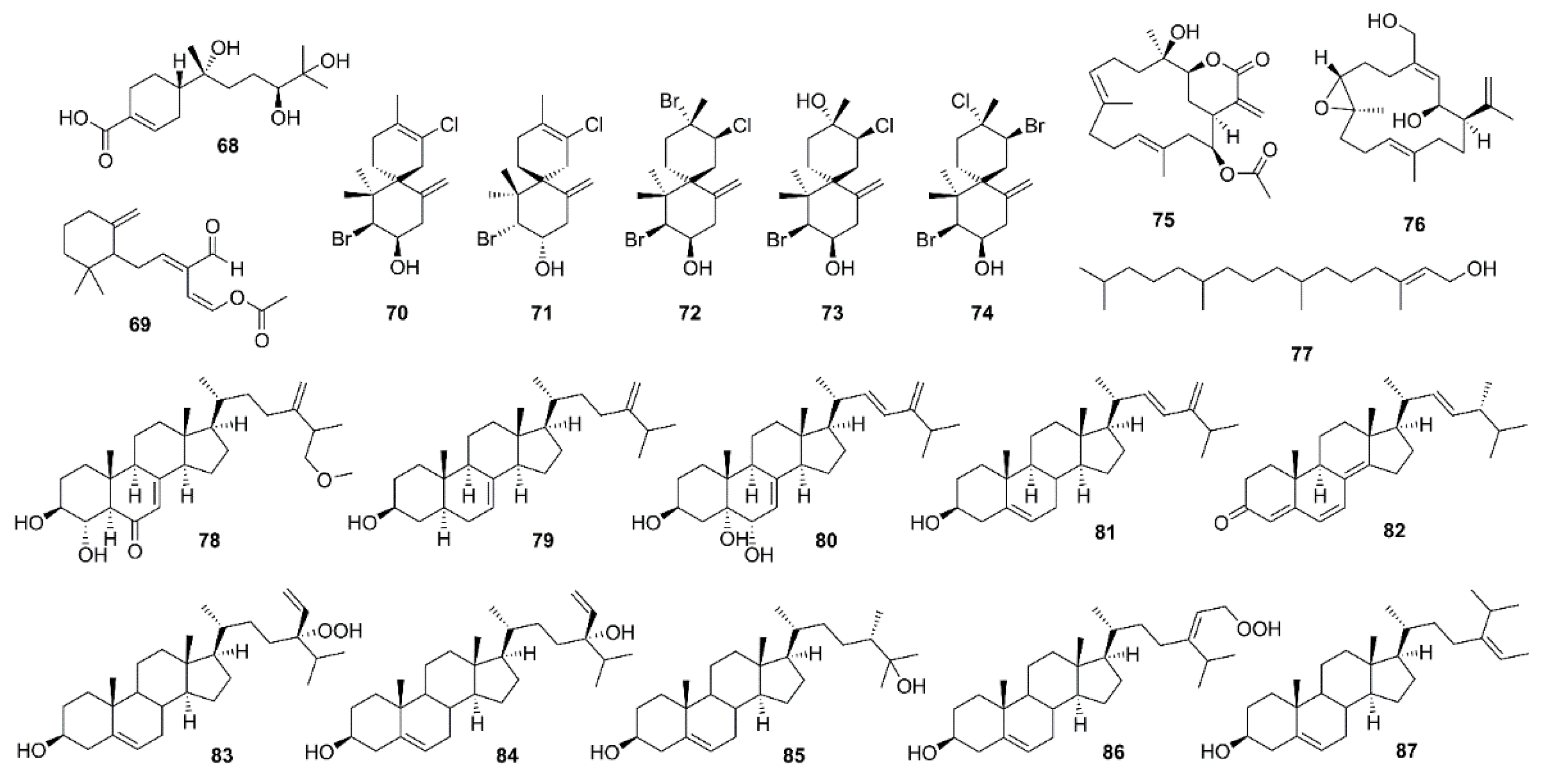
2.3. Terpenoids
2.3.1. Sesquiterpenes
2.3.2. Diterpenes
2.3.3. Sterols
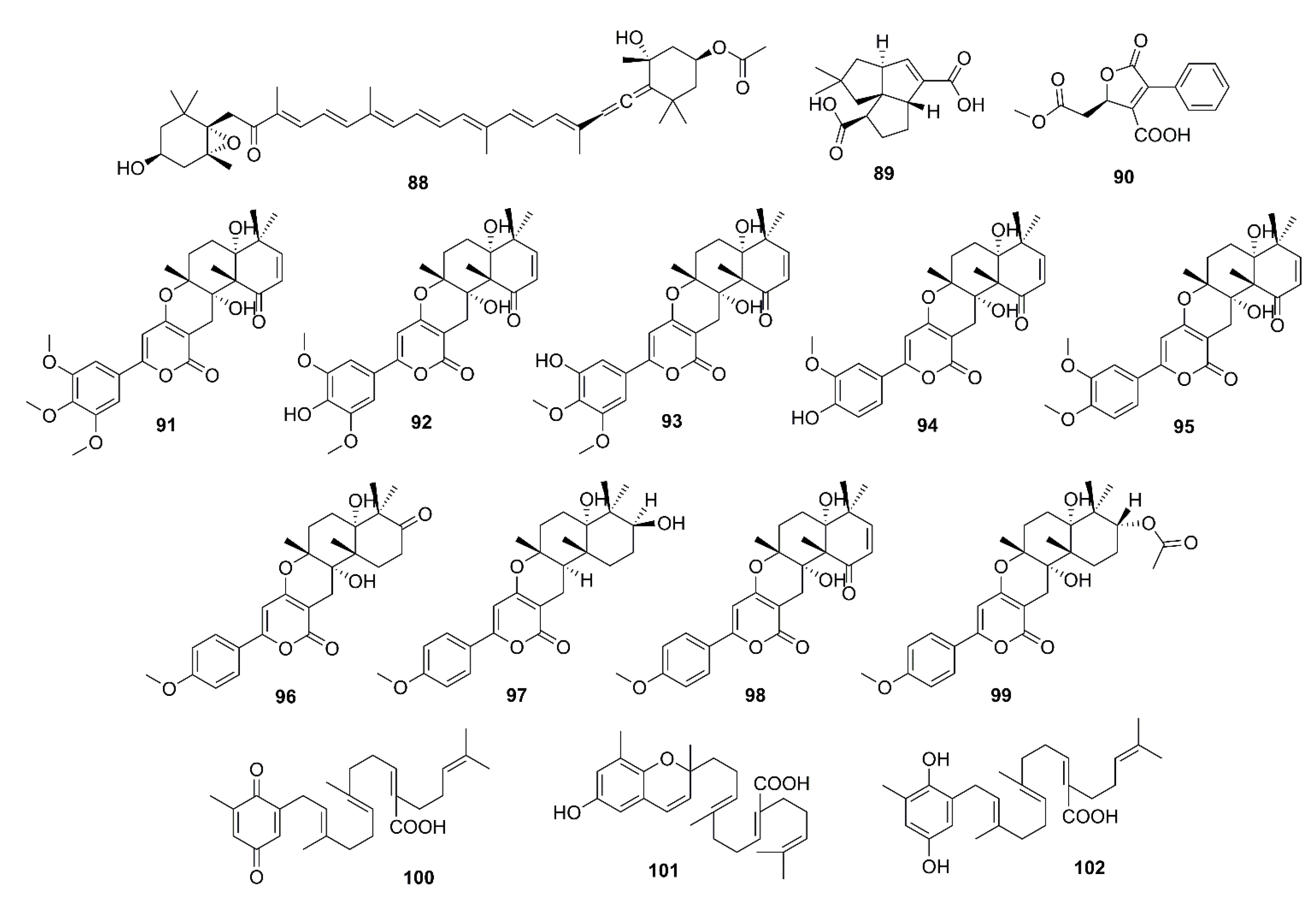
2.3.4. Tetraterpenes
2.3.5. Meroterpenoids
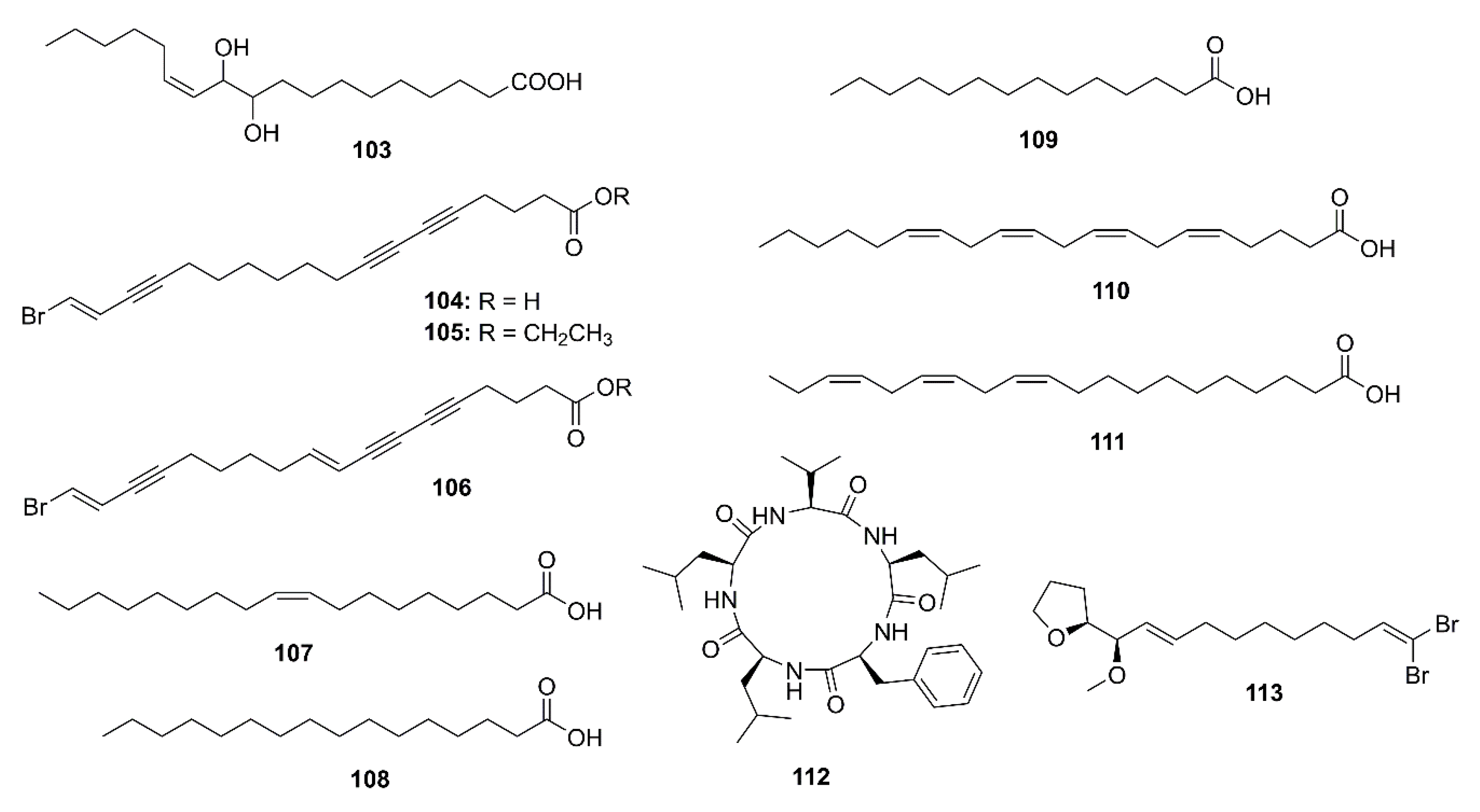
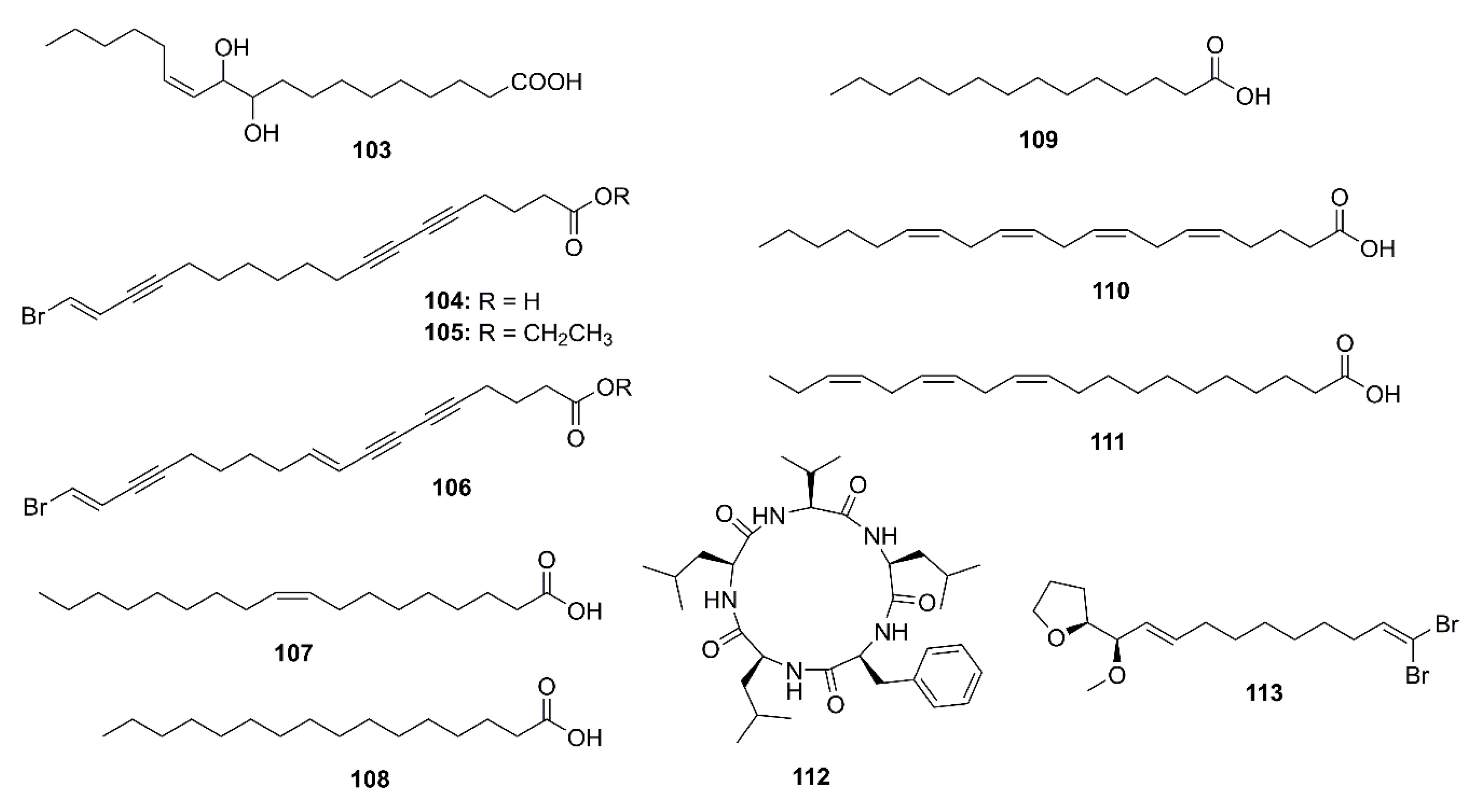
2.4. Marine Fatty Acids
2.5. Peptides
2.6. Miscellaneous Compounds
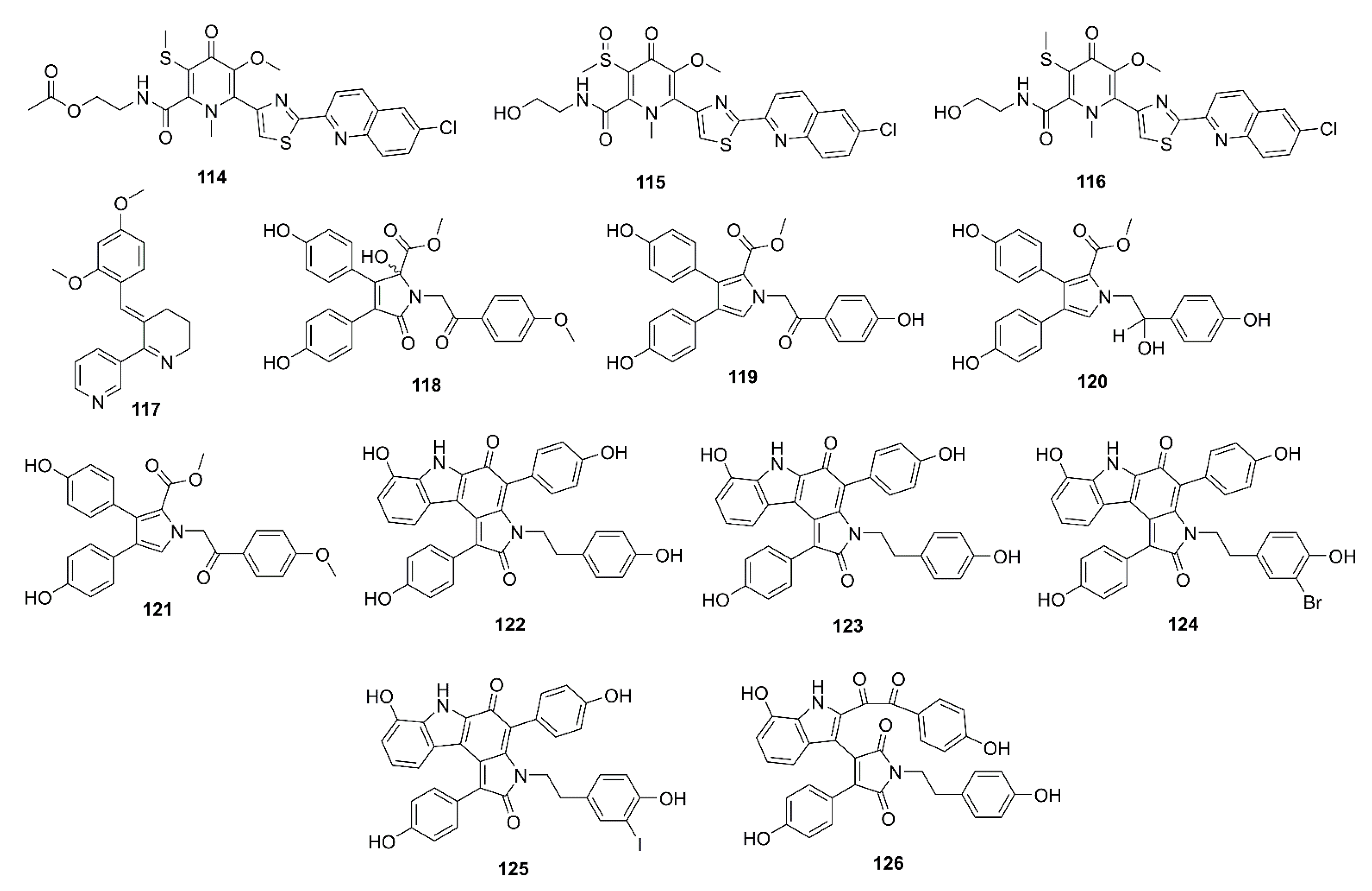
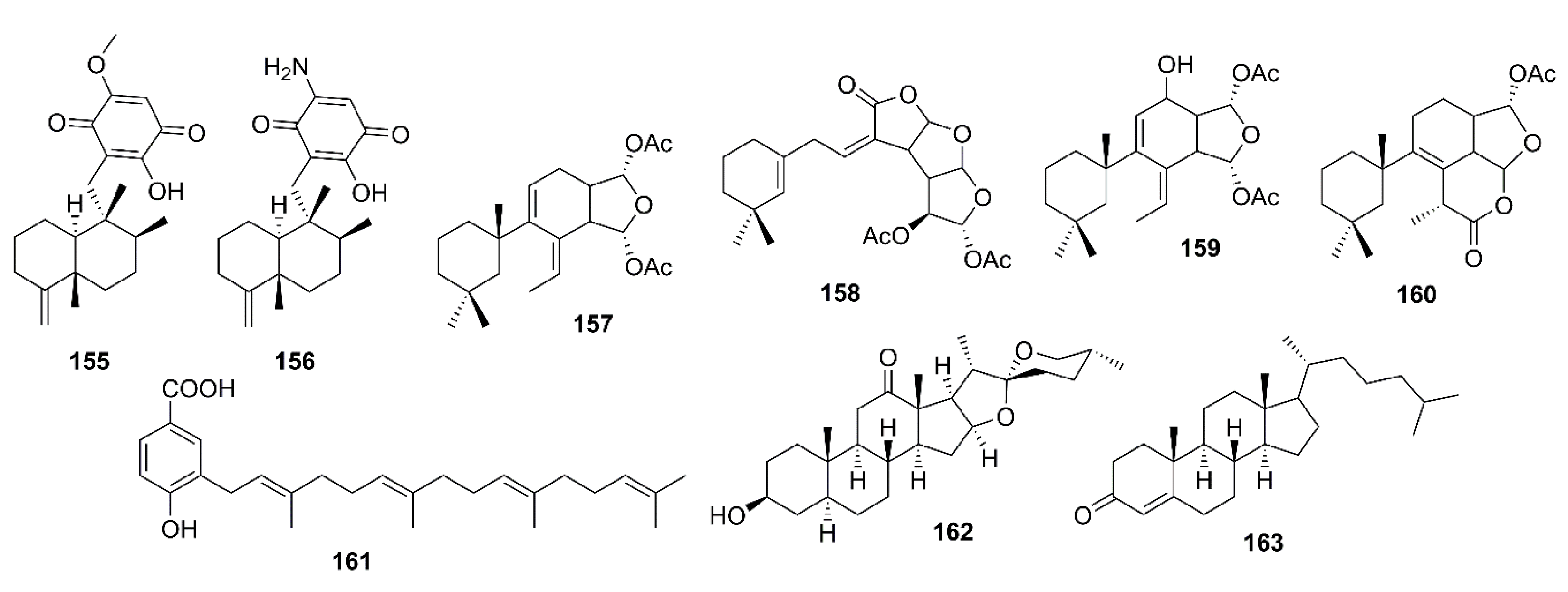
3. Marine-Derived Compounds That Inhibit β-Secretase (BACE-1) and γ-Secretase Activities
3.1. Alkaloids
3.2. Phenolics
3.3. Terpenoids
3.3.1. Sesquiterpenoids
3.3.2. Diterpenoids
3.3.3. Meroterpenoids
3.3.4. Steroids
3.3.5. Carotenoids
3.4. Peptides
3.5. Polycyclic Ethers
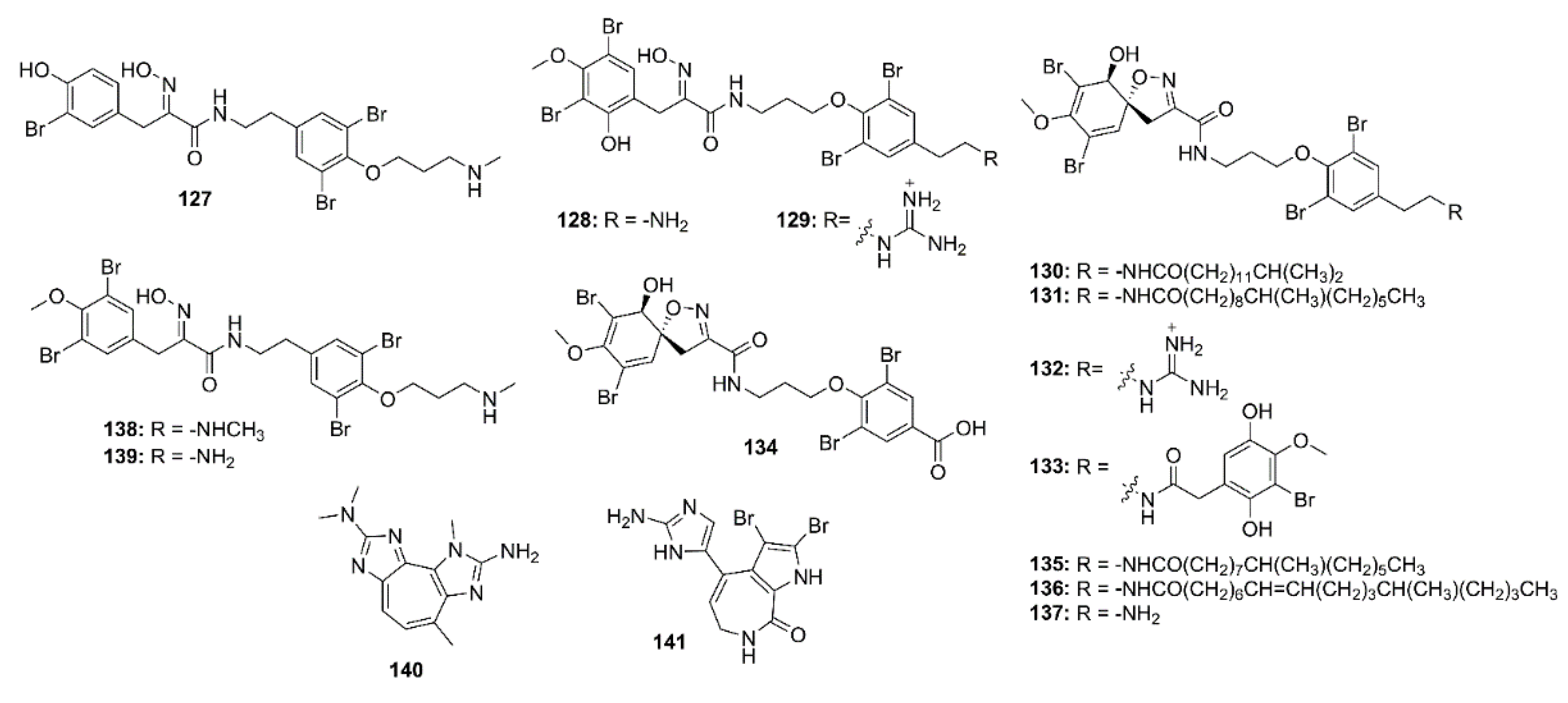
4. Marine-Derived Compounds That Inhibit Aβ Aggregation
4.1. Alkaloids
4.2. Phenolics
4.3. Terpenoids
4.3.1. Meroterpenoids
4.3.2. Steroids and Their Derivatives
4.3.3. Tetraterpenes
4.4. Fatty Acids
4.5. Peptides
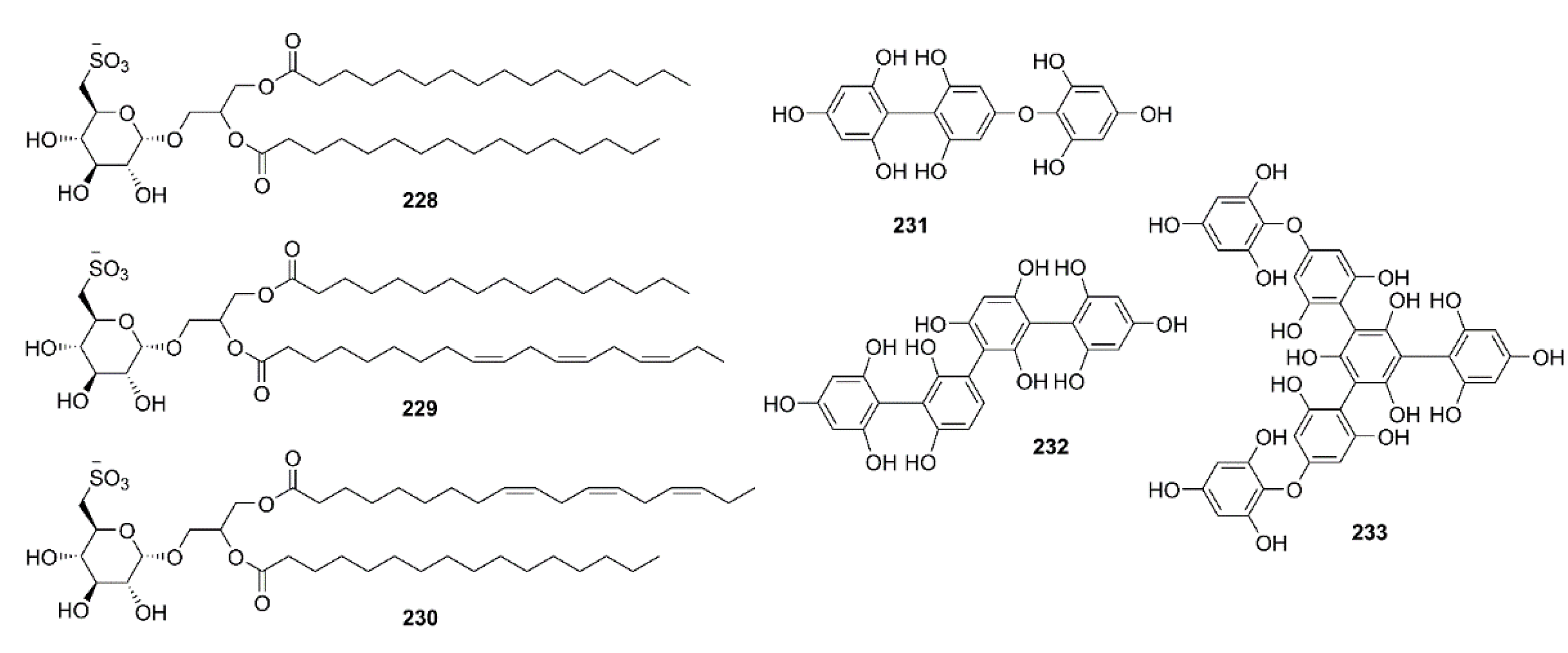
4.6. Carbohydrates
4.7. Miscellaneous
5. Marine-Derived Compounds That Inhibit Protein Kinases
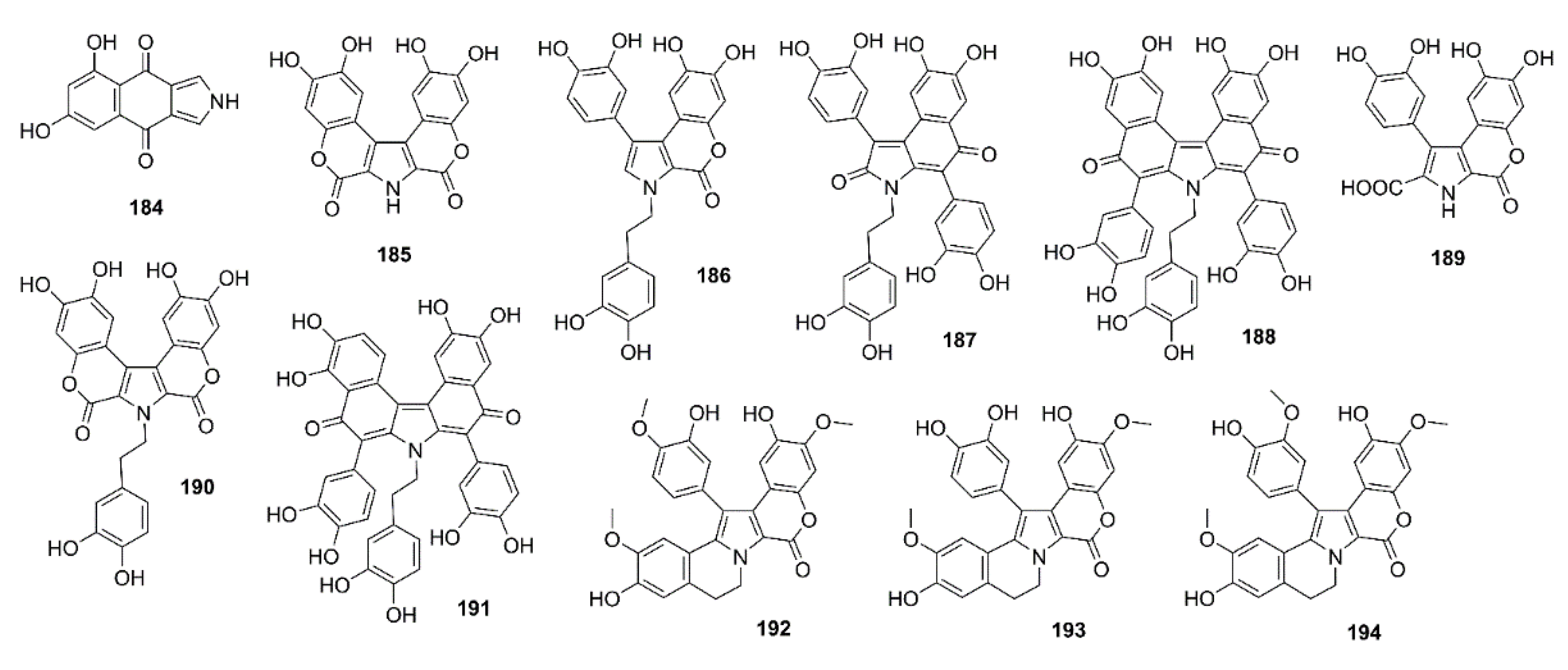
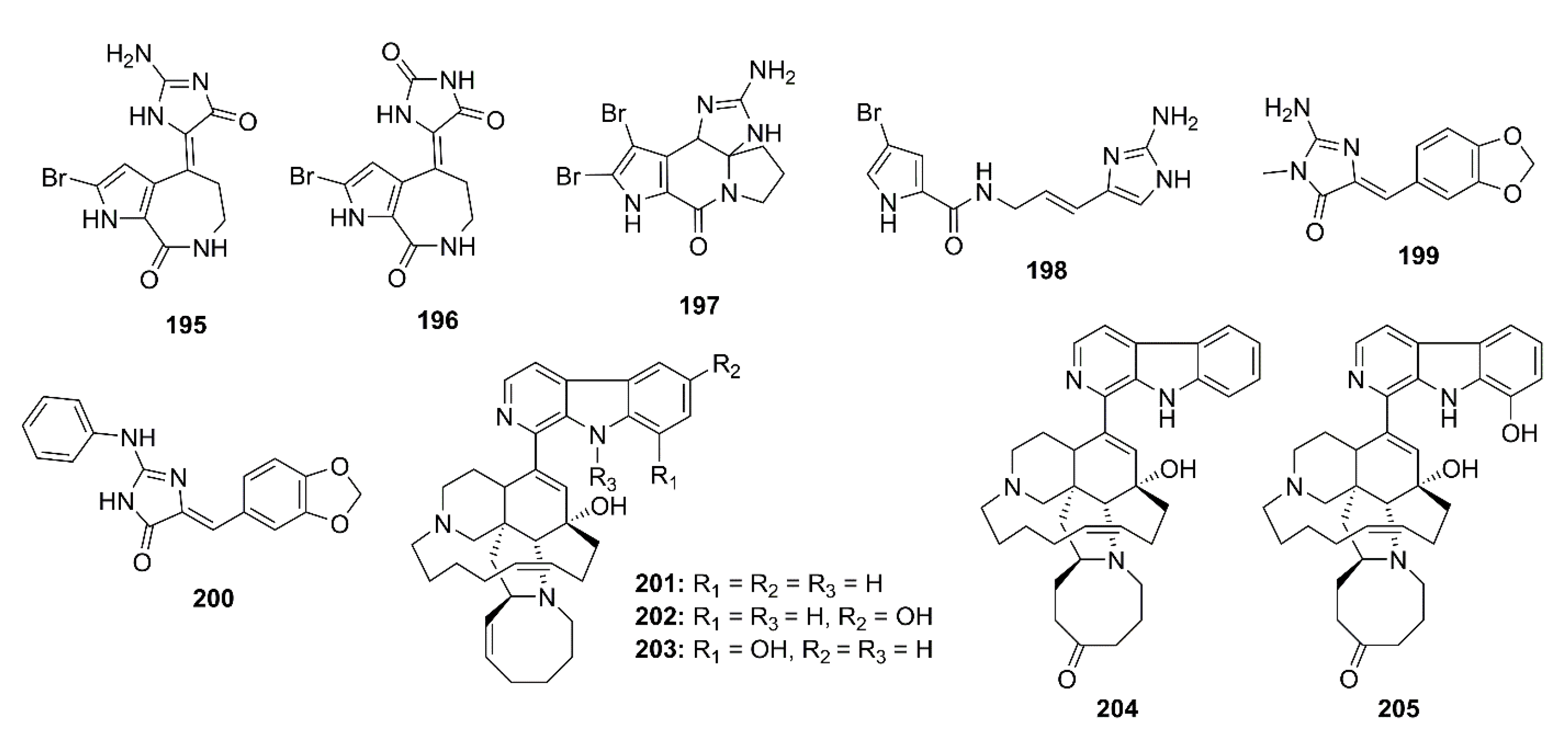
5.1. Alkaloids
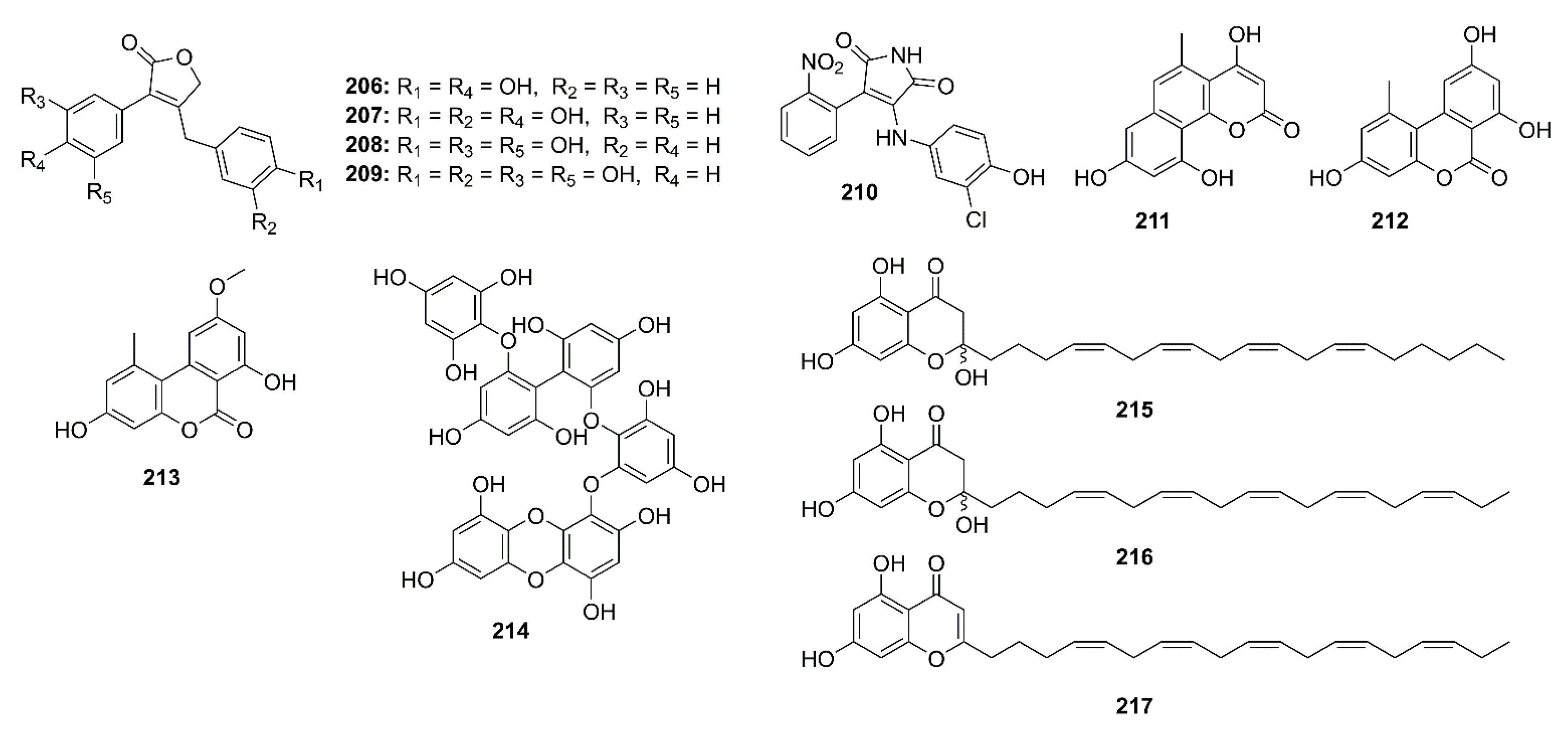
5.2. Phenolics
5.3. Terpenoids
Sesquiterpenes
5.4. Naphthoquinones
5.5. Miscellaneous Marine Natural Products
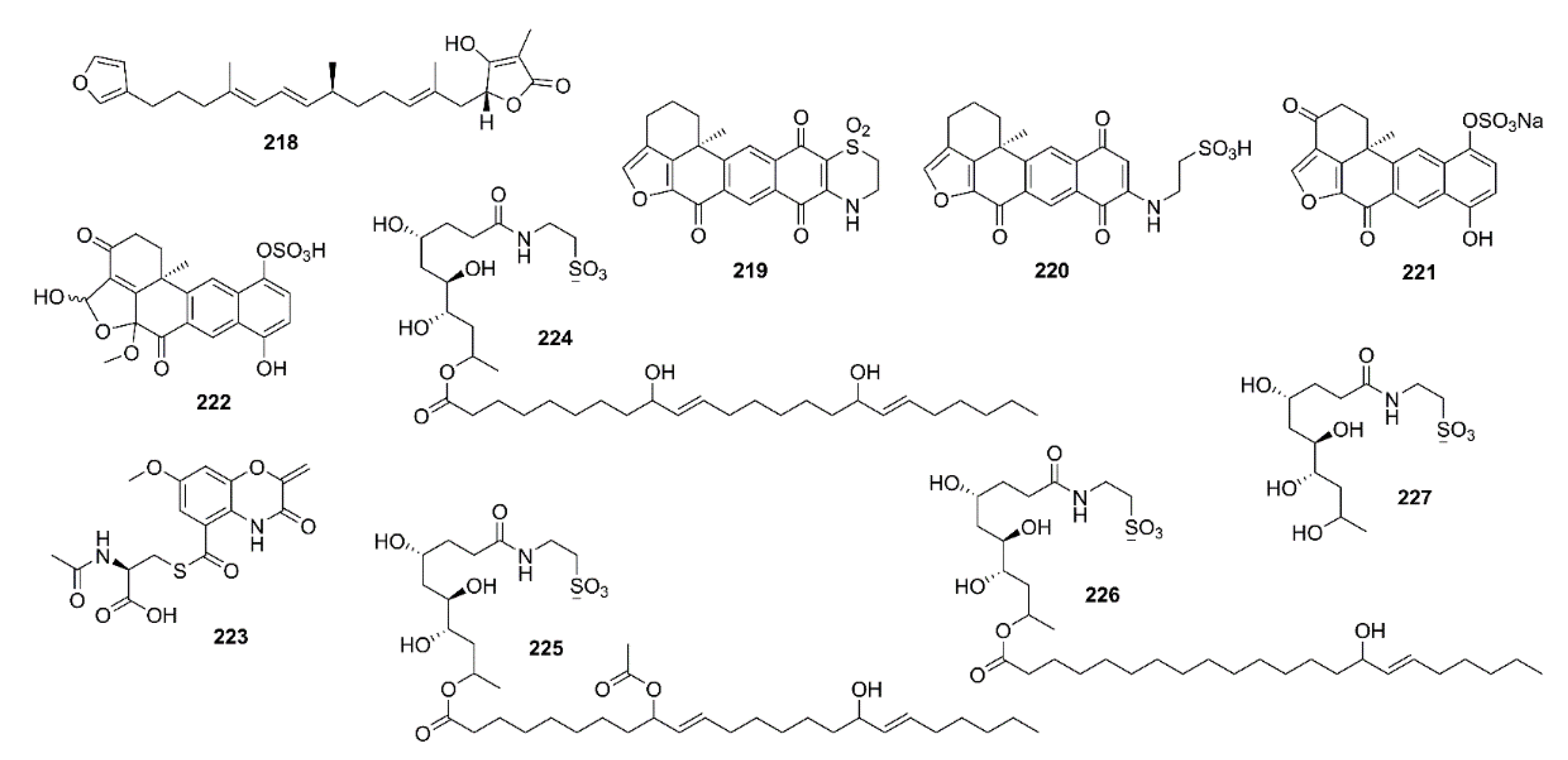
6. Marine-Derived Compounds with Miscellaneous Enzyme Inhibition
7. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association Report. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef]
- Price, D.L.; Tanzi, R.E.; Borchelt, D.R.; Sisodia, S.S. Alzheimer’s disease: Genetic studies and transgenic models. Annu. Rev. Genet. 1998, 32, 461–493. [Google Scholar] [CrossRef]
- Mucke, L. Neuroscience: Alzheimer’s disease. Nature 2009, 461, 895–897. [Google Scholar] [CrossRef]
- Giacobini, E. Cholinesterases: New roles in brain function and in Alzheimer’s disease. Neurochem. Res. 2003, 28, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Finder, V.H.; Glockshuber, R. Amyloid-β aggregation. Neurodegener. Dis. 2007, 4, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.; Dobkins, K.R.; Hansen, L.A.; Terry, R.D.; Saitoh, T. Decreased levels of protein kinase C in Alzheimer brain. Brain Res. 1988, 452, 165–174. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Pratico, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys Acta Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/antioxidant imbalance in Alzheimer’s disease: Therapeutic and diagnostic prospects. Oxid. Med. Cell Longev. 2018. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.V.; Toms, N.J. Pyruvate and oxaloacetate limit zinc-induced oxidative HT-22 neuronal cell injury. Neurotoxicology 2006, 27, 1043–1051. [Google Scholar] [CrossRef]
- Yassine, H.N.; Braskie, M.N.; Mack, W.J.; Castor, K.J.; Fonteh, A.N.; Schneider, L.S.; Harrington, M.G.; Chui, H.C. Association of docosahexaenoic acid supplementation with Alzheimer disease stage in apolipoprotein E ε4 carriers: A review. JAMA Neurol. 2017, 74, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, J.; Qiu, J.; Li, Y.; Wang, J.; Jiao, J. Intakes of fish and polyunsaturated fatty acids and mild-to-severe cognitive impairment risks: A dose-response meta-analysis of 21 cohort studies–3. Am. J. Clin. Nutr. 2015, 103, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Silvente, L.; Castells, X.; Saez, M.; Barceló, M.A.; Garre-Olmo, J.; Vilalta-Franch, J.; Capellà, D. Discontinuation, efficacy, and safety of cholinesterase inhibitors for Alzheimer’s disease: A meta-analysis and meta-regression of 43 randomized clinical trials enrolling 16 106 patients. Int. J. Neuropsychopharmacol. 2017, 20, 519–528. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Iwata, N. Memantine monotherapy for Alzheimer’s disease: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0123289. