
Microbial Oligosaccharides with Biomedical Applications

Abstract
1. Introduction
2. Acarviosine-Containing Aminooligosaccharides
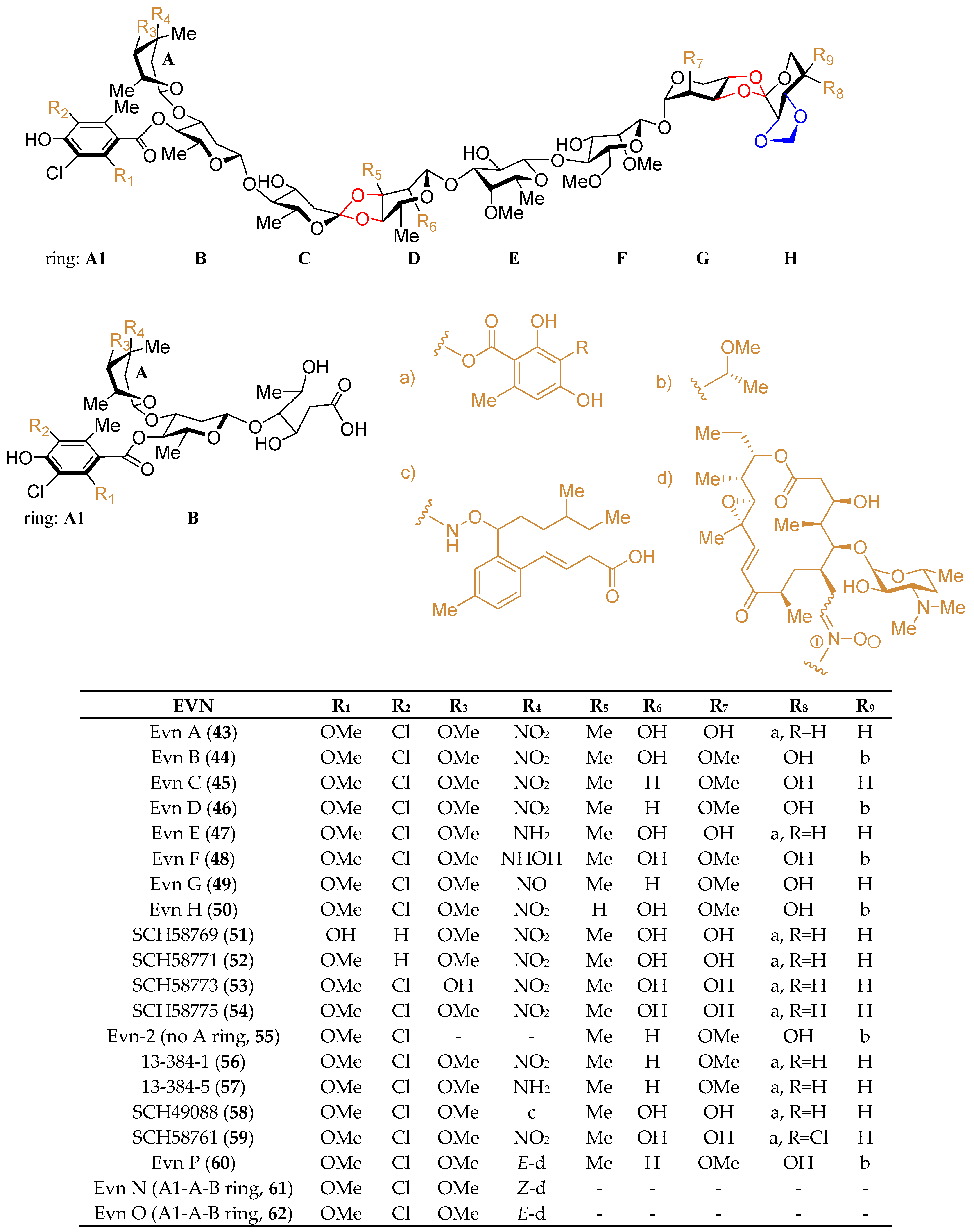
2.1. Structural Features and Biological Properties
2.2. Mode of Action
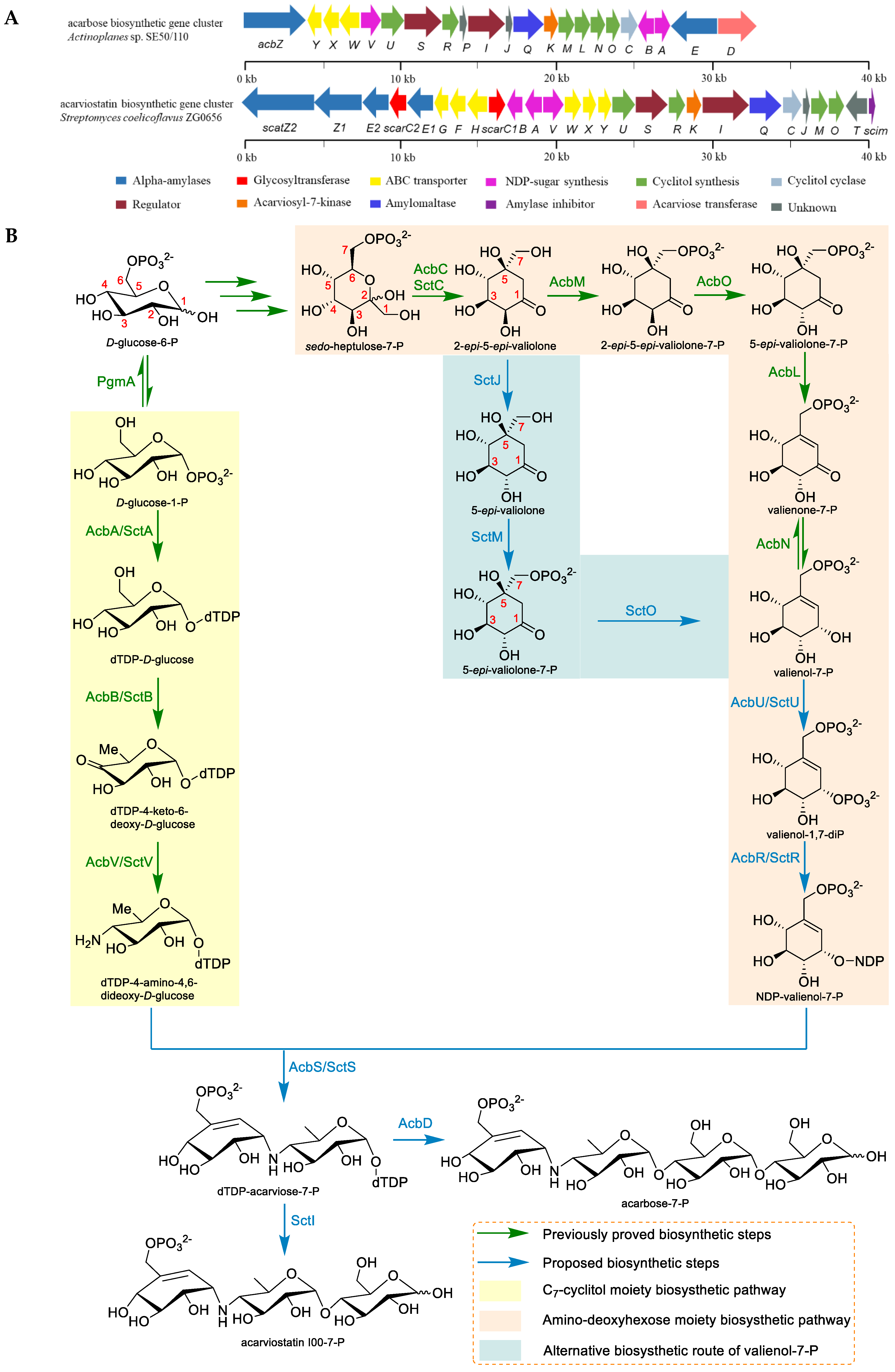
2.3. The Biosynthesis of Acarbose and the Acarviostatins
3. Saccharomicins
3.1. Structural Features and Biological Properties
3.2. Mode of Action
3.3. The Biosynthesis of Saccharomicin A
4. Orthosomycins
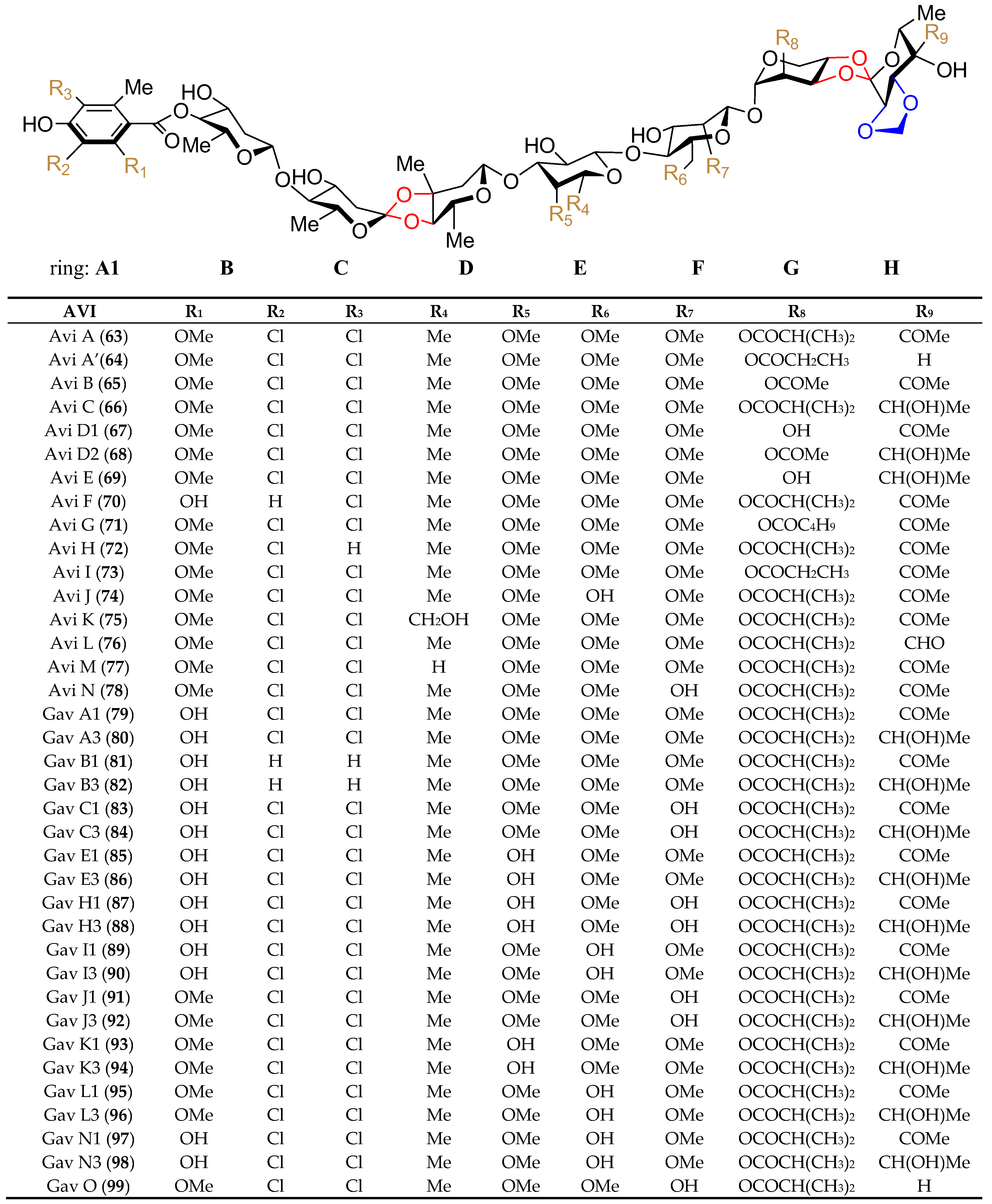
4.1. Structural Features and Biological Properties
4.2. Mode of Action
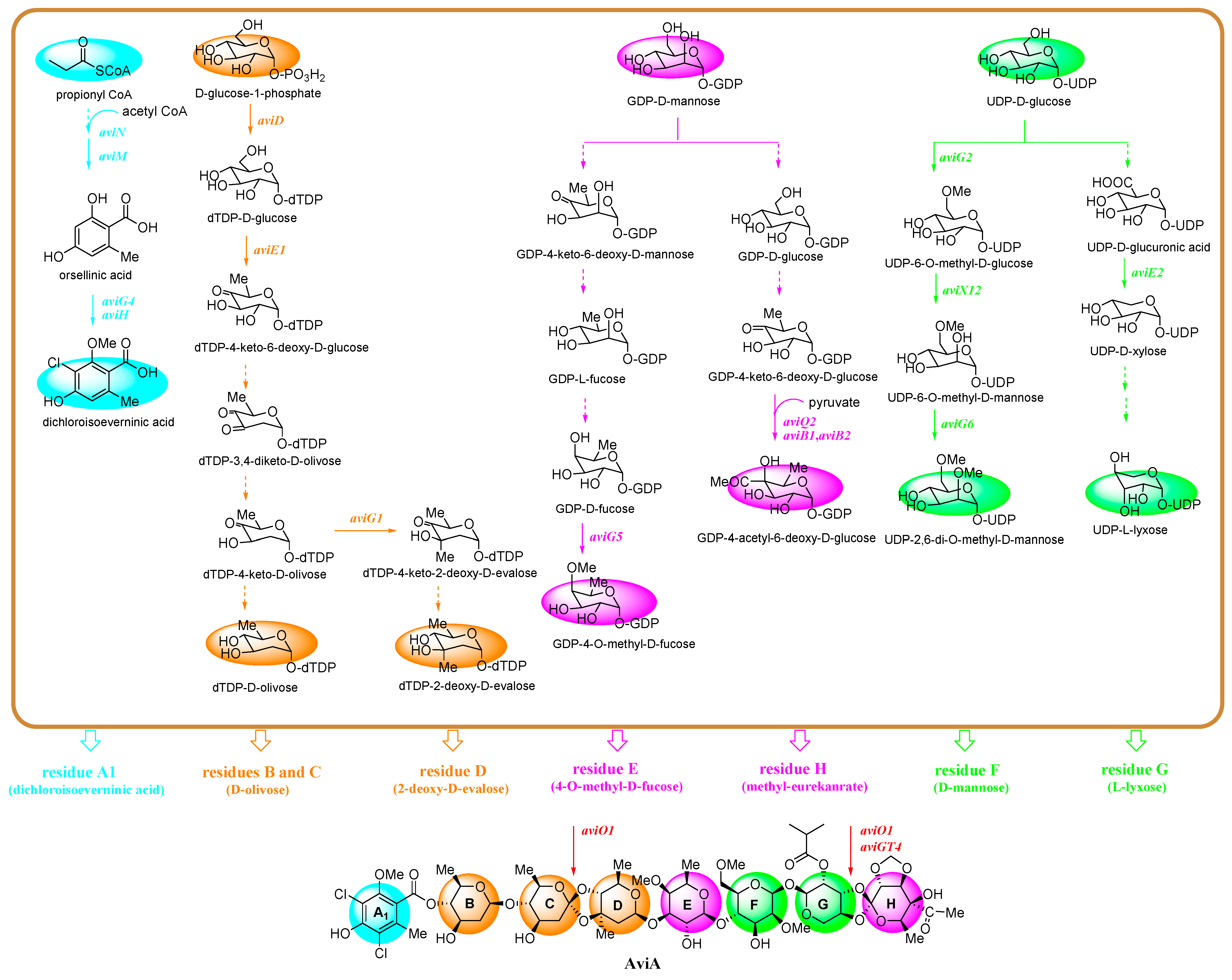
4.3. The Biosynthesis of Avi A
5. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cheong, K.L.; Qiu, H.M.; Du, H.; Liu, Y.; Khan, B.M. Oligosaccharides derived from red seaweed: Production, properties, and potential health and cosmetic applications. Molecules 2018, 23, 2451. [Google Scholar] [CrossRef]
- Elshahawi, S.I.; Shaaban, K.A.; Kharel, M.K.; Thorson, J.S. A comprehensive review of glycosylated bacterial natural products. Chem. Soc. Rev. 2015, 44, 7591–7697. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Raposo, M.F.; de Morais, A.M.B.; de Morais, R.M.S.C. Marine polysaccharides from algae with potential biomedical applications. Mar. Drugs 2015, 13, 2967–3028. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.W.; Ni, F.; Xiong, Q.; Yao, Z. Marine oligosaccharides originated from seaweeds: Source, preparation, structure, physiological activity and applications. Crit. Rev. Food Sci. Nutr. 2021, 61, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.K.; Howlader, P.; Wang, W.X.; Yin, H. Oligosaccharide is a promising natural preservative for improving postharvest preservation of fruit: A review. Food Chem. 2021, 341, 128178. [Google Scholar] [CrossRef] [PubMed]
- Truscheit, E.; Frommer, W.; Junge, B.; Müller, L.; Schmidt, D.D.; Wingender, W. Chemistry and biochemistry of microbial α-glucosidase inhibitors. Angew. Chem. Int. Ed. Engl. 1981, 20, 744–761. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Werz, D.B. Synthesis and medical applications of oligosaccharides. Nature 2007, 446, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, T. The C7N aminocyclitol family of natural products. Nat. Prod. Rep. 2003, 20, 137–166. [Google Scholar] [CrossRef]
- Carter, G.T. NP/MS since 1970: From the basement to the bench top. Nat. Prod. Rep. 2014, 31, 711–717. [Google Scholar] [CrossRef]
- Kersten, R.D.; Ziemert, N.; Gonzalez, D.J.; Duggan, B.M.; Nizet, V.; Dorrestein, P.C.; Moore, B.S. Glycogenomics as a mass spectrometry-guided genome-mining method for microbial glycosylated molecules. Proc. Natl. Acad. Sci. USA 2013, 110, E4407–E4416. [Google Scholar] [CrossRef]
- Lang, Y.Z.; Zhao, X.; Liu, L.L.; Yu, G.L. Applications of mass spectrometry to structural analysis of marine oligosaccharides. Mar. Drugs 2014, 12, 4005–4030. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Q.; Zeng, W.F.; Fang, P.; Cao, W.Q.; Liu, C.; Yan, G.Q.; Zhang, Y.; Peng, C.; Wu, J.Q.; Zhang, X.J.; et al. pGlyco 2.0 enables precision N-glycoproteomics with comprehensive quality control and one-step mass spectrometry for intact glycopeptide identification. Nat. Commun. 2017, 8, 438. [Google Scholar] [CrossRef]
