
Anti-Alzheimer’s Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Current Alzheimer’s Drug Therapy
3. Therapeutic Potential of Marine-Derived Anti-Alzheimer’s Molecules
3.1. Bryostatin-1
3.1.1. Preclinical Evidence
3.1.2. Clinical Evidence
3.2. Homotaurine
3.2.1. Preclinical Evidence
3.2.2. Clinical Evidence
3.3. Anabaseine and Its Derivative GTS-21
3.3.1. Preclinical Evidence
3.3.2. Clinical Evidence
3.4. Rifampicin
3.4.1. Preclinical Evidence
3.4.2. Clinical Evidence
3.5. Dictyostatin
3.5.1. Preclinical Evidence
3.5.2. Clinical Evidence
3.6. Anhydroexfoliamycin and Undecylprodigioisin
3.7. Gracilins
3.8. 13-Desmethyl Spirolide-C
3.9. Docosahexaenoic Acid
3.9.1. Preclinical Evidence
3.9.2. Clinical Evidence

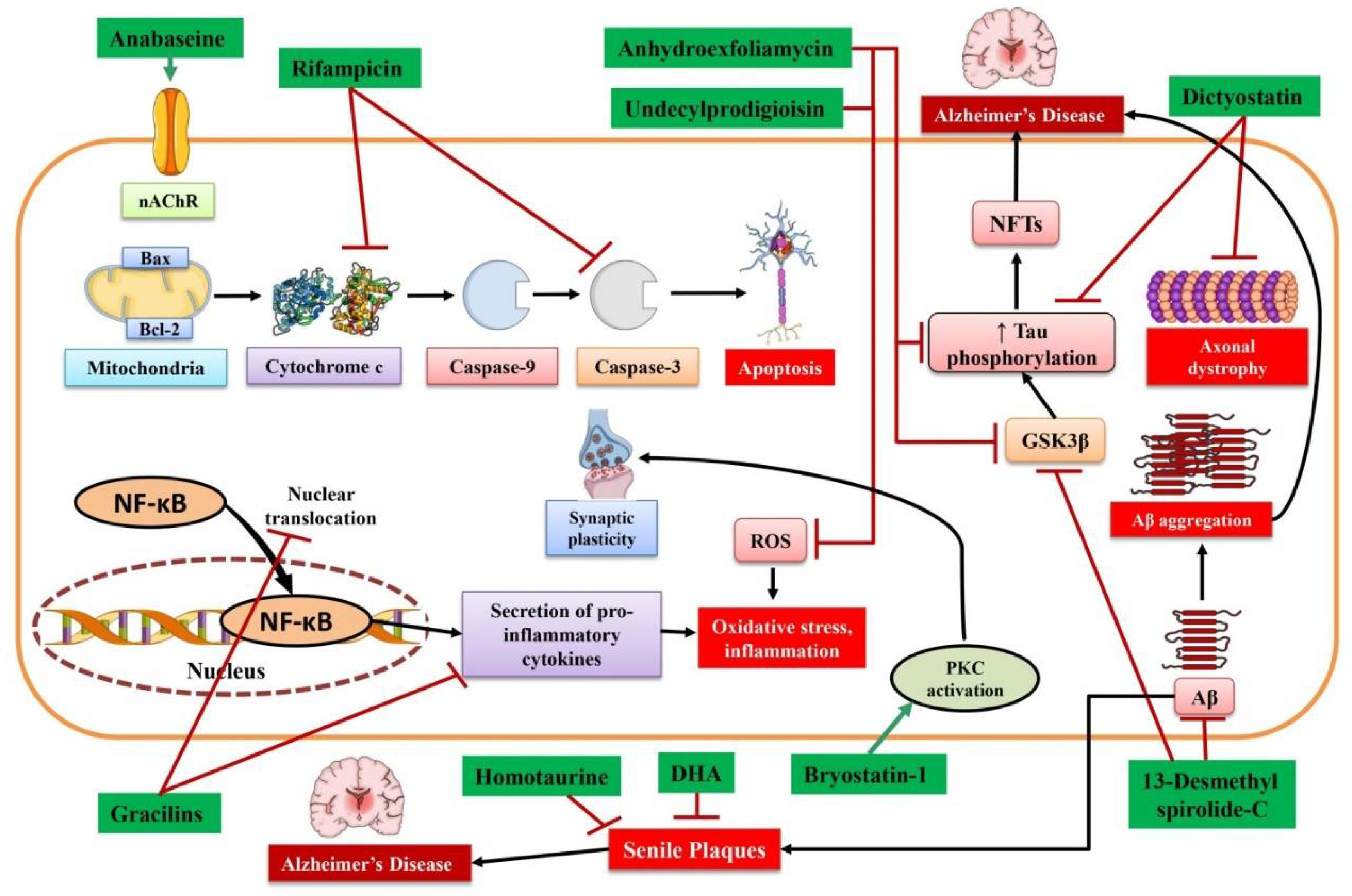{kind=link}
{kind=link}
| Compound | Marine Source | Mechanism of Action | Results of Animal Studies | Outcomes of Clinical Studies | References |
|---|---|---|---|---|---|
| Bryostatin-1 | Bugula neritina | Enhances spatial learning and memory; improves cognitive function and activities of daily living in moderate-to-severe AD; decreases Aβ level; retrieves neurotrophic activity; modulates neuronal synapses under synaptic dysfunctions | Activated PKCε in the brain and prevented Aβ accumulation of synaptic loss and memory deficit in AD transgenic mouse models; mediated preservation of synapses and improved memory in aged rat models; improved learning and memory in an AD mouse model | Increased the Mini-Mental State Examination (MMSE) score of AD patients | [34,43,53,54,58,136,137] |
| Homotaurine (tramiprosate) | Seaweed | Activates sirtuin 1; decreases the formation of Aβ oligomers and deposition of amyloid fibrils as plaques | Reduced the formation of Aβ oligomers and deposition of amyloid fibrils as plaques in an AD mouse model | Safely decreased the levels of Aβ42 in the cerebrospinal fluid (CSF) of individuals with mild-to-moderate AD | [63,65] |
| Anabaseine | Ribbon worm (Amphiporus sp.) | Potent agonist of alpha-bungarotoxin-sensitive nicotinic receptors; improves memory | Stimulated nicotinic acetylcholine receptors (especially at the neuromuscular junction) in various animal models | GTS-21 (a synthetic anabaseine derivative) exerted a statistically significant improvement in cognitive function, including episodic secondary memory, working memory, and attention | [71,73,77,138] |
| Rifamycin | Pseudoceratina clavata | Reduces neuroinflammation and free radical injury; exerts significant neuroprotective activity; suppresses fibril formation and aggregation of Aβ1–40; suppresses Aβ aggregation | Inhibited fibril formation and aggregation of synthetic Aβ1–40 and prevented neurotoxic effects in pheochromocytoma PC12 rat cells in a dose-dependent manner; exerted marked effects against the buildup of tau oligomers and Aβ in multiple transgenic mouse models | Exhibited an abnormal absence of senile plaques in the brains of leprosy patients; anti-dementia effects in 101 patients with mild-to-moderate AD | [79,83,89,96,98] |
| Dictyostatin | Spongia sp. | Improves density of microtubules; reduces levels of axonal dystrophy and tau pathology; increases survival rate of hippocampal neurons | Improved density of microtubules and reduced levels of axonal dystrophy along with a decreased level of tau pathology and a tendency towards an elevated survival rate of hippocampal neurons in mouse models | Exerted beneficial effects in mild-moderate AD individuals; showed no significant effect in case of Clinical Dementia Rating Scale Sum of Boxes (CDR-SB) | [98,104,105] |
| Anhydroexfoliamycin and undecylprodigioisin | Streptomyces | Exert antioxidant properties; decrease the levels of reactive oxygen species (ROS); elevates the glutathione levels and catalase activity; induces nuclear factor erythroid 2–related factor 2 (Nrf2); reduces the caspase-3 effect; preserves the mitochondrial membrane potential (MMP) | Undecylprodigiosin exerted poor outcomes; anhydroexfoliamycin significantly suppressed GSK3β and decreased tau phosphorylation | - | [106,107] |