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, W.J.; Grossberg, G.T. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer’s disease. Drug Des. Dev. Ther. 2016, 10, 3267. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.-Y.; Choi, H. Natural products from marine organisms with neuroprotective activity in the experimental models of Alzheimer’s disease, Parkinson’s disease and ischemic brain stroke: Their molecular targets and action mechanisms. Arch. Pharm. Res. 2015, 38, 139–170. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.; Uddin, M.; Jeandet, P.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Sayed, A.A.; Abdel-Daim, M.M.; Simal-Gandara, J. Anti-alzheimer’s molecules derived from marine life: Understanding molecular mechanisms and therapeutic potential. Mar. Drugs 2021, 19, 251. [Google Scholar] [CrossRef]
- Martins, M.; Silva, R.; MM Pinto, M.; Sousa, E. Marine natural products, multitarget therapy and repurposed agents in Alzheimer’s disease. Pharmaceuticals 2020, 13, 242. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New drugs from marine organisms in Alzheimer’s disease. Mar. Drugs 2016, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Advanced preclinical and clinical trials of natural products and related compounds from marine sources. Curr. Med. Chem. 2004, 11, 1693–1713. [Google Scholar] [CrossRef]
- Oboh, G.; Bakare, O.; Ademosun, A.; Akinyemi, A.; Olasehinde, T. Inhibition of cholinesterases and some pro-oxidant induced oxidative stress in rats brain by two tomato (Lycopersicon esculentum) varieties. Int. J. Biomed. Sci. 2015, 11, 48–53. [Google Scholar]
- Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Acetylcholinesterase-inhibiting activity of pyrrole derivatives from a novel marine gliding bacterium, Rapidithrix thailandica. Mar. Drugs 2008, 6, 578–586. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, X.-M.; Meng, L.-H.; Konuklugil, B.; Li, X.; Li, H.-L.; Wang, B.-G. Isolation and characterization of three pairs of indolediketopiperazine enantiomers containing infrequent N-methoxy substitution from the marine algal-derived endophytic fungus Acrostalagmus luteoalbus TK-43. Bioorg. Chem. 2019, 90, 103030. [Google Scholar] [CrossRef]
- He, J.-B.; Ji, Y.-N.; Hu, D.-B.; Zhang, S.; Yan, H.; Liu, X.-C.; Luo, H.-R.; Zhu, H.-J. Structure and absolute configuration of penicilliumine, a new alkaloid from Penicillium commune 366606. Tetrahedron Lett. 2014, 55, 2684–2686. [Google Scholar] [CrossRef]
- Tadesse, M.; Svenson, J.; Sepčić, K.; Trembleau, L.; Engqvist, M.; Andersen, J.H.; Jaspars, M.; Stensvåg, K.; Haug, T. Isolation and synthesis of pulmonarins A and B, acetylcholinesterase inhibitors from the colonial ascidian Synoicum pulmonaria. J. Nat. Prod. 2014, 77, 364–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, T.; Maček, P.; Šuput, D. Inhibition of acetylcholinesterase by a pseudozoanthoxanthin-like compound isolated from the zoanthid Parazoanthus axinellae (O. Schmidt). Toxicon 1995, 33, 133–142. [Google Scholar] [CrossRef]
- Vitale, R.M.; Rispoli, V.; Desiderio, D.; Sgammato, R.; Thellung, S.; Canale, C.; Vassalli, M.; Carbone, M.; Ciavatta, M.L.; Mollo, E.; et al. In Silico identification and experimental validation of novel anti-alzheimer’s multitargeted ligands from a marine source featuring a “2-aminoimidazole plus aromatic group” scaffold. ACS Chem. Neurosci. 2018, 9, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Bharate, S.B.; Manda, S.; Joshi, P.; Singh, B.; Vishwakarma, R.A. Total synthesis and anti-cholinesterase activity of marine-derived bis-indole alkaloid fascaplysin. MedChemComm. 2012, 3, 1098–1103. [Google Scholar] [CrossRef]