- McCranie, E.K.; Bachmann, B.O. Bioactive oligosaccharide natural products. Nat. Prod. Rep. 2014, 31, 1026–1042. [Google Scholar] [CrossRef]
- Wang, L.Q.; Cui, Q.X.; Hou, Y.Y.; Bai, F.; Sun, J.X.; Cao, X.F.; Liu, P.; Jiang, M.; Bai, G. An integrated strategy of ultra-high-performance liquid chromatography/quadrupole-time-of-flight mass spectrometry and virtual screening for the identification of α-glucosidase inhibitors in acarviostatin-containing complex. J. Chromatogr. A 2013, 1319, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.D.; Frommer, W.; Junge, B.; Müller, L.; Wingender, W.; Truscheit, E.; Schäfer, D. α-Glucosidase inhibitors. New complex oligosaccharides of microbial origin. Naturwissenschaften 1977, 64, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Itoh, J.; Omoto, S.; Shomura, T.; Ogino, H.; Iwamatsu, K.; Inouye, S.; Hidaka, H. Oligostatins, new antibiotics with amylase inhibitory activity. I. Production, isolation and characterization. J. Antibiot. 1981, 34, 1424–1428. [Google Scholar] [CrossRef]
- Omoto, S.; Itoh, J.; Ogino, H.; Iwamatsu, K.; Nishizawa, N.; Inouye, S. Oligostatins, new antibiotics with amylase inhibitory activity. II. Structures of oligostatins C, D, and E. J. Antibiot. 1981, 34, 1429–1433. [Google Scholar] [CrossRef]
- Yokose, K.; Ogawa, K.; Sano, T.; Watanabe, K.; Maruyama, H.B.; Suhara, Y. New α-amylase inhibitor, trestatins. I. Isolation characterization and biological activities of trestatins A, B, and C. J. Antibiot. 1983, 36, 1157–1165. [Google Scholar] [CrossRef]
- Yokose, K.; Ogawa, K.; Suzuki, Y.; Umeda, I.; Suhara, Y. New α-amylase inhibitor, trestatins. II. Structure determination of trestatins A, B and C. J. Antibiot. 1983, 36, 1166–1175. [Google Scholar] [CrossRef]
- Yokose, K.; Ogawa, M.; Ogawa, K. New alpha-amylase inhibitor, trestatins. III. Structure determination of new trestatin components Ro 09-0766, Ro 09-0767 and Ro 09-0768. J. Antibiot. 1984, 37, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Namiki, S.; Kangouri, K.; Nagate, T.; Hara, H.; Sugita, K.; Omura, S. Studies on the alpha-glucoside hydrolase inhibitor, adiposin. I. Isolation and physicochemical properties. J. Antibiot. 1982, 35, 1234–1236. [Google Scholar] [CrossRef]
- Namiki, S.; Kangouri, K.; Nagate, T.; Hara, H.; Sugita, K.; Omura, S. Studies on the alpha-glucoside hydrolase inhibitor, adiposin. II. Taxonomic studies on the producing microorganism. J. Antibiot. 1982, 35, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Kangouri, K.; Namiki, S.; Nagate, T.; Hara, H.; Sugita, K.; Omura, S. Studies on the alpha-glucoside hydrolase inhibitor, adiposin. III. alpha Glucoside hydrolase inhibitory activity and antibacterial activity in vitro. J. Antibiot. 1982, 35, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Namiki, S.; Kangouri, K.; Nagate, T.; Hara, H.; Sugita, K.; Noda, K.; Tarumoto, Y.; Omura, S. Studies on the alpha-glucoside hydrolase inhibitor, adiposin. IV. Effect of adiposin on intestinal digestion of carbohydrates in experimental animals. J. Antibiot. 1982, 35, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Vertesy, L.; Betz, J.; Fehlhaber, H.W.; Geisen, K. Oxirane Pseudooligosaccharides, A Process for Their Preparation, Their Use and Pharmaceutical Preparations. U.S. Patent 4,990,500 A, 5 February 1991. [Google Scholar]