| Gracilins | Spongionella gracilis | Shows anti-inflammatory activities; neuroprotective and antioxidant properties; suppresses BACE-1; decreases tau phosphorylation; | Provided protection to SH-SY5Y cells against hydrogen peroxide-induced injury via reducing reactive oxygen species (ROS) levels, recovering GSH content, improving MMP, and elevating cell survival; regulated the translocation of NF-κB and Nrf2 and reduced the activation of p38 in SH-SY5Y and BV2 cells | - | [109,110,111,139] |
| 13-Desmethyl spirolide C | Alexandrium ostenfeldii | Decreases intracellular Aβ accumulation and levels of hyperphosphorylated tau; reduces intracellular levels of Aβ | Reduced intracellular accumulation of Aβ and phosphorylated tau levels; decreased acetylcholine-mediated effects and elevated ACh levels; reduced the levels of 2 protein kinases linked with ERK, GSK-3β, and tau phosphorylation; eliminated the glutamate-induced neurotoxic effects; mediated positive outcomes on AD markers; increased levels of N-acetyl aspartate | - | [113,114,115] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paris, D.; Beaulieu-Abdelahad, D.; Bachmeier, C.; Reed, J.; Ait-Ghezala, G.; Bishop, A.; Chao, J.; Mathura, V.; Crawford, F.; Mullan, M. Anatabine lowers Alzheimer’s Aβ production in vitro and in vivo. Eur. J. Pharmacol. 2011, 670, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Kabir, M.T.; Rahman, M.S.; Behl, T.; Jeandet, P.; Ashraf, G.M.; Najda, A.; Bin-Jumah, M.N.; El-Seedi, H.R.; Abdel-Daim, M.M. Revisiting the Amyloid Cascade Hypothesis: From Anti-Aβ Therapeutics to Auspicious New Ways for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 5858. [Google Scholar] [CrossRef]
- Uddin, M.S.; Sumsuzzman, D.M.; Jeandet, P.; Behl, T.; Rauf, A.; Amran, M.S.; Ashraf, G.M. Deciphering the Interacting Mechanisms of Circadian Disruption and Alzheimer’s Disease. Neurochem. Res. 2021, 1–15. [Google Scholar] [CrossRef]
- Isik, A.T. Late onset Alzheimer’s disease in older people. Clin. Interv. Aging 2010, 5, 307–311. [Google Scholar] [CrossRef]
- Wisniewski, T.; Dowjat, W.K.; Buxbaum, J.D.; Khorkova, O.; Efthimiopoulos, S.; Kulczycki, J.; Lojkowska, W.; Wegiel, J.; Wisniewski, H.M.; Frangione, B. A novel polish presenilin-1 mutation (P117L) is associated with familial Alzheimer’s disease and leads to death as early as the age of 28 years. Neuroreport 1998, 9, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Painter, M.M.; Bu, G.; Kanekiyo, T. Apolipoprotein E as a Therapeutic Target in Alzheimer’s Disease: A Review of Basic Research and Clinical Evidence. CNS Drugs 2016, 30, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Sahab Uddin, M.; Fahim Bin Bashar, M.; Zaman, S.; Begum, Y.; Jahan Bulbul, I.; Siddiqul Islam, M.; Shahid Sarwar, M.; Mathew, B.; Shah Amran, M.; et al. Molecular Insight into the Therapeutic Promise of Targeting APOE4 for Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 5086250. [Google Scholar] [CrossRef]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Iqbal, K.; Del, C.; Alonso, A.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 198–210. [Google Scholar] [CrossRef]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Ashraf, G.M. Neurotoxic Aβ: Linking Extracellular and Intracellular Aβ in Alzheimer’s Disease. Curr. Protein Pept. Sci. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef]
- Pająk, B.; Kania, E.; Orzechowski, A. Killing Me Softly: Connotations to Unfolded Protein Response and Oxidative Stress in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease-linked presenilin I variants elevate aβ1- 42/1-40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T. The Carboxy Terminus of the β Amyloid Protein Is Critical for the Seeding of Amyloid Formation: Implications for the Pathogenesis of Alzheimer’s Disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Odaka, A.; Suzuki, N.; Mizusawa, H.; Nukina, N.; Ihara, Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13, 45–53. [Google Scholar] [CrossRef]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 Suppresses β-Amyloid Production by Activating the α-Secretase Gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef]