- Manda, S.; Sharma, S.; Wani, A.; Joshi, P.; Kumar, V.; Guru, S.K.; Bharate, S.S.; Bhushan, S.; Vishwakarma, R.A.; Kumar, A.; et al. Discovery of a marine-derived bis-indole alkaloid fascaplysin, as a new class of potent P-glycoprotein inducer and establishment of its structure–activity relationship. Eur. J. Med. Chem. 2016, 107, 1–11. [Google Scholar] [CrossRef]
- Pan, H.; Qiu, H.; Zhang, K.; Zhang, P.; Liang, W.; Yang, M.; Mou, C.; Lin, M.; He, M.; Xiao, X.; et al. Fascaplysin Derivatives are potent multitarget agents against Alzheimer’s disease: in vitro and in vivo evidence. ACS Chem. Neurosci. 2019, 10, 4741–4756. [Google Scholar] [CrossRef]
- Nukoolkarn, V.S.; Saen-oon, S.; Rungrotmongkol, T.; Hannongbua, S.; Ingkaninan, K.; Suwanborirux, K. Petrosamine, a potent anticholinesterase pyridoacridine alkaloid from a Thai marine sponge Petrosia n. sp. Bioorg. Med. Chem. 2008, 16, 6560–6567. [Google Scholar] [CrossRef] [PubMed]
- Botić, T.; Defant, A.; Zanini, P.; Žužek, M.C.; Frangež, R.; Janussen, D.; Kersken, D.; Knez, Ž.; Mancini, I.; Sepčić, K. Discorhabdin alkaloids from Antarctic Latrunculia spp. sponges as a new class of cholinesterase inhibitors. Eur. J. Med. Chem. 2017, 136, 294–304. [Google Scholar] [CrossRef]
- Langjae, R.; Bussarawit, S.; Yuenyongsawad, S.; Ingkaninan, K.; Plubrukarn, A. Acetylcholinesterase-inhibiting steroidal alkaloid from the sponge Corticium sp. Steroids 2007, 72, 682–685. [Google Scholar] [CrossRef]
- Khan, I.; Samad, A.; Khan, A.Z.; Habtemariam, S.; Badshah, A.; Abdullah, S.M.; Ullah, N.; Khan, A.; Zia-Ul-Haq, M. Molecular interactions of 4-acetoxy-plakinamine B with peripheral anionic and other catalytic subsites of the aromatic gorge of acetylcholinesterase: Computational and structural insights. Pharm. Biol. 2013, 51, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepčić, K.; Marcel, V.; Klaebe, A.; Turk, T.; Šuput, D.; Fournier, D. Inhibition of acetylcholinesterase by an alkylpyridinium polymer from the marine sponge, Reniera sarai. Biochim. Biophys. Acta. 1998, 1387, 217–225. [Google Scholar] [CrossRef]
- Huang, H.; Feng, X.; Xiao, Z.E.; Liu, L.; Li, H.; Ma, L.; Lu, Y.; Ju, J.; She, Z.; Lin, Y. Azaphilones and p-terphenyls from the mangrove endophytic fungus Penicillium chermesinum (ZH4-E2) isolated from the South China Sea. J. Nat. Prod. 2011, 74, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-C.; Bao, H.-Y.; Liu, Y.-Y.; Nie, Y.-Y.; Yang, J.-M.; Hong, P.-Z.; Zhang, Y. Depsidone derivatives and a cyclopeptide produced by marine fungus Aspergillus unguis under chemical induction and by its plasma induced mutant. Molecules 2018, 23, 2245. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Ohlendorf, B.; Oesker, V.; Wiese, J.; Malien, S.; Schmaljohann, R.; Imhoff, J.F. Acetylcholinesterase inhibitors from a marine fungus Talaromyces sp. strain LF458. Mar. Biotechnol. 2015, 17, 110–119. [Google Scholar] [CrossRef]
- Chen, S.; Liu, H.; Ye, W.; Li, S.; Li, D.; Liu, Z.; Zhang, W. Ochuscins A‒G, highly oxygenated usnic acid derivatives from the deep-sea-derived fungus Ochroconis sp. FS449. Tetrahedron 2020, 76, 131066. [Google Scholar] [CrossRef]
- Okamoto, Y.; Ojika, M.; Suzuki, S.; Murakami, M.; Sakagami, Y. Iantherans A and B, unique dimeric polybrominated benzofurans as Na, K-ATPase inhibitors from a marine sponge, Ianthella sp. Bioorg. Med. Chem. 2001, 9, 179–183. [Google Scholar] [CrossRef]
- Kannan, R.R.; Aderogba, M.A.; Ndhlala, A.R.; Stirk, W.A.; Van Staden, J. Acetylcholinesterase inhibitory activity of phlorotannins isolated from the brown alga, Ecklonia maxima (Osbeck) Papenfuss. Food Res. Int. 2013, 54, 1250–1254. [Google Scholar] [CrossRef]