- Kim, J.G.; Chang, H.B.; Kwon, Y.I.; Moon, S.K.; Chun, H.S.; Ahn, S.K.; Hong, C.I. Novel alpha-glucosidase inhibitors, CKD-711 and CKD-711a produced by Streptomyces sp. CK-4416. I. Taxonomy, fermentation and isolation. J. Antibiot. 2002, 55, 457–461. [Google Scholar] [CrossRef][Green Version]
- Kwon, Y.I.; Son, H.J.; Moon, K.S.; Kim, J.K.; Kim, J.G.; Chun, H.S.; Ahn, S.K.; Hong, C.I. Novel alpha-glucosidase inhibitors, CKD-711 and CKD-711a produced by Streptomyces sp. CK-4416. II. Biological properties. J. Antibiot. 2002, 55, 462–466. [Google Scholar] [CrossRef]
- Chang, H.B.; Kim, S.H.; Kwon, Y.I.; Choung, D.H.; Choi, W.K.; Kang, T.W.; Lee, S.; Kim, J.G.; Chun, H.S.; Ahn, S.K.; et al. Novel alpha-glucosidase inhibitors, CKD-711 and CKD-711a produced by Streptomyces sp. CK-4416. III. Physico-chemical properties and structure elucidation. J. Antibiot. 2002, 55, 467–471. [Google Scholar] [CrossRef][Green Version]
- Geng, P.; Bai, G. Two novel aminooligosaccharides isolated from the culture of Streptomyces coelicoflavus ZG0656 as potent inhibitors of α-amylase. Carbohydr. Res. 2008, 343, 470–476. [Google Scholar] [CrossRef]
- Geng, P.; Qiu, F.; Zhu, Y.Y.; Bai, G. Four acarviosin-containing oligosaccharides identified from Streptomyces coelicoflavus ZG0656 are potent inhibitors of α-amylase. Carbohydr. Res. 2008, 343, 882–892. [Google Scholar] [CrossRef]
- Geng, P.; Sun, T.; Zhong, Q.P.; Li, X.X.; Shi, L.Y.; Bai, F.; Bai, G. Two novel potent α-amylase inhibitors from the family of acarviostatins isolated from the culture of Streptomyces coelicoflavus ZG0656. Chem. Biodivers. 2013, 10, 452–459. [Google Scholar] [CrossRef]
- Zhong, D.; Si, D.Y.; He, W.Y.; Zhao, L.M.; Xu, Q.M. Structural revision of isovalertatins M03, M13, and M23 isolated from the culture of Streptomyces luteogriseus. Carbohydr. Res. 2001, 331, 69–75. [Google Scholar] [CrossRef]
- Si, D.Y.; Zhong, D.F.; He, W.Y.; Zhao, L.M. Structural revision of isovalertatins D03 and D23 isolated from the culture filtrate of Streptomyces luteogriseus. Chin. Chem. Lett. 2001, 12, 327–330. [Google Scholar]
- Si, D.Y.; Zhong, D.F.; Xu, Q.M. Two butylated aminooligosaccharides isolated from the culture filtrate of Streptomyces luteogriseus. Carbohydr. Res. 2001, 335, 127–132. [Google Scholar] [CrossRef]
- Liu, H.L.; E, H.-C.; Xie, D.A.; Cheng, W.B.; Tao, W.Q.; Wang, Y. Acylated aminooligosaccharides with inhibitory effects against α-amylase from Streptomyces sp. HO1518. Mar. Drugs 2018, 16, 403. [Google Scholar] [CrossRef]
- Xu, J.L.; Liu, H.L.; Liu, Z.F.; Ren, Y.H.; Wang, Y. Acylated aminooligosaccharides from the yellow sea Streptomyces sp. HO1518 as both α-glucosidase and lipase inhibitors. Mar. Drugs 2020, 18, 576. [Google Scholar] [CrossRef]
- Geng, P.; Bai, G.; Shi, Q.; Zhang, L.; Gao, Z.; Zhang, Q. Taxonomy of the Streptomyces strain ZG0656 that produces acarviostatin α-amylase inhibitors and analysis of their effects on blood glucose levels in mammalian systems. J. Appl. Microbiol. 2009, 106, 525–533. [Google Scholar] [CrossRef]
- Li, C.M.; Begum, A.; Numao, S.; Park, K.H.; Withers, S.G.; Brayer, G.D. Acarbose rearrangement mechanism implied by the kinetic and structural analysis of human pancreatic alpha-amylase in complex with analogues and their elongated counterparts. Biochemistry 2005, 44, 3347–3357. [Google Scholar] [CrossRef]