- Holsinger, R.M.D.; McLean, C.A.; Beyreuther, K.; Masters, C.L.; Evin, G. Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann. Neurol. 2002, 51, 783–786. [Google Scholar] [CrossRef]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef]
- Mamun, A.; Uddin, M.; Mathew, B.; Ashraf, G. Toxic tau: Structural origins of tau aggregation in Alzheimer’s disease. Neural Regen. Res. 2020, 15, 1417. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef]
- Lindequist, U. Marine-derived pharmaceuticals—Challenges and opportunities. Biomol. Ther. 2016, 24, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bahbah, E.I.; Ghozy, S.; Attia, M.S.; Negida, A.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Abdel-Daim, M.M.; Uddin, M.S.; Simal-Gandara, J. Molecular Mechanisms of Astaxanthin as a Potential Neurotherapeutic Agent. Mar. Drugs 2021, 19, 201. [Google Scholar] [CrossRef]
- Kong, D.X.; Jiang, Y.Y.; Zhang, H.Y. Marine natural products as sources of novel scaffolds: Achievement and concern. Drug Discov. Today 2010, 15, 884–886. [Google Scholar] [CrossRef]
- Liu, L.; Zheng, Y.-Y.; Shao, C.-L.; Wang, C.-Y. Metabolites from marine invertebrates and their symbiotic microorganisms: Molecular diversity discovery, mining, and application. Mar. Life Sci. Technol. 2019, 1, 60–94. [Google Scholar] [CrossRef]
- Nii-Trebi, N.I. Emerging and Neglected Infectious Diseases: Insights, Advances, and Challenges. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Jha, R.K.; Zi-rong, X. Biomedical Compounds from Marine organisms. Mar. Drugs 2004, 2, 123–146. [Google Scholar] [CrossRef]
- Petersen, L.-E.; Kellermann, M.Y.; Schupp, P.J. Secondary Metabolites of Marine Microbes: From Natural Products Chemistry to Chemical Ecology. In YOUMARES 9-The Oceans: Our Research, Our Future: Proceedings of the 2018 conference for YOUng MArine RESearcher in Oldenburg; Springer Nature: Berlin, Germany, 2020; pp. 159–180. [Google Scholar] [CrossRef]
- Tian, X.R.; Tang, H.F.; Tian, X.L.; Hu, J.J.; Huang, L.L.; Gustafson, K.R. Review of bioactive secondary metabolites from marine bryozoans in the progress of new drugs discovery. Future Med. Chem. 2018, 10, 1497–1514. [Google Scholar] [CrossRef]
- Nelson, T.J.; Sun, M.K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin Effects on Cognitive Function and PKCϵ in Alzheimer’s Disease Phase IIa and Expanded Access Trials. J. Alzheimers Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Tayeb, H.O.; Yang, H.D.; Price, B.H.; Tarazi, F.I. Pharmacotherapies for Alzheimer’s disease: Beyond cholinesterase inhibitors. Pharmacol. Ther. 2012, 134, 8–25. [Google Scholar] [CrossRef]
- Dekker, M.J.H.J.; Bouvy, J.C.; O’Rourke, D.; Thompson, R.; Makady, A.; Jonsson, P.; Gispen-de Wied, C.C. Alignment of European Regulatory and Health Technology Assessments: A Review of Licensed Products for Alzheimer’s Disease. Front. Med. 2019, 6, 73. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Begum, M.M.; Thangapandiyan, S.; Rahman, M.S.; Aleya, L.; Mathew, B.; Ahmed, M.; Ashraf, G.M.; Barreto, G.E. Cholinesterase Inhibitors for Alzheimer’s Disease: Multitargeting Strategy based on Anti-Alzheimer’s Drugs Repositioning. Curr. Pharm. Des. 2019, 25, 3519–3535. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Rolinski, M.; Fox, C.; Maidment, I.; Mcshane, R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst. Rev. 2012, 2012. [Google Scholar] [CrossRef]
- Kabir, M.T.; Abu Sufian, M.; Uddin, M.S.; Begum, M.M.; Akhter, S.; Islam, A.; Mathew, B.; Islam, M.S.; Amran, M.S.; Md Ashraf, G. NMDA Receptor Antagonists: Repositioning of Memantine as Multitargeting Agent for Alzheimer’s Therapy. Curr. Pharm. Des. 2019, 25, 3506–3518. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New drugs from marine organisms in Alzheimer’s disease. Mar. Drugs 2016, 14, 5. [Google Scholar] [CrossRef]