- Lee, B.H.; Choi, B.W.; Lee, S.Y. Isolation of 6,6’-bieckol from Grateloupia elliptica and its antioxidative and anti-cholinesterase activity. Ocean Polar Res. 2017, 39, 45–49. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Park, H.J.; Jung, H.A.; Choi, J.S. Anti-Alzheimer’s disease activity of bromophenols from a red alga, Symphyocladia latiuscula (Harvey) Yamada. ACS Omega 2019, 4, 12259–12270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.-L.; Dai, Y.; She, J.-L.; Zeng, Y.-B.; Dai, H.-F.; Ou, S.-L.; Zhou, X.-F.; Liu, Y.-H. Bisabolanoic acid A, a new polychiral sesquiterpene with AChE inhibitory activity from a mangrove-derived fungus Colletotrichum sp. J. Asian Nat. Prod. Res. 2021, 1–8. [Google Scholar] [CrossRef]
- Abramson, S.N.; Radic, Z.; Manker, D.; Faulkner, D.J.; Taylor, P. Onchidal: A naturally occurring irreversible inhibitor of acetylcholinesterase with a novel mechanism of action. Mol. Pharmacol. 1989, 36, 349–354. [Google Scholar] [PubMed]
- Stoddard, S.V.; Hamann, M.T.; Wadkins, R.M. Insights and ideas garnered from marine metabolites for development of dual-function acetylcholinesterase and amyloid-β aggregation inhibitors. Mar. Drugs 2014, 12, 2114–2131. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, K.G.; da Silva, L.L.; Soares, A.R.; Romeiro, N.C. Acetylcholinesterase as a target of halogenated marine natural products from Laurencia dendroidea. Algal Res. 2020, 52, 102130. [Google Scholar] [CrossRef]
- Tello, E.; Castellanos, L.; Arevalo-Ferro, C.; Duque, C. Cembranoid diterpenes from the Caribbean sea whip Eunicea knighti. J. Nat. Prod. 2009, 72, 1595–1602. [Google Scholar] [CrossRef]
- Castellanos, F.; Amaya-García, F.; Tello, E.; Ramos, F.A.; Umaña, A.; Puyana, M.; Resende, J.A.; Castellanos, L. Screening of acetylcholinesterase inhibitors in marine organisms from the Caribbean Sea. Nat. Prod. Res. 2019, 33, 3533–3540. [Google Scholar] [CrossRef]
- Syad, A.N.; Devi, K.P. Assessment of anti-amyloidogenic activity of marine red alga G. acerosa against Alzheimer’s beta-amyloid peptide 25–35. Neurol. Res. 2015, 37, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Syad, A.N.; Rajamohamed, B.S.; Shunmugaiah, K.P.; Kasi, P.D. Neuroprotective effect of the marine macroalga Gelidiella acerosa: Identification of active compounds through bioactivity-guided fractionation. Pharm. Biol. 2016, 54, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Qiao, M.F.; Ji, N.Y.; Miao, F.P.; Yin, X.L. Steroids and an oxylipin from an algicolous isolate of Aspergillus flavus. Magn. Reson. Chem. 2011, 49, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lu, Y.; Lin, X.; Yang, B.; Yang, X.; Liu, Y. Brominated aliphatic hydrocarbons and sterols from the sponge Xestospongia testudinaria with their bioactivities. Chem. Phys. Lipids 2011, 164, 703–706. [Google Scholar] [CrossRef]
- Castro-Silva, E.; Bello, M.; Hernández-Rodríguez, M.; Correa-Basurto, J.; Murillo-Alvarez, J.; Rosales-Hernandez, M.; Muñoz-Ochoa, M. In vitro and in silico evaluation of fucosterol from Sargassum horridum as potential human acetylcholinesterase inhibitor. J. Biomol. Struct. Dyn. 2018, 37, 3259–3268. [Google Scholar] [CrossRef]
- Miyashita, K. Function of marine carotenoids. In Food Factors for Health Promotion; Yoshikawa, T., Ed.; Forum Nutr. Basel: Karger, Switzerland, 2009; Volume 61, pp. 136–146. [Google Scholar] [CrossRef]
- Lin, J.; Huang, L.; Yu, J.; Xiang, S.; Wang, J.; Zhang, J.; Yan, X.; Cui, W.; He, S.; Wang, Q. Fucoxanthin, a marine carotenoid, reverses scopolamine-induced cognitive impairments in mice and inhibits acetylcholinesterase in vitro. Mar. Drugs 2016, 14, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, C.-L.; Kong, F.-D.; Li, Y.; Ma, Q.-Y.; Xie, Q.-Y.; Yuan, J.-Z.; Zhou, L.-M.; Dai, H.-F.; Yu, Z.-F.; Zhao, Y.-X. Asperpenes D and E from the fungus Aspergillus sp. SCS-KFD66 isolated from a bivalve mollusk, Sanguinolaria chinensis. J. Asian Nat. Prod. Res. 2020, 23, 1–6. [Google Scholar] [CrossRef]