- Qin, X.H.; Ren, L.M.; Yang, X.; Bai, F.; Wang, L.L.; Geng, P.; Bai, G.; Shen, Y.Q. Structures of human pancreatic α-amylase in complex with acarviostatins: Implications for drug design against type II diabetes. J. Struct. Biol. 2011, 174, 196–202. [Google Scholar] [CrossRef]
- Schwientek, P.; Szczepanowski, R.; Rückert, C.; Kalinowski, J.; Klein, A.; Selber, K.; Wehmeier, U.F.; Stoye, J.; Pühler, A. The complete genome sequence of the acarbose producer Actinoplanes sp. SE50/110. BMC Genom. 2012, 13, 112. [Google Scholar] [CrossRef]
- Ortseifen, V.; Kalinowski, J.; Pühler, A.; Rückert, C. The complete genome sequence of the actinobacterium Streptomyces glaucescens GLA.O (DSM 40922) carrying gene clusters for the biosynthesis of tetracenomycin C, 5′-hydroxy streptomycin, and acarbose. J. Biotechnol. 2017, 262, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Geng, P.; Bai, F.; Bai, G.; Sun, T.; Li, X.; Shi, L.; Zhong, Q. Draft genome sequence of Streptomyces coelicoflavus ZG0656 reveals the putative biosynthetic gene cluster of acarviostatin family α-amylase inhibitors. Lett. Appl. Microbiol. 2012, 55, 162–169. [Google Scholar] [CrossRef]
- Degwert, U.; van Hülst, R.; Pape, H.; Herrold, R.E.; Beale, J.M.; Keller, P.J.; Lee, J.P.; Floss, H.G. Studies on the biosynthesis of the alpha-glucosidase inhibitor acarbose: Valienamine, a m-C7N unit not derived from the shikimate pathway. J. Antibiot. 1987, 40, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, T.; Tornus, I.; Egelkrout, E.; Wolf, E.; Uy, C.; Floss, H.G.; Lee, S. Biosynthetic studies on the α-glucosidase inhibitor acarbose in Actinoplanes sp.: 2-epi-5-epi-valiolone is the direct precursor of the valienamine moiety. J. Am. Chem. Soc. 1999, 121, 6973–6983. [Google Scholar] [CrossRef]
- Stratmann, A.; Mahmud, T.; Lee, S.; Distler, J.; Floss, H.G.; Piepersberg, W. The AcbC protein from Actinoplanes species is a C7-cyclitol synthase related to 3-dehydroquinate synthases and is involved in the biosynthesis of the alpha-glucosidase inhibitor acarbose. J. Biol. Chem. 1999, 274, 10889–10896. [Google Scholar] [CrossRef]
- Zhang, C.S.; Stratmann, A.; Block, O.; Brückner, R.; Podeschwa, M.; Altenbach, H.J.; Wehmeier, U.F.; Piepersberg, W. Biosynthesis of the C7-cyclitol moiety of acarbose in Actinoplanes species SE50/110. 7-O-phosphorylation of the initial cyclitol precursor leads to proposal of a new biosynthetic pathway. J. Biol. Chem. 2002, 277, 22853–22862. [Google Scholar] [CrossRef]
- Zhao, Q.Q.; Luo, Y.C.; Zhang, X.; Kang, Q.J.; Zhang, D.; Zhang, L.L.; Bai, L.Q.; Deng, Z.X. A severe leakage of intermediates to shunt products in acarbose biosynthesis. Nat. Commun. 2020, 11, 1468. [Google Scholar] [CrossRef]
- Wehmeier, U.F.; Piepersberg, W. Biotechnology and molecular biology of the α-glucosidase inhibitor acarbose. Appl. Microbiol. Biotechnol. 2004, 63, 613–625. [Google Scholar] [CrossRef]
- Wehmeier, U.F. The biosynthesis and metabolism of acarbose in Actinoplanes sp. SE 50/110: A progress report. Biocatal. Biotransfor. 2003, 21, 279–284. [Google Scholar] [CrossRef]