- Deardorff, W.J.; Grossberg, G.T. A fixed-dose combination of memantine extended-release and donepezil in the treatment of moderate-to-severe Alzheimer’s disease. Drug Des. Dev. Ther. 2016, 10, 3267–3279. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.S.; Mamun, A.A.; Jeandet, P.; Aleya, L.; Mansouri, R.A.; Ashraf, G.M.; Mathew, B.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Combination Drug Therapy for the Management of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3272. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Kabir, M.T.; Ashraf, G.M.; Bin-Jumah, M.N.; Abdel-Daim, M.M. Multi-Target Drug Candidates for Multifactorial Alzheimer’s Disease: AChE and NMDAR as Molecular Targets. Mol. Neurobiol. 2021, 58, 281–303. [Google Scholar] [CrossRef]
- Mizuno, S.; Iijima, R.; Ogishima, S.; Kikuchi, M.; Matsuoka, Y.; Ghosh, S.; Miyamoto, T.; Miyashita, A.; Kuwano, R.; Tanaka, H. AlzPathway: A comprehensive map of signaling pathways of Alzheimer’s disease. BMC Syst. Biol. 2012, 6. [Google Scholar] [CrossRef]
- Kabir, M.T.; Uddin, M.S.; Mathew, B.; Das, P.K.; Perveen, A.; Ashraf, G.M. Emerging Promise of Immunotherapy for Alzheimer’s Disease: A New Hope for the Development of Alzheimer’s Vaccine. Curr. Top. Med. Chem. 2020, 20, 1214–1234. [Google Scholar] [CrossRef] [PubMed]
- Pul, R.; Dodel, R.; Stangel, M. Antibody-based therapy in Alzheimer’s disease. Expert Opin. Biol. Ther. 2011, 11, 343–357. [Google Scholar] [CrossRef]
- Trindade-Silva, A.E.; Lim-Fong, G.E.; Sharp, K.H.; Haygood, M.G. Bryostatins: Biological context and biotechnological prospects. Curr. Opin. Biotechnol. 2010, 21, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Nelson, T.J.; Alkon, D.L. Towards universal therapeutics for memory disorders. Trends Pharmacol. Sci. 2015, 36, 384–394. [Google Scholar] [CrossRef]
- Yi, P.; Schrott, L.; Castor, T.P.; Alexander, J.S. Bryostatin-1 vs. TPPB: Dose-dependent APP processing and PKC-α, -δ, and -ε Isoform activation in SH-SY5Y neuronal cells. J. Mol. Neurosci. 2012, 48, 234–244. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Tan, M.; Dewachtert, I.; Kuipéri, C.; Van Der Auwera, I.; Wera, S.; Qiao, L.; Bank, B.; Nelson, T.J.; Kozikowski, A.P.; et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 11141–11146. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKC ε activation prevents synaptic loss, Aβ elevation, and cognitive deficits in alzheimer’s disease transgenic mice. J. Neurosci. 2011, 31, 630–643. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Xu, C.; Sen, A.; Nelson, T.J.; Alkon, D.L. PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol. Dis. 2013, 55, 44–62. [Google Scholar] [CrossRef]
- Sun, M.K.; Hongpaisan, J.; Nelson, T.J.; Alkon, D.L. Poststroke neuronal rescue and synaptogenesis mediated in vivo by protein kinase C in adult brains. Proc. Natl. Acad. Sci. USA 2008, 105, 13620–13625. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Hongpaisan, J.; Alkon, D.L. Rescue of synaptic phenotypes and spatial memory in Young Fragile X Mice. J. Pharmacol. Exp. Ther. 2016, 357, 300–310. [Google Scholar] [CrossRef]
- Schrott, L.M.; Jackson, K.; Yi, P.; Dietz, F.; Johnson, G.S.; Basting, T.F.; Purdum, G.; Tyler, T.; Rios, J.D.; Castor, T.P.; et al. Acute Oral Bryostatin-1 Administration Improves Learning Deficits in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Smith, M.D.; Shirazi, H.A.; Calabresi, P.A.; Snyder, S.H.; Kim, P.M. Bryostatin-1 alleviates experimental multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 2186–2191. [Google Scholar] [CrossRef]
- Farlow, M.R.; Thompson, R.E.; Wei, L.J.; Tuchman, A.J.; Grenier, E.; Crockford, D.; Wilke, S.; Benison, J.; Alkon, D.L.; Moreira, P. A randomized, double-blind, placebo-controlled, phase II study assessing safety, tolerability, and efficacy of bryostatin in the treatment of moderately severe to severe Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 555–570. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study of Bryostatin in Moderately Severe to Severe Alzheimers Disease Subjects Not On Memantine. Available online: https://clinicaltrials.gov/ct2/show/NCT03560245 (accessed on 20 March 2021).