- Bunbamrung, N.; Intaraudom, C.; Dramae, A.; Komwijit, S.; Laorob, T.; Khamsaeng, S.; Pittayakhajonwut, P. Antimicrobial, antimalarial and anticholinesterase substances from the marine-derived fungus Aspergillus terreus BCC51799. Tetrahedron 2020, 76, 131496. [Google Scholar] [CrossRef]
- Nong, X.-H.; Wang, Y.-F.; Zhang, X.-Y.; Zhou, M.-P.; Xu, X.-Y.; Qi, S.-H. Territrem and butyrolactone derivatives from a marine-derived fungus Aspergillus terreus. Mar. Drugs 2014, 12, 6113–6124. [Google Scholar] [CrossRef]
- Ding, B.; Wang, Z.; Huang, X.; Liu, Y.; Chen, W.; She, Z. Bioactive α-pyrone meroterpenoids from mangrove endophytic fungus Penicillium sp. Nat. Prod. Res. 2016, 30, 2805–2812. [Google Scholar] [CrossRef]
- Choi, B.W.; Ryu, G.; Park, S.H.; Kim, E.S.; Shin, J.; Roh, S.S.; Shin, H.C.; Lee, B.H. Anticholinesterase activity of plastoquinones from Sargassum sagamianum: Lead compounds for Alzheimer’s disease therapy. Phytother. Res. 2007, 21, 423–426. [Google Scholar] [CrossRef]
- Seong, S.H.; Ali, M.Y.; Kim, H.-R.; Jung, H.A.; Choi, J.S. BACE1 inhibitory activity and molecular docking analysis of meroterpenoids from Sargassum serratifolium. Bioorg. Med. Chem. 2017, 25, 3964–3970. [Google Scholar] [CrossRef]
- Yang, W.C.; Zhang, Y.Y.; Li, Y.J.; Nie, Y.Y.; Liang, J.Y.; Liu, Y.Y.; Liu, J.S.; Zhang, Y.P.; Song, C.; Qian, Z.J. Chemical composition and anti-Alzheimer’s disease-related activities of a functional oil from the edible seaweed Hizikia fusiforme. Chem. Biodivers. 2020, 17, e2000055. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Wiese, J.; Schmaljohann, R.; Imhoff, J.F. Biscogniauxone, a new isopyrrolonaphthoquinone compound from the fungus Biscogniauxia mediterranea isolated from deep-sea sediments. Mar. Drugs 2016, 14, 204. [Google Scholar] [CrossRef] [Green Version]
- Rafiquzzaman, S.M.; Kim, E.Y.; Kim, Y.R.; Nam, T.-J.; Kong, I.S. Antioxidant activity of glycoprotein purified from Undaria pinnatifida measured by an in vitro digestion model. Int. J. Biol. Macromol. 2013, 62, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Rafiquzzaman, S.; Kim, E.Y.; Lee, J.M.; Mohibbullah, M.; Alam, M.B.; Moon, I.S.; Kim, J.-M.; Kong, I.-S. Anti-Alzheimers and anti-inflammatory activities of a glycoprotein purified from the edible brown alga Undaria pinnatifida. Food Res. Int. 2015, 77, 118–124. [Google Scholar] [CrossRef]
- Vassar, R.; Kandalepas, P.C. The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 20. [Google Scholar] [CrossRef]
- Le, T.C.; Yim, C.-Y.; Park, S.; Katila, N.; Yang, I.; Song, M.C.; Yoon, Y.J.; Choi, D.-Y.; Choi, H.; Nam, S.-J.; et al. Lodopyridones B and C from a marine sediment-derived bacterium Saccharomonospora sp. Bioorg. Med. Chem. Lett. 2017, 27, 3123–3126. [Google Scholar] [CrossRef] [PubMed]
- Zawieja, P.; Kornprobst, J.M.; Métais, P. 3-(2,4-Dimethoxybenzylidene)-anabaseine: A promising candidate drug for Alzheimer’s disease? Geriatr. Gerontol. Int. 2012, 12, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Amamiya, T.; Mizoguchi, H.; Kawanishi, S.; Kuroda, E.; Kitamura, R.; Ito, A.; Saito, Y.; Tawa, M.; Nagasawa, T. Alpha7 nicotinic acetylcholine receptor-specific agonist DMXBA (GTS-21) attenuates Aβ accumulation through suppression of neuronal γ-secretase activity and promotion of microglial amyloid-β phagocytosis and ameliorates cognitive impairment in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 62, 197–209. [Google Scholar] [CrossRef]
- Zhang, H.; Conte, M.M.; Huang, X.-C.; Khalil, Z.; Capon, R.J. A search for BACE inhibitors reveals new biosynthetically related pyrrolidones, furanones and pyrroles from a southern Australian marine sponge, Ianthella sp. Org. Biomol. Chem. 2012, 10, 2656–2663. [Google Scholar] [CrossRef]
- Zhang, H.; Conte, M.M.; Khalil, Z.; Huang, X.-C.; Capon, R.J. New dictyodendrins as BACE inhibitors from a southern Australian marine sponge, Ianthella sp. RSC Adv. 2012, 2, 4209–4214. [Google Scholar] [CrossRef]