- Rockser, Y.; Wehmeier, U.F. The gac-gene cluster for the production of acarbose from Streptomyces glaucescens GLA.O: Identification, isolation and characterization. J. Biotechnol. 2009, 140, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Meng, X.S.; Bai, G.; Luo, G.A. Profiling of acarviostatin family secondary metabolites secreted by Streptomyces coelicoflavus ZG0656 using ultraperformance liquid chromatography coupled with electrospray ionization mass spectrometry. Anal. Chem. 2008, 80, 7554–7561. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.M.; Zhao, N.; Siegel, M.M.; Janota, K.; Ashcroft, J.S.; Koehn, F.E.; Borders, D.B.; Carter, G.T. Saccharomicins, novel heptadecaglycoside antibiotics effective against multidrug-resistant bacteria. J. Am. Chem. Soc. 1998, 120, 13301–13311. [Google Scholar] [CrossRef]
- Singh, M.P.; Petersen, P.J.; Weiss, W.J.; Kong, F.; Greenstein, M. Saccharomicins, novel heptadecaglycoside antibiotics produced by Saccharothrix espanaensis: Antibacterial and mechanistic activities. Antimicrob. Agents Chemother. 2000, 44, 2154–2159. [Google Scholar] [CrossRef] [PubMed]
- Strobel, T.; Al-Dilaimi, A.; Blom, J.; Gessner, A.; Kalinowski, J.; Luzhetska, M.; Pühler, A.; Szczepanowski, R.; Bechthold, A.; Rückert, C. Complete genome sequence of Saccharothrix espanaensis DSM 44229T and comparison to the other completely sequenced Pseudonocardiaceae. BMC Genom. 2012, 13, 465. [Google Scholar] [CrossRef] [PubMed]
- Berner, M.; Krug, D.; Bihlmaier, C.; Vente, A.; Muller, R.; Bechthold, A. Genes and enzymes involved in caffeic acid biosynthesis in the actinomycete Saccharothrix espanaensis. J. Bacteriol. 2006, 188, 2666–2673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.F.; Mo, T.L.; Li, X.H.; Ding, W.; Zhang, Q. Dissection of the glycosylation in the biosynthesis of the heptadecaglycoside antibiotic saccharomicin A. J. Org. Chem. 2021. [Google Scholar] [CrossRef]
- Mann, R.L.; Bromer, W.W. The isolation of a second antibiotic from Streptomyces hygroscopicus. J. Am. Chem. Soc. 1958, 80, 2714–2716. [Google Scholar] [CrossRef]
- Wright, D.E. The Orthosomycins, a new family of antibiotics. Tetrahedron 1979, 35, 1207–1237. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Sarre, O.Z.; Greeves, D.; Morton, J. Structure of everninomicin D. J. Am. Chem. Soc. 1975, 97, 1982–1985. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, A.K.; Sarre, O.; Girijavallabhan, V.M. The structure of new oligosaccharide antibiotics, 13-384 components 1 and 5. Heterocycles 1989, 28, 83–88. [Google Scholar] [CrossRef]
- Saksena, A.K.; Jao, E.; Murphy, B.; Schumacher, D.; Chan, T.M.; Puar, M.S.; Jenkins, J.K.; Maloney, D.; Cordero, M.; Pramanik, B.D.; et al. Structure elucidation of SCH 49088, a novel everninomicin antibiotic containing an unusual hydroxylamino-ether sugar, everhydroxylaminose. Tetrahedron Lett. 2000, 39, 8441–8444. [Google Scholar] [CrossRef]
- Chu, M.; Mierzwa, R.; Jenkins, J.; Chan, T.; Das, P.; Pramanik, B.N.; Patel, M.; Gullo, V. Isolation and characterization of novel oligosaccharides related to Ziracin. J. Nat. Prod. 2002, 65, 1588–1593. [Google Scholar] [CrossRef]
- Mertz, J.L.; Peloso, J.S.; Barker, B.J.; Babbitt, G.E.; Occolowitz, J.L.; Simson, V.L.; Kline, R.M. Isolation and structural identification of nine avilamycins. J. Antibiot. 1986, 39, 877–887. [Google Scholar] [CrossRef]