- Tsolaki, M. Future strategies of management of Alzheimer’s Disease. The role of homotaurine. Hell. J. Nucl. Med. 2019, 22, 82–94. [Google Scholar]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]
- Santa-Maria, I.; Hernández, F.; Del Rio, J.; Moreno, F.J.; Avila, J. Tramiprosate, a drug of potential interest for the treatment of Alzheimer’s disease, promotes an abnormal aggregation of tau. Mol. Neurodegener. 2007, 2. [Google Scholar] [CrossRef]
- Aisen, P.S.; Saumier, D.; Briand, R.; Laurin, J.; Gervais, F.; Tremblay, P.; Garceau, D. A Phase II study targeting amyloid-β with 3APS in mild-to-moderate Alzheimer disease. Neurology 2006, 67, 1757–1763. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—A randomized, double-blind, placebo-controlled, multi-centre study (the alphase study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef]
- Abushakra, S.; Porsteinsson, A.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Hendrix, S.; Wang, P.; Shen, L.; et al. Clinical Benefits of Tramiprosate in Alzheimer’s Disease Are Associated with Higher Number of APOE4 Alleles: The “APOE4 Gene-Dose Effect”. J. Prev. Alzheimers Dis. 2016, 3, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N. Clinical Effects of Oral Tramiprosate in APOE4/4 Homozygous Patients with Mild Alzheimer’s Disease Suggest Disease Modification. J. Prev. Alzheimers Dis. 2017, 4, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Aisen, P.S.; Ferris, S.H.; Saumier, D.; Duong, A.; Haine, D.; Garceau, D.; Suhy, J.; Oh, J.; Lau, W.; et al. Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: Exploratory analyses of the MRI sub-group of the Alphase study. J. Nutr. Health Aging 2009, 13, 550–557. [Google Scholar] [CrossRef]
- Saumier, D.; Aisen, P.S.; Gauthier, S.; Vellas, B.; Ferris, S.H.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.; Garceau, D.; et al. Lessons learned in the use of volumetric MRI in therapeutic trials in Alzheimer’s disease: The ALZHEMEDTM (tramiprosate) experience. J. Nutr. Health Aging 2009, 13, 370–372. [Google Scholar] [CrossRef]
- Kem, W.; Soti, F.; Wildeboer, K.; LeFrancois, S.; MacDougall, K.; Wei, D.-Q.; Chou, K.-C.; Arias, H.R. The Nemertine Toxin Anabaseine and Its Derivative DMXBA (GTS-21): Chemical and Pharmacological Properties. Mar. Drugs 2006, 4, 255–273. [Google Scholar] [CrossRef]
- Schaller, S.J.; Nagashima, M.; Schönfelder, M.; Sasakawa, T.; Schulz, F.; Khan, M.A.S.; Kem, W.R.; Schneider, G.; Schlegel, J.; Lewald, H.; et al. GTS-21 attenuates loss of body mass, muscle mass, and function in rats having systemic inflammation with and without disuse atrophy. Pflugers Arch. Eur. J. Physiol. 2018, 470, 1647–1657. [Google Scholar] [CrossRef]
- Kem, W.R.; Mahnir, V.M.; Papke, R.L.; Lingle, C.J. Anabaseine Is a Potent Agonist on Muscle and Neuronal Alpha-Bungarotoxin-Sensitive Nicotinic Receptors. J. Pharmacol. Exp. Ther. 1997, 283, 979–992. [Google Scholar]
- Briggs, C.A.; Anderson, D.J.; Brioni, J.D.; Buccafusco, J.J.; Buckley, M.J.; Campbell, J.E.; Decker, M.W.; Donnelly-Roberts, D.; Elliott, R.L.; Gopalakrishnan, M.; et al. Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol. Biochem. Behav. 1997, 57, 231–241. [Google Scholar] [CrossRef]
- Meyer, E.M.; Tay, E.T.; Papke, R.L.; Meyers, C.; Huang, G.L.; De Fiebre, C.M. 3-[2,4-dimethoxybenzylidene]anabaseine (DMXB) selectively activates rat α 7 receptors and improves memory-related behaviors in a mecamylamine- sensitive manner. Brain Res. 1997, 768, 49–56. [Google Scholar] [CrossRef]
- Russo, P.; Bufalo, A.; Frustaci, A.; Fini, M.; Cesario, A. Beyond Acetylcholinesterase Inhibitors for Treating Alzheimer&aposs Disease:.α7-nAChR Agonists in Human Clinical Trials. Curr. Pharm. Des. 2014, 20, 6014–6021. [Google Scholar] [CrossRef]
- Kitagawa, H.; Takenouchi, T.; Azuma, R.; Wesnes, K.A.; Kramer, W.G.; Clody, D.E.; Burnett, A.L. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology 2003, 28, 542–551. [Google Scholar] [CrossRef]
- Kim, T.K.; Hewavitharana, A.K.; Shaw, P.N.; Fuerst, J.A. Discovery of a new source of rifamycin antibiotics in marine sponge actinobacteria by phylogenetic prediction. Appl. Environ. Microbiol. 2006, 72, 2118–2125. [Google Scholar] [CrossRef]
- Kilic, Ü.; Kilic, E.; Lingor, P.; Yulug, B.; Bähr, M. Rifampicin inhibits neurodegeneration in the optic nerve transection model in vivo and after 1-methyl-4-phenylpyridinium intoxication in vitro. Acta Neuropathol. 2004, 108, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Dyrks, T.; Dyrks, E.; Masters, C.L.; Beyreuther, K. Amyloidogenicity of rodent and human βA4 sequences. FEBS Lett. 1993, 324, 231–236. [Google Scholar] [CrossRef]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Asano, S.; Suwa, Y.; Morita, T.; Kataoka, K.I.; Mori, H.; Endo, N. Rifampicin prevents the aggregation and neurotoxicity of amyloid β protein in vitro. Biochem. Biophys. Res. Commun. 1994, 204, 76–83. [Google Scholar] [CrossRef]