- Dai, J.; Parrish, S.M.; Yoshida, W.Y.; Yip, M.R.; Turkson, J.; Kelly, M.; Williams, P. Bromotyrosine-derived metabolites from an Indonesian marine sponge in the family Aplysinellidae (Order Verongiida). Bioorg. Med. Chem. Lett. 2016, 26, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Le, T.C.; Katila, N.; Park, S.; Lee, J.; Yang, I.; Choi, H.; Choi, D.-Y.; Nam, S.-J. Two new secondary metabolites, saccharochlorines A and B, from a marine bacterium Saccharomonospora sp. KCTC-19160. Bioorg. Med. Chem. Lett. 2020, 30, 127145. [Google Scholar] [CrossRef] [PubMed]
- Millán-Aguiñaga, N.; Soria-Mercado, I.E.; Williams, P. Xestosaprol D and E from the Indonesian marine sponge Xestospongia sp. Tetrahedron Lett. 2010, 51, 751–753. [Google Scholar] [CrossRef]
- Dai, J.; Sorribas, A.; Yoshida, W.Y.; Kelly, M.; Williams, P.G. Xestosaprols from the Indonesian marine sponge Xestospongia sp. J. Nat. Prod. 2010, 73, 1188–1191. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Byun, H.-G. A novel BACE inhibitor isolated from Eisenia bicyclis exhibits neuroprotective activity against β-amyloid toxicity. Fish. Aquat. Sci. 2018, 21, 38. [Google Scholar] [CrossRef] [Green Version]
- Neupane, R.P.; Parrish, S.M.; Bhandari Neupane, J.; Yoshida, W.Y.; Yip, M.; Turkson, J.; Harper, M.K.; Head, J.D.; Williams, P.G. Cytotoxic sesquiterpenoid quinones and quinols, and an 11-membered heterocycle, kauamide, from the Hawaiian marine sponge Dactylospongia elegans. Mar. Drugs 2019, 17, 423. [Google Scholar] [CrossRef] [Green Version]
- Leirós, M.; Alonso, E.; Rateb, M.E.; Houssen, W.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. Gracilins: Spongionella-derived promising compounds for Alzheimer disease. Neuropharmacol. 2015, 93, 285–293. [Google Scholar] [CrossRef] [PubMed]
- López-Ogalla, J.; García-Palomero, E.; Sánchez-Quesada, J.; Rubio, L.; Delgado, E.; García, P.; Medina, M.; Castro, A.; Muñoz, P. Bioactive prenylated phenyl derivatives derived from marine natural products: Novel scaffolds for the design of BACE inhibitors. MedChemComm 2014, 5, 474–488. [Google Scholar] [CrossRef]
- Zhu, Y.-Z.; Liu, J.-W.; Wang, X.; Jeong, I.-H.; Ahn, Y.-J.; Zhang, C.-J. Anti-BACE1 and antimicrobial activities of steroidal compounds isolated from marine Urechis unicinctus. Mar. Drugs 2018, 16, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.A.; Ali, M.Y.; Choi, R.J.; Jeong, H.O.; Chung, H.Y.; Choi, J.S. Kinetics and molecular docking studies of fucosterol and fucoxanthin, BACE1 inhibitors from brown algae Undaria pinnatifida and Ecklonia stolonifera. Food Chem. Toxicol. 2016, 89, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. The isolation and structure elucidation of tasiamide B, a 4-amino-3-hydroxy-5-phenylpentanoic acid containing peptide from the marine cyanobacterium Symploca sp. J. Nat. Prod. 2003, 66, 1006–1009. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, W.; Li, L.; Salvador, L.A.; Chen, T.; Chen, W.; Felsenstein, K.M.; Ladd, T.B.; Price, A.R.; Golde, T.E. Cyanobacterial peptides as a prototype for the design of potent β-secretase inhibitors and the development of selective chemical probes for other aspartic proteases. J. Med. Chem. 2012, 55, 10749–10765. [Google Scholar] [CrossRef]
- Al-Awadhi, F.H.; Ratnayake, R.; Paul, V.J.; Luesch, H. Tasiamide F, a potent inhibitor of cathepsins D and E from a marine cyanobacterium. Bioorg. Med. Chem. 2016, 24, 3276–3282. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Li-Chan, E.C.; Byun, H.-G. Characterization of β-secretase inhibitory peptide purified from skate skin protein hydrolysate. Eur. Food Res. Technol. 2015, 240, 129–136. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, S.R.; Byun, H.-G. Purification and characterization of β-secretase inhibitory peptide from sea hare (Aplysia kurodai) by enzymatic hydrolysis. Fish. Aquat. Sci. 2018, 21, 13. [Google Scholar] [CrossRef]