- Nakashio, S.; Iwasawa, H.; Dun, F.Y.; Kanemitsu, K.; Shimada, J. Everninomicin, a new oligosaccharide antibiotic: Its antimicrobial activity, post-antibiotic effect and synergistic bactericidal activity. Drugs Exp. Clin. Res. 1995, 21, 7–16. [Google Scholar]
- Foster, D.R.; Rybak, M.J. Pharmacologic and bacteriologic properties of SCH27899 (Ziracin), an investigational antibiotic from the everninomicin family. Pharmacotherapy 1999, 19, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.J.; Mowrey, D.H.; Anderson, D.B.; Wellenreiter, R.H. Effect of various levels of avilamycin on the performance of growing-finishing swine. J. Anim. Sci. 1987, 65, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Shryock, T.R. Will avilamycin convert ziracine into zerocine? Emerg. Infect. Dis. 2001, 7, 488–489. [Google Scholar] [CrossRef]
- Arenz, S.; Juette, M.F.; Graf, M.; Nguyen, F.; Huter, P.; Polikanov, Y.S.; Blanchard, S.C.; Wilson, D.N. Structures of the orthosomycin antibiotics avilamycin and evernimicin in complex with the bacterial 70S ribosome. Proc. Natl. Acad. Sci. USA 2016, 113, 7527–7532. [Google Scholar] [CrossRef]
- Krupkin, M.; Wekselman, I.; Matzov, D.; Eyal, Z.; Posner, Y.D.; Rozenberg, H.; Zimmerman, E.; Bashan, A.; Yonath, A. Avilamycin and evernimicin induce structural changes in rProteins uL16 and CTC that enhance the inhibition of A-site tRNA binding. Proc. Natl. Acad. Sci. USA 2016, 113, E6796–E6805. [Google Scholar] [CrossRef]
- Limbrick, E.M.; Graf, M.; Derewacz, D.K.; Nguyen, F.; Spraggins, J.M.; Wieland, M.; Ynigez-Gutierrez, A.E.; Reisman, B.J.; Zinshteyn, B.; McCulloch, K.M.; et al. Bifunctional nitrone-conjugated secondary metabolite targeting the ribosome. J. Am. Chem. Soc. 2020, 142, 18369–18377. [Google Scholar] [CrossRef] [PubMed]
- Gaisser, S.; Trefzer, A.; Stockert, S.; Kirschning, A.; Bechthold, A. Cloning of an avilamycin biosynthetic gene cluster from Streptomyces viridochromogenes Tü57. J. Bacteriol. 1997, 179, 6271–6278. [Google Scholar] [CrossRef]
- Weitnauer, G.; Muhlenweg, A.; Trefzer, A.; Hoffmeister, D.; Sussmuth, R.D.; Jung, G.; Welzel, K.; Vente, A.; Girreser, U.; Bechthold, A. Biosynthesis of the orthosomycin antibiotic avilamycin A: Deductions from the molecular analysis of the avi biosynthetic gene cluster of Streptomyces viridochromogenes Tü57 and production of new antibiotics. Chem. Biol. 2001, 8, 569–581. [Google Scholar] [CrossRef]
- Weitnauer, G.; Gaisser, S.; Kellenberger, L.; Leadlay, P.F.; Bechthold, A. Analysis of a C-methyltransferase gene (aviG1) involved in avilamycin biosynthesis in Streptomyces viridochromogenes Tü57 and complementation of a Saccharopolyspora erythraea eryBIII mutant by aviG1. Microbiology 2002, 148, 373–379. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Treede, I.; Hauser, G.; Muhlenweg, A.; Hofmann, C.; Schmidt, M.; Weitnauer, G.; Glaser, S.; Bechthold, A. Genes involved in formation and attachment of a two-carbon chain as a component of eurekanate, a branched-chain sugar moiety of avilamycin A. Appl. Environ. Microbiol. 