- Mindermann, T.; Landolt, H.; Zimmerli, W.; Rajacic, Z.; Gratzl, O. Penetration of rifampicin into the brain tissue and cerebral extracellular space of rats. J. Antimicrob. Chemother. 1993, 31, 731–737. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tomiyama, T.; Kaneko, H.; Kataoka, K.I.; Asano, S.; Endo, N. Rifampicin inhibits the toxicity of pre-aggregated amyloid peptides by binding to peptide fibrils and preventing amyloid-cell interaction. Biochem. J. 1997, 322, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Findeis, M.A. Approaches to discovery and characterization of inhibitors of amyloid β-peptide polymerization. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 76–84. [Google Scholar] [CrossRef]
- Lieu, V.H.; Wu, J.W.; Wang, S.S.S.; Wu, C.H. Inhibition of amyloid fibrillization of hen egg-white lysozymes by rifampicin and p-benzoquinone. Biotechnol. Prog. 2007, 23, 698–706. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Yoshiike, Y.; Takashima, A.; Yamada, M.; Naiki, H. Nordihydroguaiaretic acid potently breaks down pre-formed Alzheimer’s β-amyloid fibrils in vitro. J. Neurochem. 2002, 81, 434–440. [Google Scholar] [CrossRef]
- Ono, K.; Hamaguchi, T.; Naiki, H.; Yamada, M. Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 575–586. [Google Scholar] [CrossRef]
- Umeda, T.; Ono, K.; Sakai, A.; Yamashita, M.; Mizuguchi, M.; Klein, W.L.; Yamada, M.; Mori, H.; Tomiyama, T. Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain 2016, 139, 1568–1586. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-β peptides—Implications for the mechanisms of Aβ clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-Î2 clearance in Alzheimerâ€TMs disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced brain amyloid-β clearance by rifampicin and caffeine as a possible protective mechanism against alzheimer’s disease. J. Alzheimers Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Sodhi, R.K. Memory recuperative potential of rifampicin in aluminum chloride-induced dementia: Role of pregnane X receptors. Neuroscience 2015, 288, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Kawatsu, K.; Izumi, S.; Ueki, A.; Ikeda, K. Neurofibrillary tangles and senile plaques in brain of elderly leprosy patients. Lancet 1992, 340, 978. [Google Scholar] [CrossRef]
- Endoh, M.; Kunishita, T.; Tabira, T. No effect of anti-leprosy drugs in the prevention of Alzheimer’s disease and β-amyloid neurotoxicity. J. Neurol. Sci. 1999, 165, 28–30. [Google Scholar] [CrossRef]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A Randomized, Controlled Trial of Doxycycline and Rifampin for Patients with Alzheimer’s Disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Morimoto, K.; Sasaki, Y.; Kameyama, M.; Kurashima, A.; Hayasaka, K.; Ogata, H.; Goto, H. Preventive Effect of Rifampicin on Alzheimer Disease Needs at Least 450 mg Daily for 1 Year: An FDG-PET Follow-Up Study. Dement. Geriatr. Cogn. Dis. Extra 2017, 7, 204–214. [Google Scholar] [CrossRef]
- Umeda, T.; Tanaka, A.; Sakai, A.; Yamamoto, A.; Sakane, T.; Tomiyama, T. Intranasal rifampicin for Alzheimer’s disease prevention. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 304–313. [Google Scholar] [CrossRef]
- Paterson, I.; Britton, R.; Delgado, O.; Gardner, N.M.; Meyer, A.; Naylor, G.J.; Poullennec, K.G. Total synthesis of (-)-dictyostatin, a microtubule-stabilising anticancer macrolide of marine sponge origin. Tetrahedron 2010, 66, 6534–6545. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Frontiers Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef]
- Makani, V.; Zhang, B.; Han, H.; Yao, Y.; Lassalas, P.; Lou, K.; Paterson, I.; Lee, V.M.Y.; Trojanowski, J.Q.; Ballatore, C.; et al. Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol. Commun. 2016, 4, 106. [Google Scholar] [CrossRef]
- Molloy, D.W.; Standish, T.I.; Zhou, Q.; Guyatt, G. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int. J. Geriatr. Psychiatry 2013, 28, 463–470. [Google Scholar] [CrossRef]
- Leirós, M.; Alonso, E.; Sanchez, J.A.; Rateb, M.E.; Ebel, R.; Houssen, W.E.; Jaspars, M.; Alfonso, A.; Botana, L.M. Mitigation of ROS insults by streptomyces secondary metabolites in primary cortical neurons. ACS Chem. Neurosci. 2014, 5, 71–80. [Google Scholar] [CrossRef]