- Lee, J.K.; Li-Chan, E.C.; Cheung, I.W.; Jeon, Y.-J.; Ko, J.-Y.; Byun, H.-G. Neuroprotective effect of β-secretase inhibitory peptide from Pacific hake (Merluccius productus) fish protein hydrolysate. Curr. Alzheimer Res. 2019, 16, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, S.R.; Byun, H.-G. Characterization of β-secretase inhibitory peptide purified from Blackfin flounder (Glyptocephalus stelleri) protein hydrolysate. J. Mar. Biosci. Biotechnol. 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Alonso, E.; Fuwa, H.; Vale, C.; Suga, Y.; Goto, T.; Konno, Y.; Sasaki, M.; LaFerla, F.M.; Vieytes, M.R.; Giménez-Llort, L.; et al. Design and synthesis of skeletal analogues of gambierol: Attenuation of amyloid-β and tau pathology with voltage-gated potassium channel and N-methyl-D-aspartate receptor implications. J. Am. Chem. Soc. 2012, 134, 7467–7479. [Google Scholar] [CrossRef]
- Alonso, E.; Vieira, A.C.; Rodriguez, I.; Alvariño, R.; Gegunde, S.; Fuwa, H.; Suga, Y.; Sasaki, M.; Alfonso, A.; Cifuentes, J.M.; et al. Tetracyclic truncated analogue of the marine toxin gambierol modifies NMDA, tau, and amyloid β expression in mice brains: Implications in AD pathology. ACS Chem. Neurosci. 2017, 8, 1358–1367. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.-Q.; Mao, S.-C.; Zhang, H.-Y.; Yu, X.-Q.; Feng, M.-T.; Wang, B.; Feng, L.-H.; Guo, Y.-W. Racemosins A and B, two novel bisindole alkaloids from the green alga Caulerpa racemosa. Fitoterapia 2013, 91, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Harms, H.; Kehraus, S.; Nesaei-Mosaferan, D.; Hufendieck, P.; Meijer, L.; König, G.M. Aβ-42 lowering agents from the marine-derived fungus Dichotomomyces cejpii. Steroids 2015, 104, 182–188. [Google Scholar] [CrossRef]
- Seong, S.H.; Paudel, P.; Jung, H.A.; Choi, J.S. Identifying phlorofucofuroeckol-A as a dual inhibitor of amyloid-β25-35 self-aggregation and insulin glycation: Elucidation of the molecular mechanism of action. Mar. Drugs 2019, 17, 600. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Liu, D.-Q.; Liang, T.-J.; Li, J.; Zhang, H.-Y.; Liu, A.-H.; Guo, Y.-W.; Mao, S.-C. Bioactive constituents from the green alga Caulerpa racemosa. Bioorg. Med. Chem. 2015, 23, 38–45. [Google Scholar] [CrossRef]
- Xiang, S.; Liu, F.; Lin, J.; Chen, H.; Huang, C.; Chen, L.; Zhou, Y.; Ye, L.; Zhang, K.; Jin, J. Fucoxanthin inhibits β-amyloid assembly and attenuates β-amyloid oligomer-induced cognitive impairments. J. Agric. Food Chem. 2017, 65, 4092–4102. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Li, H.; Yang, C.; Xie, W.; Wang, Y.; Zhang, J.; Mao, Z.; Xie, W.; Lü, T. DHA attenuates Aβ-induced necroptosis through the RIPK1/RIPK3 signaling pathway in THP-1 monocytes. Biomed. Pharmacother. 2020, 126, 110102. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yao, M.; Lu, J.-H.; Wang, Y.; Yin, X.; Liang, M.; Yuan, E.; Ren, J. Identification of novel oligopeptides from the simulated digestion of sea cucumber (Stichopus japonicus) to alleviate Aβ aggregation progression. J. Funct. Foods 2019, 60, 103412. [Google Scholar] [CrossRef]
- Hu, J.; Geng, M.; Li, J.; Xin, X.; Wang, J.; Tang, M.; Zhang, J.; Zhang, X.; Ding, J. Acidic oligosaccharide sugar chain, a marine-derived acidic oligosaccharide, inhibits the cytotoxicity and aggregation of amyloid beta protein. J. Pharmacol. Sci. 2004, 95, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdinia, A.; Sheijooni Fumani, N.; Baradaran Kayyal, T.; Ghaderiardakani, F. Homotaurine of marine macroalgae of the Persian Gulf as a potential treatment agent for Alzheimer. J. Persian Gulf 2018, 9, 1–8. Available online: http://jpg.inio.ac.ir/article-1-594-en.html (accessed on 25 July 2020).