2005, 71, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Weitnauer, G.; Hauser, G.; Hofmann, C.; Linder, U.; Boll, R.; Pelz, K.; Glaser, S.J.; Bechthold, A. Novel avilamycin derivatives with improved polarity generated by targeted gene disruption. Chem. Biol. 2004, 11, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Boll, R.; Heitmann, B.; Hauser, G.; Clemens, D.; Frerich, A.; Weitnauer, G.; Glaser, S.J.; Bechthold, A. Genes encoding enzymes responsible for biosynthesis of L-lyxose and attachment of eurekanate during avilamycin biosynthesis. Chem. Biol. 2005, 12, 1137–1143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boll, R.; Hofmann, C.; Heitmann, B.; Hauser, G.; Glaser, S.; Koslowski, T.; Friedrich, T.; Bechthold, A. The active conformation of avilamycin A is conferred by AviX12, a radical AdoMet enzyme. J. Biol. Chem. 2006, 281, 14756–14763. [Google Scholar] [CrossRef]
- McCulloch, K.M.; McCranie, E.K.; Smith, J.A.; Sarwar, M.; Mathieu, J.L.; Gitschlag, B.L.; Du, Y.; Bachmann, B.O.; Iverson, T.M. Oxidative cyclizations in orthosomycin biosynthesis expand the known chemistry of an oxygenase superfamily. Proc. Natl. Acad. Sci. USA 2015, 112, 11547–11552. [Google Scholar] [CrossRef]
- Weitnauer, G.; Gaisser, S.; Trefzer, A.; Stockert, S.; Westrich, L.; Quiros, L.M.; Mendez, C.; Salas, J.A.; Bechthold, A. An ATP-binding cassette transporter and two rRNA methyltransferases are involved in resistance to avilamycin in the producer organism Streptomyces viridochromogenes Tü57. Antimicrob. Agents Chemother. 2001, 45, 690–695. [Google Scholar] [CrossRef]
- Rebets, Y.; Boll, R.J.; Horbal, L.; Fedorenko, V.; Bechthold, A. Production of avilamycin A is regulated by AviC1and AviC2, two transcriptional activators. J. Antibiot. 2009, 62, 461–464. [Google Scholar] [CrossRef]
- Limbrick, E.M.; Ynigez-Gutierrez, A.E.; Dulin, C.C.; Derewacz, D.K.; Spraggins, J.M.; McCulloch, K.M.; Iverson, T.M.; Bachmann, B.O. Methyltransferase contingencies in the pathway of everninomicin D antibiotics and analogues. ChemBioChem 2020, 21, 3349–3358. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Amino Acids | Identity/Similarity to the acb-Cluster | Proposed Function |
|---|---|---|---|
| SctZ2 | 1619 | 50%/63% homologous to AcbZ | α-amylase |
| SctZ1 | 1042 | 47%/59% homologous to AcbZ | α-amylase |
| SctE2 | 565 | 25%/39% homologous to AcbD | α-amylase |
| SctC2 | 352 | no homologue | LacI family transcriptional regulator |
| SctE1 | 553 | 25%/36% homologous to AcbE | α-glucosidase |
| SctG | 301 | 26%/51% homologous to AcbG | ABC transporter membrane protein |
| SctF | 335 | 29%/50% homologous to AcbF | ABC transporter membrane protein |
| SctH | 424 | 25%/37% homologous to AcbH | ABC transporter membrane protein |
| SctC1 | 342 | no homologue | malR-like regulator |
| SctB | 325 | 59%/70% homologous to AcbB | dTDP-glucose 4,6-dehydratase |
| SctA | 357 | 43%/59% homologous to AcbA | dTDP-glucose synthase |
| SctV | 417 | 75%/83% homologous to AcbV | dTDP-4-keto-6-deoxy-glucose 4-aminotransferase |
| SctW | 341 | 62%/77% homologous to AcbW | ABC transporter permease protein |
| SctX | 274 | 49%/68% homologous to AcbX | ABC transporter permease protein |
| SctY | 268 | 57%/75% homologous to AcbY | ABC transporter permease protein |
| SctU | 487 | 42%/51% homologous to AcbU | 1-epi-valienol-7-phosphate-1-kinase |
| SctS | 691 | 62%/71% homologous to AcbS | glycosyltransferase |
| SctR | 354 | 70%/80% homologous to AcbR | 1-epi-valienol-1,7-bisphosphate-1-adenylyltransferase |