- Leirós, M.; Alonso, E.; Rateb, M.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. The Streptomyces metabolite anhydroexfoliamycin ameliorates hallmarks of Alzheimer’s disease in vitro and in vivo. Neuroscience 2015, 305, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Gegunde, S.; Alfonso, A.; Alonso, E.; Alvariño, R.; Botana, L.M. Gracilin-Derivatives as Lead Compounds for Anti-inflammatory Effects. Cell. Mol. Neurobiol. 2020, 40, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, N.; Praba, G.O.; Velmurugan, D. Modeling studies on phospholipase A2-inhibitor complexes. Indian J. Biochem. Biophys. 2008, 45, 256–262. [Google Scholar]
- Leirós, M.; Sánchez, J.A.; Alonso, E.; Rateb, M.E.; Houssen, W.E.; Ebel, R.; Jaspars, M.; Alfonso, A.; Botana, L.M. Spongionella secondary metabolites protect mitochondrial function in cortical neurons against oxidative stress. Mar. Drugs 2014, 12, 700–718. [Google Scholar] [CrossRef] [PubMed]
- Abbasov, M.E.; Alvariño, R.; Chaheine, C.M.; Alonso, E.; Sánchez, J.A.; Conner, M.L.; Alfonso, A.; Jaspars, M.; Botana, L.M.; Romo, D. Simplified immunosuppressive and neuroprotective agents based on gracilin A. Nat. Chem. 2019, 11, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Alvariño, R.; Alonso, E.; Abbasov, M.E.; Chaheine, C.M.; Conner, M.L.; Romo, D.; Alfonso, A.; Botana, L.M. Gracilin A Derivatives Target Early Events in Alzheimer’s Disease: In Vitro Effects on Neuroinflammation and Oxidative Stress. ACS Chem. Neurosci. 2019, 10, 4102–4111. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Do Carmo Pereira, M. Natural compounds for alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Alonso, E.; Vale, C.; Vieytes, M.R.; Laferla, F.M.; Giménez-Llort, L.; Botana, L.M. 13-Desmethyl spirolide-C is neuroprotective and reduces intracellular Aβ and hyperphosphorylated tau in vitro. Neurochem. Int. 2011, 59, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Otero, P.; Vale, C.; Alfonso, A.; Antelo, A.; Gimenez-Llort, L.; Chabaud, L.; Guillou, C.; M Botana, L. Benefit of 13-desmethyl Spirolide C Treatment in Triple Transgenic Mouse Model of Alzheimer Disease: Beta-Amyloid and Neuronal Markers Improvement. Curr. Alzheimer Res. 2013, 10, 279–289. [Google Scholar] [CrossRef]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease. JAMA J. Am. Med. Assoc. 2010, 304, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Calder, P.C. Dietary α-linolenic acid and health-related outcomes: A metabolic perspective. Nutr. Res. Rev. 2006, 19, 26–52. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Amos, E.C.; Canick, J.; Ackerley, M.; Raji, C.; Fiala, M.; Ahdidan, J. Reversal of cognitive decline in Alzheimer’s disease. Aging (Albany NY) 2016, 8, 1250–1258. [Google Scholar] [CrossRef]
- Lim, A.S.P.; Yu, L.; Kowgier, M.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A. Modification of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013, 70, 1544–1551. [Google Scholar] [CrossRef]
- Smith, J.C.; Nielson, K.A.; Woodard, J.L.; Seidenberg, M.; Durgerian, S.; Hazlett, K.E.; Figueroa, C.M.; Kandah, C.C.; Kay, C.D.; Matthews, M.A.; et al. Physical activity reduces hippocampal atrophy in elders at genetic risk for Alzheimer’s disease. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef]
- Green, K.N.; Martinez-Coria, H.; Khashwji, H.; Hall, E.B.; Yurko-Mauro, K.A.; Ellis, L.; LaFerla, F.M. Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-β and tau pathology via a mechanism involving presenilin 1 levels. J. Neurosci. 2007, 27, 4385–4395. [Google Scholar] [CrossRef]
- Pifferi, F.; Roux, F.; Langelier, B.; Alessandri, J.M.; Vancassel, S.; Jouin, M.; Lavialle, M.; Guesnet, P. (n-3) polyunsaturated fatty acid deficiency reduces the expression of both isoforms of the brain glucose transporter GLUT1 in rats. J. Nutr. 2005, 135, 2241–2246. [Google Scholar] [CrossRef]
- Da Silva, A.X.; Lavialle, F.; Gendrot, G.; Guesnet, P.; Alessandri, J.M.; Lavialle, M. Glucose transport and utilization are altered in the brain of rats deficient in n-3 polyunsaturated fatty acids. J. Neurochem. 2002, 81, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, F.; Jouin, M.; Alessandri, J.M.; Haedke, U.; Roux, F.; Perrière, N.; Denis, I.; Lavialle, M.; Guesnet, P. n-3 Fatty acids modulate brain glucose transport in endothelial cells of the blood-brain barrier. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Brenna, J.T.; Diau, G.Y. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Freund Levi, Y.; Vedin, I.; Cederholm, T.; Basun, H.; Faxén Irving, G.; Eriksdotter, M.; Hjorth, E.; Schultzberg, M.; Vessby, B.; Wahlund, L.O.; et al. Transfer of omega-3 fatty acids across the blood-brain barrier after dietary supplementation with a docosahexaenoic acid-rich omega-3 fatty acid preparation in patients with Alzheimer’s disease: The OmegAD study. J. Intern. Med. 2014, 275, 428–436. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxén-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.O.; Palmblad, J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: A randomized double-blind trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Basun, H.; Cederholm, T.; Faxén-Irving, G.; Garlind, A.; Grut, M.; Vedin, I.; Palmblad, J.; Wahlund, L.O.; Eriksdotter-Jönhagen, M. Omega-3 supplementation in mild to moderate Alzheimer’s disease: Effects on neuropsychiatric symptoms. Int. J. Geriatr. Psychiatry 2008, 23, 161–169. [Google Scholar] [CrossRef]