- Miyazawa, K.; Ito, K.; Matsumoto, F. Occurrence of d-2-hydroxy-3-aminopropane sulfonic acid and 3-aminopropane sulfonic acid in a red alga, Grateloupia livida. Bull. Jap. Soc. Sci. Fish. 1970, 36, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Y.; Li, C.; Zheng, Q.; Tian, J.; Li, Z.; Huang, T.Y.; Zhang, W.; Xu, H. Inhibition of PKCδ reduces amyloid-β levels and reverses Alzheimer disease phenotypesPKCδ modulates Aβ production in Alzheimer disease. J. Exp. Med. 2018, 215, 1665–1677. [Google Scholar] [CrossRef] [Green Version]
- Eldar-Finkelman, H. Glycogen synthase kinase 3: An emerging therapeutic target. Trends Mol. Med. 2002, 8, 126–132. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Plisson, F.; Conte, M.; Khalil, Z.; Huang, X.C.; Piggott, A.M.; Capon, R.J. Kinase inhibitor scaffolds against neurodegenerative diseases from a Southern Australian ascidian, Didemnum sp. ChemMedChem 2012, 7, 983–990. [Google Scholar] [CrossRef]
- Cimino, G.; De Rosa, S.; De Stefano, S.; Mazzarella, L.; Puliti, R.; Sodano, G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 1982, 23, 767–768. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.-M.; White, A.; Garnier, M.; Nikolic, M.; Tsai, L.; Walter, J.; Cleverley, K.; Salinas, P.; Wu, Y.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Plisson, F.; Prasad, P.; Xiao, X.; Piggott, A.M.; Huang, X.-C.; Khalil, Z.; Capon, R.J. Callyspongisines A–D: Bromopyrrole alkaloids from an Australian marine sponge, Callyspongia sp. Org. Biomol. Chem. 2014, 12, 1579–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, G.W.; Mong, S.; Hemling, M.E.; Freyer, A.J.; Offen, P.H.; DeBrosse, C.W.; Sarau, H.M.; Westley, J.W. New leukotriene B4 receptor antagonist: Leucettamine A and related imidazole alkaloids from the marine sponge Leucetta microraphis. J. Nat. Prod. 1993, 56, 116–121. [Google Scholar] [CrossRef]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Hu, J.-F.; Hamann, M.T.; Hill, R.; Kelly, M. The manzamine alkaloids. Alkaloids-Chem. Biol. 2003, 60, 207–286. [Google Scholar] [CrossRef]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; Del Monte-Millán, M.; Medina, M.; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure–activity relationship (SAR) studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef]
- Schulz, D.; Ohlendorf, B.; Zinecker, H.; Schmaljohann, R.; Imhoff, J.F. Eutypoids B− E produced by a Penicillium sp. strain from the North Sea. J. Nat. Prod. 2011, 74, 99–101. [Google Scholar] [CrossRef]
- Wiese, J.; Imhoff, J.F.; Gulder, T.A.; Labes, A.; Schmaljohann, R. Marine fungi as producers of benzocoumarins, a new class of inhibitors of glycogen-synthase-kinase 3β. Mar. Drugs 2016, 14, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zheng, J.; Huang, C.; Zhao, J.; Lin, J.; Zhou, X.; Naman, C.B.; Wang, N.; Gerwick, W.H.; Wang, Q.; et al. Eckmaxol, a phlorotannin extracted from Ecklonia maxima, produces anti-β-amyloid oligomer neuroprotective effects possibly via directly acting on glycogen synthase kinase 3β. ACS Chem. Neurosci. 2018, 9, 1349–1356. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, X.; Conte, M.M.; Khalil, Z.; Capon, R.J. Spiralisones A–D: Acylphloroglucinol hemiketals from an Australian marine brown alga, Zonaria spiralis. Org. Biomol. Chem. 2012, 10, 9671–9676. [Google Scholar] [CrossRef] [PubMed]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Mai, L.H.; Longeon, A.; Copp, B.R.; Loaëc, N.; Bescond, A.; Meijer, L.; Bourguet-Kondracki, M.-L. Novel adociaquinone derivatives from the Indonesian sponge Xestospongia sp. Mar. Drugs 2015, 13, 2617–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachtigall, J.; Schneider, K.; Bruntner, C.; Bull, A.T.; Goodfellow, M.; Zinecker, H.; Imhoff, J.F.; Nicholson, G.; Irran, E.; Süssmuth, R.D.; et al. Benzoxacystol, a benzoxazine-type enzyme inhibitor from the deep-sea strain Streptomyces sp. NTK 935. J. Antibiot. 2011, 64, 453–457. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, M.W.; Bugni, T.S.; Concepcion, G.P.; Coombs, G.S.; Harper, M.K.; Kaur, S.; Mangalindan, G.C.; Mutizwa, M.M.; Veltri, C.A.; Virshup, D.M.; et al. Carteriosulfonic Acids A−C, GSK-3β Inhibitors from a Carteriospongia sp. J. Nat. Prod. 2009, 72, 1651–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezura, Y.; Kajita, M.; Ishida, R.; Yoshida, S.; Yoshida, H.; Suzuki, T.; Hosoi, T.; Inoue, S.; Shiraki, M.; Orimo, H.; et al. Association of multiple nucleotide variations in the pituitary glutaminyl cyclase gene (QPCT) with low radial BMD in adult women. J. Bone Miner. Res. 2004, 19, 1296–1301. [Google Scholar] [CrossRef] [Green Version]
- Cynis, H.; Scheel, E.; Saido, T.C.; Schilling, S.; Demuth, H.-U. Amyloidogenic processing of amyloid precursor protein: Evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-β. Biochemistry 2008, 47, 7405–7413. [Google Scholar] [CrossRef]
- Höfling, C.; Indrischek, H.; Höpcke, T.; Waniek, A.; Cynis, H.; Koch, B.; Schilling, S.; Morawski, M.; Demuth, H.-U.; Roßner, S.; et al. Mouse strain and brain region-specific expression of the glutaminyl cyclases QC and isoQC. Int. J. Dev. Neurosci. 2014, 36, 64–73. [Google Scholar] [CrossRef]
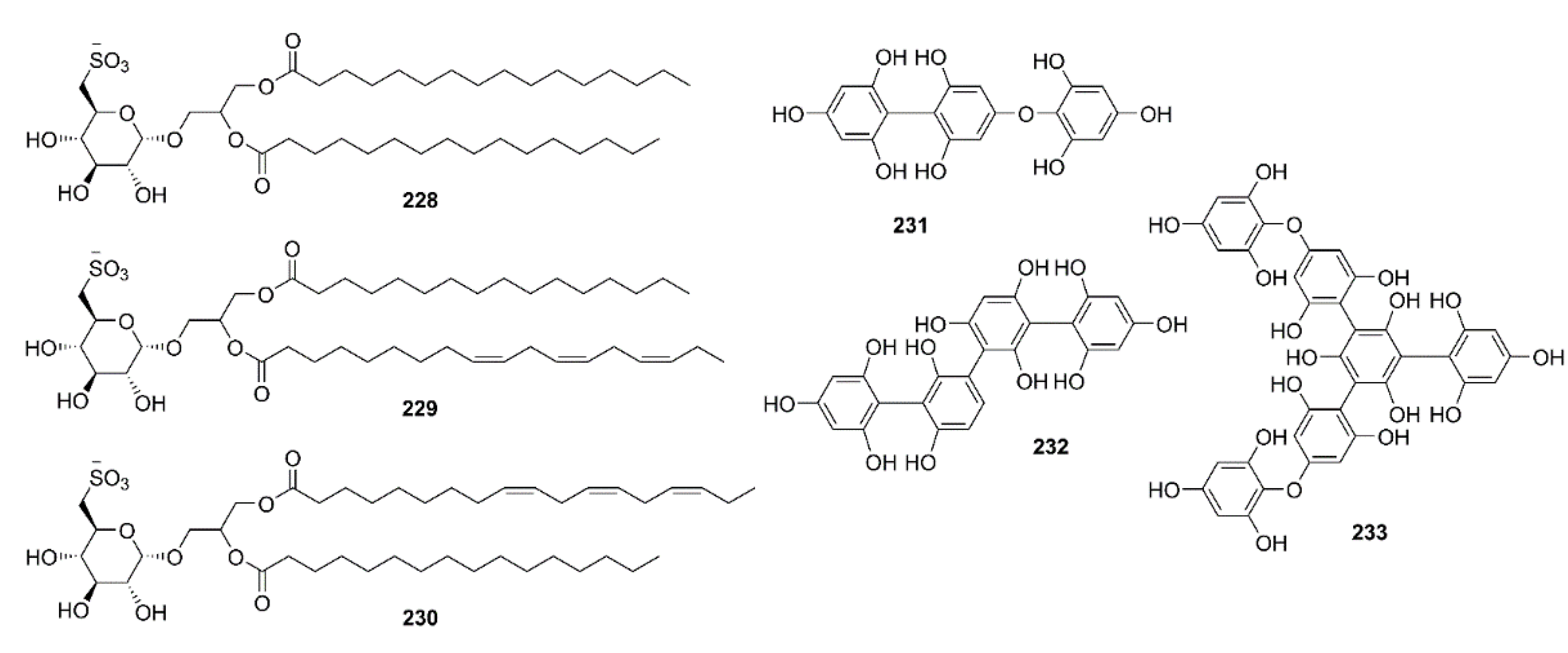
- Hielscher-Michael, S.; Griehl, C.; Buchholz, M.; Demuth, H.-U.; Arnold, N.; Wessjohann, L.A. Natural products from microalgae with potential against Alzheimer’s disease: Sulfolipids are potent glutaminyl cyclase inhibitors. Mar. Drugs 2016, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Münch, G.; Thome, J.; Foley, P.; Schinzel, R.; Riederer, P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res. Rev. 1997, 23, 134–143. [Google Scholar] [CrossRef]
- Liu, H.; Gu, L. Phlorotannins from brown algae (Fucus vesiculosus) inhibited the formation of advanced glycation endproducts by scavenging reactive carbonyls. J. Agric. Food Chem. 2012, 60, 1326–1334. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafez Ghoran, S.; Kijjoa, A. Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities. Mar. Drugs 2021, 19, 410. https://doi.org/10.3390/md19080410
Hafez Ghoran S, Kijjoa A. Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities. Marine Drugs. 2021; 19(8):410. https://doi.org/10.3390/md19080410
Chicago/Turabian StyleHafez Ghoran, Salar, and Anake Kijjoa. 2021. "Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities" Marine Drugs 19, no. 8: 410. https://doi.org/10.3390/md19080410
APA StyleHafez Ghoran, S., & Kijjoa, A. (2021). Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities. Marine Drugs, 19(8), 410. https://doi.org/10.3390/md19080410