| SctK | 308 | 48%/60% homologous to AcbK | acarbose-7-kinase |
| SctI | 1027 | 47%/59% homologous to AcbI | putative glycosyltransferase |
| SctQ | 702 | 55%/66% homologous to AcbQ | putative acarbose 4-alpha-glucanotransferase |
| SctC | 393 | 39%/52% homologous to AcbC | 2-epi-5-epi-valiolone synthase |
| SctJ | 138 | no homologue | 2-epimerase |
| SctM | 350 | 31%/43% homologous to AcbM | C7-cyclitol-7-kinase |
| SctO | 324 | no homologue | putative 2-epi-5-epi-valiolone dehydratase |
| SctT | 464 | no homologue | unknown |
| Scim | 112 | no homologue | α-amylase |
| Genes | Encoded Enzymes | Proposed Function |
|---|---|---|
| avi N | ketoacyl synthase III homologues | controlling the starter unit for orstainic acid biosynthesis |
| avi M | type I polyketide synthase | orstainic acid biosynthesis |
| avi D | dTDP-glucose synthase | starter enzyme for residues B, C, D biosynthesis |
| avi E1, aviS | dTDP-glucose-4,6-dehydratase | residues B, C, D biosynthesis |
| avi E2 | UDP-glucuronic acid decarboxylase | starter enzyme for residue G biosynthesis |
| avi E3 | GDP-mannose-4,6-dehydratase | starter enzyme for residue E biosynthesis |
| avi B1, avi B2 | pyruvate dehydrogenase (α2β2 chains) | residue H biosynthesis |
| avi G1 | C-methyltransferase | residue D biosynthesis |
| avi G5 | O-methyltransferase | C-4 methylation of residue E |
| avi G2, avi G6 | O-methyltransferase | C-2, C-6 methylation of residue F |
| avi G3 | O-methyltransferase | residue H methylation |
| avi G4 | O-methyltransferase | residue N methylation |
| avi Q1-avi Q3 | UDP-glucose-4-epimerase | epimerization of oligosaccharides |
| avi Z1 | ketoreductase | residues D and E synthesis |
| avi T | ketoreductase | residues B-D synthesis |
| avi Z3 | ketoreductase | residues B and C synthesis |
| avi Z2 | ketoreductase | residues D and E synthesis |
| avi H | halogenase | halogenated residue A |
| avi X12 | S-adenosylmethioninase | activated residues F and G |
| avi O1, avi O3 | hydroxylase | formation of orthoester bond and glycosidic bond |
| Avi O2 | hydroxylase | residue H biosynthesis |
| avi GT1-avi GT3 | glycosyltransferase | assembly of heptasaccharide chain |
| avi GT4 | glycosyltransferase | conjugation between residues G and H |
| avi C1, avi C2 | regulator | AVI positive regulator |
| avi Ra, avi Rb | rRNA methyltransferase | AVI resistance |
| avi ABC1, avi ABC2 | ABC transporter, ATP binding protein | AVI antibiotic transport |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.-L.; Liu, Z.-F.; Zhang, X.-W.; Liu, H.-L.; Wang, Y. Microbial Oligosaccharides with Biomedical Applications. Mar. Drugs 2021, 19, 350. https://doi.org/10.3390/md19060350
Xu J-L, Liu Z-F, Zhang X-W, Liu H-L, Wang Y. Microbial Oligosaccharides with Biomedical Applications. Marine Drugs. 2021; 19(6):350. https://doi.org/10.3390/md19060350
Chicago/Turabian StyleXu, Jian-Lin, Zhi-Feng Liu, Xiao-Wei Zhang, Hai-Li Liu, and Yong Wang. 2021. "Microbial Oligosaccharides with Biomedical Applications" Marine Drugs 19, no. 6: 350. https://doi.org/10.3390/md19060350
APA StyleXu, J.-L., Liu, Z.-F., Zhang, X.-W., Liu, H.-L., & Wang, Y. (2021). Microbial Oligosaccharides with Biomedical Applications. Marine Drugs, 19(6), 350. https://doi.org/10.3390/md19060350