- Vedin, I.; Cederholm, T.; Freund-Levi, Y.; Basun, H.; Garlind, A.; Irving, G.F.; Eriksdotter-Jönhagen, M.; Wahlund, L.O.; Dahlman, I.; Palmblad, J. Effects of DHA- rich n-3 fatty acid supplementation on gene expression in blood mononuclear leukocytes: The omegAD study. PLoS ONE 2012, 7, e35425. [Google Scholar] [CrossRef]
- Vedin, I.; Cederholm, T.; Freund-Levi, Y.; Basun, H.; Garlind, A.; Irving, G.F.; Jönhagen, M.E.; Vessby, B.; Wahlund, L.O.; Palmblad, J. Effects of docosahexaenoic acid-rich n-3 fatty acid supplementation on cytokine release from blood mononuclear leukocytes: The OmegAD study. Am. J. Clin. Nutr. 2008, 87, 1616–1622. [Google Scholar] [CrossRef]
- Carrie, I.; Van Kan, G.A.; Gillette-Guyonnet, S.; Andrieu, S.; Dartigues, J.F.; Touchon, J.; Dantoine, T.; Rouaud, O.; Bonnefoy, M.; Robert, P.; et al. Recruitment strategies for preventive trials. the MAPT study (Multidomain Alzheimer preventive trial). J. Nutr. Health Aging 2012, 16, 355–359. [Google Scholar] [CrossRef]
- Gillette-Guyonnet, S.; Andrieu, S.; Dantoine, T.; Dartigues, J.F.; Touchon, J.; Vellas, B. Commentary on “A roadmap for the prevention of dementia II. Leon Thal Symposium 2008.” The Multidomain Alzheimer Preventive Trial (MAPT): A new approach to the prevention of Alzheimer’s disease. Alzheimers Dement. 2009, 5, 114–121. [Google Scholar] [CrossRef]
- Andrieu, S.; Guyonnet, S.; Coley, N.; Cantet, C.; Bonnefoy, M.; Bordes, S.; Bories, L.; Cufi, M.N.; Dantoine, T.; Dartigues, J.F.; et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): A randomised, placebo-controlled trial. Lancet Neurol. 2017, 16, 377–389. [Google Scholar] [CrossRef]
- Delrieu, J.; Payoux, P.; Carrié, I.; Cantet, C.; Weiner, M.; Vellas, B.; Andrieu, S. Multidomain intervention and/or omega-3 in nondemented elderly subjects according to amyloid status. Alzheimers Dement. 2019, 15, 1392–1401. [Google Scholar] [CrossRef]
- Hooper, C.; Coley, N.; De Souto Barreto, P.; Payoux, P.; Salabert, A.S.; Andrieu, S.; Weiner, M.; Vellas, B. Cortical β-Amyloid in Older Adults Is Associated with Multidomain Interventions with and without Omega 3 Polyunsaturated Fatty Acid Supplementation. J. Prev. Alzheimers Dis. 2020, 7, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; Burns, J.M.; Gorelick, K.J.; Crockford, D.R.; Grenier, E.; Wilke, S.; Cooper, E.C.; Alkon, D.L. Bryostatin-1 improves cognition and daily living tasks in moderate to severe Alzheimer’s disease: Preliminary report of a phase 2 study. Alzheimers Dement. 2017, 13, P1476. [Google Scholar] [CrossRef]
- Blanco, F.A.; Pany, S.; Ghosh, A.A.; You, Y.; Das, J. Novel Bryostatin-1 Targets: Mammalian Unc13-1 and Unc13-2 Isoforms. FASEB J. 2017, 31, 760.11. [Google Scholar] [CrossRef]
- Zawieja, P.; Kornprobst, J.-M.; Métais, P. 3-(2,4-Dimethoxybenzylidene)-anabaseine: A promising candidate drug for Alzheimer’s disease? Geriatr. Gerontol. Int. 2012, 12, 365–371. [Google Scholar] [CrossRef]
- Rateb, M.E.; Houssen, W.E.; Schumacher, M.; Harrison, W.T.A.; Diederich, M.; Ebel, R.; Jaspars, M. Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod. 2009, 72, 1471–1476. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabir, M.T.; Uddin, M.S.; Jeandet, P.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Sayed, A.A.; Abdel-Daim, M.M.; Simal-Gandara, J. Anti-Alzheimer’s Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential. Mar. Drugs 2021, 19, 251. https://doi.org/10.3390/md19050251
Kabir MT, Uddin MS, Jeandet P, Emran TB, Mitra S, Albadrani GM, Sayed AA, Abdel-Daim MM, Simal-Gandara J. Anti-Alzheimer’s Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential. Marine Drugs. 2021; 19(5):251. https://doi.org/10.3390/md19050251
Chicago/Turabian StyleKabir, Md. Tanvir, Md. Sahab Uddin, Philippe Jeandet, Talha Bin Emran, Saikat Mitra, Ghadeer M. Albadrani, Amany A. Sayed, Mohamed M. Abdel-Daim, and Jesus Simal-Gandara. 2021. "Anti-Alzheimer’s Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential" Marine Drugs 19, no. 5: 251. https://doi.org/10.3390/md19050251
APA StyleKabir, M. T., Uddin, M. S., Jeandet, P., Emran, T. B., Mitra, S., Albadrani, G. M., Sayed, A. A., Abdel-Daim, M. M., & Simal-Gandara, J. (2021). Anti-Alzheimer’s Molecules Derived from Marine Life: Understanding Molecular Mechanisms and Therapeutic Potential. Marine Drugs, 19(5), 251. https://doi.org/10.3390/md19050251